
Fig. 3.
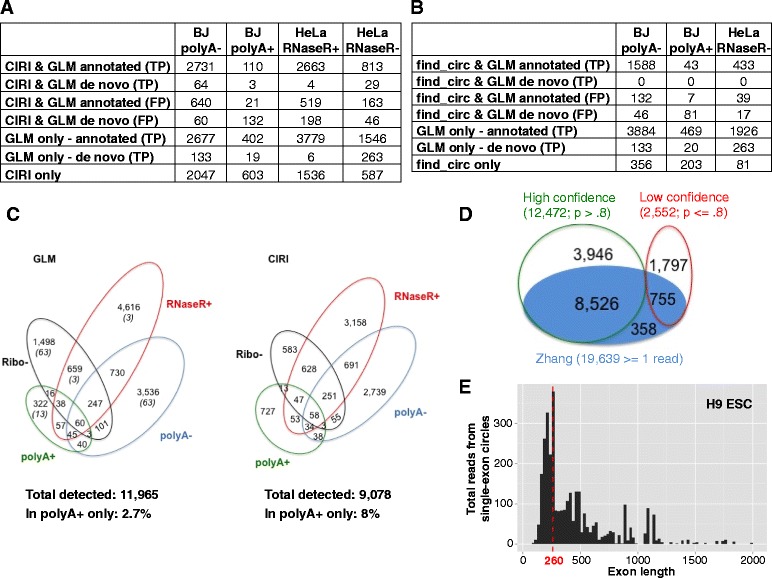
Statistical algorithm improves the sensitivity of circular RNA detection. a, b Circular RNA detected by both algorithms are divided into false positives (FP; flagged as false positives due to low posterior probability) or true positives (TP; our posterior probability ≥ 0.9). a Number of circular RNAs detected by our GLM or CIRI in ENCODE BJ poly(A)+/− data and HeLa RNase-R+/− data generated by Gao et al. [23]. CIRI results are based on all default parameters except the -E flag set to exclude false positives resulting from identical colinear exons. b Number of circular RNAs detected by our GLM or find_circ in ENCODE BJ poly(A)+/− data and HeLa RNase-R- data generated by Gao et al. [23]. c Circular RNAs detected in HeLa RNase-R+ and Ribo- data generated by Gao et al. [23] and poly(A)+, and poly(A)- data generated by ENCODE. Number of circular RNAs detected by our GLM method (one or more reads, posterior probability ≥ 0.9) compared with CIRI (default parameters except -E). For GLM results, the first number is the total number of circles and the number of those which were detected by the de novo portion of the algorithm are listed in parentheses. d Venn diagram comparing the number of putative circular RNAs identified by our annotation-dependent algorithm in Rnase-R-treated H9 cells and the results published by Zhang et al. [22]. Green circles and red circles show circular RNA identified by our algorithm with high and low confidence, respectively; the blue circle shows those identified by Zhang et al. e Total junctional reads for circles comprised of a single exon (posterior probability ≥ 0.9, read count > 1) shown by size for same data as in panel (d). Median exon length is shown in red. The x-axis is truncated at 2000 excluding 31 long exons, all but one with total read counts < 50