

Abstract
Fluorescent semiconductor nanoparticles, or quantum dots, have become a promising platform for the engineering of biofunctional probes for a variety of biomedical applications, ranging from multicolor imaging to single-molecule tracking to traceable drug delivery. Advances in organometallic synthesis have enabled preparation of hydrophobic quantum dots with high quantum yields and narrow size distribution, offering bright optical materials with narrow size-tunable emission profiles. At the same time, polymer encapsulation procedures provided a simple and versatile methodology for transferring hydrophobic nanoparticles into physiologically-relevant aqueous buffers. Taken together, hydrophobic nanoparticle platforms and polymer encapsulation should offer great flexibility for implementation of novel probe designs. However, the success of the encapsulation and purification depends on many factors often overlooked in the scientific literature, such as close match between nanoparticle and polymer physicochemical properties and dimensions, slow dynamics of polymer arrangement on the nanoparticle surface, and the size and charge similarity of resultant polymer-coated quantum dots and empty byproduct polymer micelles. To make this general hydrophobic nanoparticle modification strategy accessible by a broad range of biomedical research groups, we focus on the important technical aspects of nanoparticle polymer encapsulation, purification, bioconjugation, and characterization.
Keywords: quantum dot, polymer encapsulation, bioconjugation, fluorescence, nanoparticle
1. Introduction
Advances in bio-nanotechnology are introducing novel nanoscale materials with unique chemical and physical features potentially useful for advancing existing and creating new biomedical applications. Quantum dots (QDots), fluorescent semiconductor nanoparticles introduced to biomedical research nearly two decades ago,[1] have catalyzed development of such directions as single-cell molecular profiling,[2,3] real-time molecule tracking,[4] in vivo molecular imaging,[5] and traceable drug delivery.[6,7] This rich functionality stems from a number of unique photo-physical and chemical properties possessed by QDots. Most notably, narrow size-tunable emission profiles featured by nanoparticles of the same composition, efficient light absorption over a broad spectral range, outstanding photostability, and relatively small size comparable to that of large proteins make QDots a versatile and resourceful imaging probe for examination of biological systems.[8]
Despite a number of attractive features and innovative proof-of-concept studies published to date, QDot technology has made little impact on biomedical discoveries. One factor contributing to the lack of technology adoption is complexity of QDot probe engineering and preparation. A number of water-soluble QDots currently available from commercial sources offer a simple off-the-shelf solution to this issue, but only cover basic imaging and detection applications and often prove sub-optimal for implementation of custom probe designs and development of novel methodologies. In this regard, high-quality QDots synthesized via organometallic procedure[9] in non-polar solvents and stabilized with hydrophobic surface ligands represent a more versatile platform. The hydrophobic nature makes such nanoparticles incompatible with biologically-relevant assay conditions and requires further surface modification to render nanoparticles water-soluble. One approach, polymer encapsulation,[10,11] provides a desirable probe design flexibility, as custom hydrophilic coatings can be tailored to specific parameters and applications. However, many important aspects of QDot probe preparation have not been well described. In particular, non-intuitive size and charge similarity between polymer-encapsulated QDots and byproduct empty polymer micelles complicates probe purification and downstream application.
Given the lack of expertise working with nanoparticles in the biomedical research community, further discussion is warranted. To facilitate implementation of novel QDot probes by a broad range of biomedical research groups, we highlight critical steps in probe preparation, purification, bioconjugation, characterization, and purity control, which are often overlooked in the scientific literature.
2. Results and Discussion
2.1. Preparation of Water-Soluble QDots
Hydrophobic QDots were rendered hydrophilic via encapsulation with an amphiphilic polymer poly(maleic anhydride-alt-1-tetradecene) (PMAT, MW=9,000 Da), a robust nanoparticle polymer encapsulation procedure described by Pellegrino et al.[11] The general procedure consisted of three main steps (Figure 1): polymer encapsulation of hydrophobic QDots with PMAT, cross-linking of a portion of the maleic anhydrides in the polymer shell, and rendering particles hydrophilic via hydrolysis of the remaining maleic anhydride moieties into negatively-charged carboxylic acid groups. Through application of this procedure for the preparation of stable water-soluble QDots, several key technical aspects lacking detailed examination in the scientific literature were identified.
Figure 1.
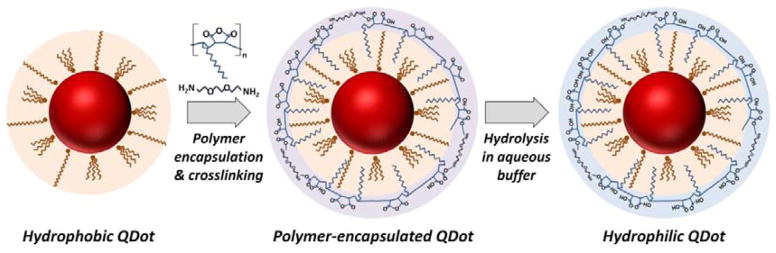
Schematic of QDot polymer encapsulation procedure. Bright monodisperse QDots are stabilized by organic surface ligands, which render nanoparticles hydrophobic. Incubation of such nanoparticles with a size-matched amphiphilic polymer drives the polymer self-assembly onto QDot surface via hydrophobic interaction of QDot surface ligands and polymer aliphatic side chains. Spontaneous reaction between maleic anhydride groups and di-amine compounds cross-links adjacent polymer chains together, yielding a stable coating. Finally, hydrolysis of the remaining anhydride groups in an aqueous buffer produces a highly negatively-charged hydrophilic shell around the nanoparticle optically active core, which is protected from the environment by a hydrophobic bilayer.
Polymer encapsulation was achieved by dissolving hydrophobic QDots in chloroform, mixing with excess of PMAT, and evaporating the solvent. Up to 10% methanol was added to aid in dissolution of the polymer anhydride groups and promote QDot-PMAT self-assembly upon chloroform evaporation. Consisting of alternating aliphatic side-chains on a maleic anhydride backbone, PMAT self-assembled onto the hydrophobic QDot core as the polarity of the solvent increased. As complex dynamic self-assembly of a linear polymer on a 3-dimensional nanoparticle surface had to occur for proper encapsulation, extended reaction time and slow solvent evaporation were beneficial. In contrast to prevailing practice of employing sonication for enhanced nanoparticle/polymer mixing, we determined this technique inappropriate for handling of hydrophobic QDots at any stage of the polymer encapsulation procedure, as even brief treatment in a bath sonicator led to severe loss of QDot fluorescence (Figure S1) and subsequent particle aggregation. We attribute this effect to the disruption of labile small-ligand passivation layer on the QDot surface and exposure of surface trap sites,[12] which lead to fluorescence quenching, nanocrystal degradation, and loss of colloidal stability. Brief mild mixing proved more suitable for preparation of polymer-coated QDots.
Cross-linking of individual polymer chains on the QDot surface was performed via a spontaneous reaction between anhydride groups and a diamine cross-linker 2,2′-(Ethylenedioxy)bis(ethylamine). PMAT shell cross-linking was determined to be an essential step in preparation of stable water-soluble QDots amenable to a wide range of conditions used for further bioconjugation and bioassays. While polymer encapsulation without cross-linking could also produce water-soluble particles, downstream chemical modification of such particles often resulted in severe aggregation due to exposure of hydrophobic patches and unbound PMAT chains on the QDot surface. Cross-linking of individual polymer chains together presumably formed a more stable network-like shell around each nanoparticle, preventing dissociation of polymer chains and providing a more complete coverage of the hydrophobic surface.
Rendering of polymer-coated QDots water-soluble was achieved by hydrolysis of maleic anhydride groups in an aqueous buffer with basic pH (50 mM Borate buffer, pH 8.5). Buffers with neutral and acidic pH failed to provide a sufficient rate of hydrolysis, driving slow dissolution of QDots with hydrophobic surface patches into an aqueous environment, which eventually led to QDot aggregation. A similar effect was also observed with highly basic buffers when quick dissolution of polymer-coated QDots was forced by rigorous vortexing or sonication, likely due to insufficient time provided for complete hydrolysis of anhydride groups and proper re-arrangement of the polymer on the QDot surface. Therefore, slow QDot-PMAT dissolution in a basic aqueous buffer under mild shaking was determined to be the only acceptable route for preparation of well-stabilized single QDots. Following complete hydrolysis and dissolution polymer-coated QDots could be successfully transferred to variety of aqueous buffers ranging from pH 4 to pH 9, offering good stability in common bio-conjugation procedures and bioassays.
Retention of the native hydrophobic surface coating beneath the polymer shell proved beneficial for shielding of the nanoparticle core from the aqueous environment and preservation of the QDot optical properties. Spectrophotometry and fluorometry analysis confirmed the unperturbed light absorption and photoluminescence QDot characteristics (Figure 2a). Notably, PMAT-coated QDots typically featured nearly unchanged quantum yield (QY) of 50–55%, as compared to 60% QY recorded for hydrophobic QDots in chloroform.
Figure 2.

Characterization of polymer-encapsulated QDots. (a) Polymer encapsulation successfully preserved QDot optical properties, yielding consistent light absorbance (solid blue and red lines) and fluorescence emission (dotted blue and red lines) profiles between stock QDots in chloroform and polymer-encapsulated QDots in an aqueous buffer, respectively. Normalized absorbance and emission profiles are shown. (b) Stock hydrophobic QDots analyzed by TEM featured an average core diameter of 5–6 nm, which matched well the native PMAT micelle size in an aqueous buffer of about 5.5 nm measured by DLS (c, red curve). Interestingly, PMAT dissolved in chloroform failed to produce consistent DLS measurements (c, blue curve), likely due to lack of stable micelle formation in a non-polar solvent. (d) DLS analysis of polymer-encapsulated QDots confirmed good particle stability in an aqueous buffer and lack of aggregates, measuring a hydrodynamic size of PMAT-coated QDots of about 13.3 nm (solid line) and cross-linked PMAT-coated QDots of about 15.3 nm (dotted line).
Preparation of single stable QDots via the polymer encapsulation approach relies on the selection of a compatible polymer. In addition to amphiphilic surfactant-like properties, such polymers should feature a suitable hydrophobic interface for a strong non-disrupting interaction with QDot surface ligands and have physical dimensions consistent with a nanoparticle size. PMAT satisfied these requirements when used for encapsulation of hydrophobic QDots stabilized by aliphatic surface ligands (such as trioctylphosphine oxide, TOPO, and octadecylphosphonic acid, ODPA, used here[13]). In particular, the 12-carbon atom side-chains of PMAT matched well the 8-carbon chains of TOPO and 18-carbon chains of ODPA, forming a tight hydrophobic interface via chain intercalation when exposed to an aqueous environment. At the same time, the nanoparticle physical dimensions (about 5–6 nm core size for red-emitting QDots studied here, Figure 2b) were matched by the native PMAT micelle size (Figure 2c), facilitating natural arrangement of the polymer around single nanoparticles. Resulting water-soluble particles featured a hydrodynamic diameter of about 13–15 nm (Figure 2d), which was consistent with the expected size of single polymer-coated QDots. Additionally, high-magnification fluorescence microscopy was employed to verify the absence of QDot aggregation, as single QDots can be clearly identified by their characteristic “blinking” behavior[14] (i.e. single QDots exhibit intermittency in fluorescence emission upon continuous illumination, while QDot aggregates on average stay always “on”). In contrast, larger polymers with a similar chemical structure (such as commonly used poly(maleic anhydride-alt-1-octadecene), PMAO, Mn=30,000–50,000 Da) often bridge multiple nanoparticles together, producing small QDot clusters, rather than individual polymer-coated QDots, and, therefore, might represent a suitable material of choice for encapsulation of larger nanoparticles.
2.2. QDot Purification and Purity Control
QDot probe engineering often involves further surface modification and bioconjugation. Therefore, the polymer-coated QDot solution must be completely purified of excess polymer and cross-linker. Surprisingly, purification methods based on separation by size commonly referred to in the scientific literature, such as fractionation with Superdex-75 gel column and ultrafiltration with 100,000 Da molecular weight cut-off concentrators, failed to remove excess polymer from the sample, even though such methods were believed to be suitable for removing relatively small polymer molecules. Instead, ultra-centrifugation was found to be the most suitable method for preparation of highly pure QDot-PMAT samples. The substantially higher density of the nanoparticle inorganic core led to efficient segregation of QDots into a soft pellet upon ultra-centrifugation at 45,000 rpm for 60 minutes, leaving excess polymer and cross-linker in the supernatant, which could then be easily discarded.
Unexpectedly poor performance of conventional purification methods based on fractionation by size and/or charge might be explained by the spontaneous formation of stable high-molecular weight polymer micelles with physicochemical characteristics similar to those of PMAT-coated QDots (Figure 3a). In fact, gel electrophoresis and dynamic light scattering (DLS) studies appeared to corroborate this conclusion. When examined with agarose gel electrophoresis, free PMAT traveled slightly faster, yet very closely to PMAT-coated QDots (Figure 3b), consistent with the expectation that both species have similar surface charge density and the smaller hydrodynamic size of PMAT micelles (Figure 2c,d). Similarly, DLS analysis of non-purified QDot-PMAT samples yielded size distribution curves dominated by PMAT micelles and failed to accurately determine QDot size (Figure 3c).
Figure 3.

Purification of polymer-coated QDots and purity control strategies. (a) Polymer encapsulation procedure produces polymer-coated QDots along with an excess of “empty” polymer micelles, which feature surface functional groups similar to QDots and, therefore, compete with polymer-coated QDots in downstream bioconjugation and bioassays. Purification strategies taking this mimicry into account are required. (b) Detection of free PMAT micelles with agarose gel electrophoresis. Featuring similar surface charge density, but smaller size, PMAT traveled slightly faster than QDots. Presence of PMAT was detected in a non-purified sample by staining with SYBR Gold (lane 2), whereas purified QDots (lane 1) lacked a PMAT band. (c) Empty PMAT micelles also shifted DLS size distribution of a non-purified sample toward smaller size, hampering accurate measurement of nanoparticle size. Typical PMAT micelle and QDot sizes are indicated by green and red arrows, respectively. (d) Optical properties of PMAT in chloroform (blue curves) and an aqueous 50 mM Borate buffer (red curves). Polymer showed absorbance of light in 300–400 nm range (solid lines) and blue fluorescence emission (dotted lines) peaking at 430 nm when excited at 350 nm. This feature proved instrumental in detecting free PMAT and evaluating the QDot purity. While fluorescence spectrum of polymer-coated QDots remained unchanged after purification (e), clearly detectable PMAT fluorescence peak in a non-purified sample disappeared following purification (f). Interestingly, PMAT fluorescence intensity also dropped following deposition onto QDots (f, blue vs. purple curve), indicating suppression of the PMAT fluorescence by QDots in close proximity.
It should be noted, that QDots could be readily detected and tracked by the bright red fluorescence upon illumination with a UV or blue-light lamp, yet weak PMAT fluorescence did not permit direct observation of the contaminating polymer in the QDot sample, further hindering assessment of sample purity. We identified SYBR Gold Nucleic Acid stain, commonly used for DNA and RNA staining, as a suitable reporter for detection of PMAT. Analogous to nucleic acid labeling, SYBR Gold produced bright green fluorescence upon binding to hydrolyzed PMAT, presumably due to electrostatic interaction with a negatively-charged polymer backbone. As a result, this methodology proved instrumental for a straightforward analysis of QDot sample purity with gel electrophoresis, a technique widely accessible to biomedical research laboratories.
Fluorometry was employed as a more sensitive tool for QDot purity control purposes. In developing this method, we took advantage of PMAT producing weak, yet characteristic fluorescence peak at 430 nm when excited by a 350 nm source (Figure 3d). Since fluorescence of the polymer absorbed onto QDot surface was suppressed, free contaminating PMAT could be selectively detected with sensitivity down to 10 μM in both non-polar solvents and aqueous buffers. At the same time, spectral separation of PMAT and QDot fluorescence peaks allowed for accurate polymer detection in a QDot/PMAT sample, despite QDots featuring 4–5 orders of magnitude brighter photoluminescence under identical concentration and measurement parameters. Using the fluorometry-based purity control strategy we confirmed the preservation of QDot optical properties (fluorescence intensity and profile) before and after purification (Figure 3e), detected presence of a free PMAT peak in a non-purified QDot-PMAT sample, and demonstrated its complete disappearance after proper purification via ultra-centrifugation (Figure 3f).
2.3. Shielding of the QDot Surface Charge
Biological assays often involve analysis of complex specimens featuring a wide range of molecular interactions. As a result, high negative surface charge might hamper use of PMAT-coated QDots in such applications. For example, fluorescence imaging studies often involve labeling of molecular targets in complex biological specimens, such as fixed cells and tissues, and require access to intracellular as well as cell-surface targets. Such access can be achieved by permeabilization of fixed specimens with charged (e.g. DTAC and SDS) and non-ionic (e.g. Triton X-100 and Tween-20) detergents, but removal of natural lipid barriers eliminates the negative charge on the cell surface and opens access to a variety of electrostatic and hydrophobic interactions, rendering PMAT-coated QDots inapplicable for staining of fully processed specimens. This issue is commonly resolved by the deposition of a layer of non-fouling material (most often poly(ethylene glycol), PEG) for shielding of the negatively-charged QDot surface,[15] which, however, results in the increased nanoparticle size and impeded bioconjugation efficiency. Zwitterionic QDot surface coatings,[16] on the other hand, impede off-target binding to the biological specimens by creating thin richly hydrated shells, thus offering a potentially superior surface modification strategy. Yet, implementation of zwitterionic surface coatings via polymer encapsulation procedure has not been examined in details.
To produce QDot-PMAT particles with a zwitterionic surface we took advantage of the high density of carboxylic acid groups on the nanoparticle surface, which served as anchors for covalent conjugation of a number of ligands containing primary amines. Specifically, a fraction of carboxylic acid groups was converted to positively-charged quaternary amine moieties via EDC-mediated conjugation with (2-Aminoethyl) trimethylammonium chloride (Figure S2a), yielding nanoparticles with an overall neutral or slightly negative charge (Figure S2b). Due to excellent stability of cross-linked QDot-PMAT particles, high excess of reagents and extended incubation time could be used to achieve nearly 50% modification yield in an aqueous buffer. Over-modification with charge inversion was rarely observed, indicating that the reaction could self-terminate upon reaching a certain level of modification, likely due to steric hindrance and electrostatic repulsion between positively-charged quaternary amines. In contrast to unmodified PMAT-coated QDots, which exhibited strong off-target binding to detergent-treated formalin-fixed cells (Figure S2c), zwitterionic particles featured a dramatically reduced interaction with the specimen (Figure S2d), thus presenting a more optimal nanoparticle platform for this application.
In an alternative approach, we adapted the PMAT encapsulation procedure to a zwitterionic amphiphilic polymer PMAL-C8 (poly(maleic anhydride-alt-1-decene) substituted with 3-(dimethylamino)propylamine), which featured not only a pre-modified zwitterionic surface, but also shorter C8 hydrophobic side-chains (Figure S3a), yielding compact deposition of the polymer on the QDot surface and producing stable neutrally-charged particles without further cross-linking and modification (Figure S3b). Preparation of stable PMAL-coated QDots relied on the close structure and size similarity between the two polymers. Featuring nearly identical native micelle dimensions (Figure S3c), both PMAT and PMAL-C8 produced hydrophilic QDots with closely matching hydrodynamic size distributions (Figure S3d). Interestingly, zwitterionic nanoparticles exhibited outstanding colloidal stability in polar organic solvents, such as 100% DMF and DMSO, enabling exploration of alternative bioconjugation procedures in a water-free environment. Consistent with the expected non-fouling behavior of zwitterionic surfaces, PMAL-coated QDots demonstrated only a minimal off-target binding to detergent-treated formalin-fixed cells (Figure S3e), providing an alternative platform for preparation of hydrophilic QDots for cell and tissue staining applications.
2.4. Preparation of Universal Biofunctional QDot Probes
PMAT-coated QDots provide a stable and flexible platform for preparation of biofunctional probes via direct covalent conjugation of amine-containing ligands to carboxylic acid groups. Despite the limitations discussed above, negatively-charged nanoparticles have demonstrated utility in a variety of applications, including immunoassays,[17] fluorescence resonance energy transfer (FRET) studies,[18] and live cell imaging,[7,19] producing target-specific labeling on predominantly uncharged or negatively charged specimens. Target recognition functionality is imparted by decorating QDots with targeting moieties, most commonly immunoglobulin G (IgG) antibodies. Covalent QDot-IgG bioconjugation routinely employed for this purpose,[3,20] however, often proves cost-prohibitive and complex. Instead, universal biofunctional QDot probes featuring a straightforward methodology and reduced costs might be more adaptable for a broad range of biomedical researchers. Here, we employed covalent conjugation with protein G (PrG) to make QDots capable of binding variety of intact antibodies via PrG/antibody self-assembly[21] for on-demand preparation of targeted QDot-antibody probes (Figure 4a).
Figure 4.

Bioconjugation of polymer-encapsulated QDots. (a) Schematic of QDot-PrG covalent bioconjugation and QDot-PrG-IgG non-covalent self-assembly procedure for preparation of targeted QDot probes. (b–d) Characterization of quantum dot biofunctionalization with agarose gel electrophoresis. (b) Covalent conjugation of highly negatively-charged QDots with uncharged SA (lane 3) and PrG (lane 4) resulted in reduction in QDot gel motility. Interestingly, agarose gel produced distinct “ladder” for QDots with different number of SA molecules attached in lane 3, but failed to resolve bioconjugation stoichiometry for QDot-PrG conjugates, which migrated as a single broader band in lane 4. Reference unconjugated QDots were loaded to lane 1, and control QDots treated with EDC alone were loaded to lane 2. (c) Bioconjugation with increasing PrG:QDot excess from 1:1 (lane 2) to 5:1 (lane 3), 10:1 (lane 4), and 20:1 (lane 5) deposited increasing number of PrG molecules onto each QDot, as evident from slow-down in gel motility of QDot-PrG bioconjugates. Reference unconjugated QDots were loaded to lane 1. (d) QDot functionalization with an IgG via PrG-IgG binding further reduced QDot-PrG-IgG probe motility (lane 3) in comparison to QDot-PrG probes (lane 2) and unconjugated QDots (lane 1). Loading well position is indicated by blue arrowhead on the left. Migration of QDots in the gel is indicated by an arrow on the right.
Universal QDot probes were prepared by covalently linking adaptor PrG molecules to the surface of PMAT-coated QDots at various PrG-to-QDot ratios. The abundance of carboxylic acid groups facilitates straightforward EDC-mediated deposition of variety of proteins onto QDot surface under gentle reaction conditions in aqueous buffers (Figure 4b). Importantly, even slight molar excess of PrG-to-QDot of 5:1 produced PrG-decorated nanoparticles (Figure 4c), reducing waste of reagents and aiding in probe purification. Further increase in molar excess of PrG in a reaction mixture directly translated into increased number of PrG molecules conjugated to each QDot, as could be observed from the slower migration of QDot-PrG probes in agarose gel (Figure 4c). At the same time, excessive QDot surface decoration by PrG led to an undesirable increase of the probe hydrodynamic size (up to 30 nm in diameter) and, thus, was avoided. Antibody-binding functionality of the universal QDot-PrG probes was confirmed by QDot self-assembly with rabbit anti-mouse secondary antibodies and detection of the larger QDot-PrG-IgG assembly by gel electrophoresis (Figure 4d).
Biofunctionality of the universal QDot-PrG probes was evaluated by immunofluorescence labeling of the clinically-relevant cell surface target, prostate-specific membrane antigen (PSMA), on PSMA-positive formalin-fixed prostate cancer LNCap cells (Figure 5). The specimen was first incubated with primary mouse anti-PSMA antibodies, which specifically recognized this protein on the cell surface. Then, cell-bound primary antibodies were labeled by QDots decorated with anti-mouse secondary antibodies for examination with fluorescence microscopy. Test QDot probes were produced by self-assembly between PMAT-coated QDot-PrG bioconjugates and unmodified rabbit anti-mouse IgG via simple mixing prior to cell labeling. The reference sample was labeled with commercial PEGylated QDots covalently functionalized with goat anti-mouse F(ab′)2 fragments. Notably, both test self-assembled QDot-PrG-IgG probes (Figure 5a) and reference covalent QDot-F(ab′)2 conjugates (Figure 5b) produced identical cell membrane staining characteristic for PSMA cellular localization. At the same time, neither QDot-PrG-IgG (Figure 5c) nor reference QDot-F(ab′)2 (Figure 5d) probes produced any detectable off-target binding in control specimens (not treated by PSMA-targeting primary antibodies), confirming high staining specificity. Cell staining results highlighted the utility of universal PMAT-coated QDot-PrG probes for specific labeling of surface targets in formalin-fixed non-permeabilized cells despite having a negatively-charged nanoparticle surface, expanding the scope of potential applications employing QDot-based detection and imaging.
Figure 5.

Labeling of cell surface targets with QDot-PrG-IgG probes. Prostate cancer LNCap cells were grown on glass coverslides and briefly fixed with formaldehyde to preserve fragile cell surface proteins. Cells were first treated with primary mouse anti-PSMA antibodies and then incubated with either QDot-PrG probes pre-assembled with rabbit anti-mouse antibodies (a) or reference anti-mouse QDot-2′Ab conjugates (b). Both probes successfully produced characteristic cell membrane staining. At the same time, control specimens not treated with a primary antibody lacked any non-specific staining by either self-assembled anti-mouse QDot-PrG-IgG (c) or reference anti-mouse QDot-2′Ab probes (d). Scale bar, 50 μm.
3. Conclusion
The availability of a stable and versatile biofunctional QDot platform is essential for successful probe development satisfying design criteria of existing and emerging biomedical applications. One version of such a platform presented here employs a robust polymer-encapsulation methodology supplemented by surface stabilization and modification strategies, offering a simple general approach to preparation of water-soluble nanoparticles. Further QDot decoration with an adaptor protein, such as protein G, converts “inert” nanoparticles into universal fluorescent probes, which upon self-assembly with primary antibodies yield target-specific probes applicable for molecular detection and imaging in a variety of bioassays. The success of the overall procedure depends on many factors often overlooked in the scientific literature, such as close match between QDot and polymer physicochemical properties and dimensions, gentle handling of unstable hydrophobic nanoparticles, slow dynamics of polymer arrangement on the nanoparticle surface, and use of purification methods based on fractionation by particle weight rather than size or charge (as these parameters are similar between polymer-coated nanoparticles and empty polymer micelles). In-depth examination of these technical aspects should make QDot probe preparation broadly accessible to a biomedical research community and facilitate development of novel bioassays employing unique features of the QDot technology.
4. Experimental Section
QDot Encapsulation with PMAT
Hydrophilic biofunctional QDots were prepared from high-quality hydrophobic CdSe/CdS/ZnS nanoparticles obtained from a commercial source, Ocean Nanotech. QDot powder was dissolved in chloroform by a mild agitation in the dark to a final concentration of 1–5 μM. Due to labile nature of surface ligands on hydrophobic nanoparticles, sonication and prolonged exposure to light were avoided. An amphiphilic polymer poly(maleic anhydride-alt-1-tetradecene) (PMAT, MW=9,000 Da, Sigma-Aldrich) was dissolved in 9:1 chloroform/methanol to a final concentration of 10 mM. Polymer encapsulation was performed by combining QDot solution with a 1,000 molar excess of PMAT, mixing by mild vortexing, and slowly drying overnight under mild vacuum. Then, dry QDot/PMAT film was dissolved in 9:1 chloroform/methanol, mixed with 1,000 molar excess of a cross-linker (2,2′-(Ethylenedioxy)bis(ethylamine), Sigma Aldrich), and allowed to slowly dry under mild vacuum overnight. Finally, cross-linked QDot/PMAT film was slowly dissolved in 50 mM Borate buffer (pH 8.5) under mild agitation, producing highly negatively-charged water-soluble nanoparticles.
QDot Encapsulation with PMAL-C8
Hydrophilic QDots with a zwitterionic surface were prepared by encapsulation of hydrophobic nanoparticles with an amphiphilic polymer poly(maleic anhydride-alt-1-decene) substituted with 3-(dimethylamino)propylamine (PMAL-C8, MW=18,500 Da, Anatrace). Hydrophobic QDot powder was dissolved in chloroform by a mild agitation in the dark to a final concentration of 1–5 μM. A stock solution of 10 mM PMAL-C8 was prepared by dissolving polymer powder in chloroform. Polymer encapsulation was performed by combining QDot solution with a 1,000 molar excess of PMAL-C8, mixing by mild vortexing, and slowly drying overnight under mild vacuum. QDot/PMAL film was then slowly dissolved in 50 mM Borate buffer (pH 8.5) under mild agitation, producing stable water-soluble particles with an overall nearly neutral surface charge.
Modification of QDot-PMAT Surface Charge
The high negative surface charge of PMAT-coated QDots was efficiently reduced by partial modification of carboxylic acid groups with positively-charged quaternary amine groups. Specifically, 1μM PMAT-coated QDots were reacted with 10,000 or 100,000 molar excess of (2-Aminoethyl) trimethylammonium chloride (Sigma-Aldrich) in the presence of 100,000 molar excess of (N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (Sigma-Aldrich) overnight, yielding stable water-soluble particles with weakly negatively-charged or neutrally-charged surface coatings.
QDot Purification
Polymer-coated QDots were purified by 2–3 rounds of ultracentrifugation at 45,000 rpm for 60 minutes each using Beckman Coulter Optima TLX ultracentrifuge and TLA110 rotor. Soft pellet (100–150 μL) was collected each time and re-suspended in 50 mM Borate buffer, while the supernatant was discarded. Finally, QDots were filtered through a 0.22 μm syringe filter and stored at about 1μM stock concentration in Borate buffer. Sample purity was confirmed by agarose gel electrophoresis (lack of PMAT band) and fluorometry (lack of 430 nm PMAT fluorescence peak upon 350 nm excitation). The overall yield was determined to be 50–65%.
Bioconjugation with a Universal Protein G Adaptor
PMAT-coated QDot probes were functionalized with universal protein G adators (PrG, recombinant PrG expressed in E. coli, Sigma-Aldrich) via covalent cross-linking of the carboxylic acid groups on the QDot surface with exposed primary amine moieties on protein G. QDots were briefly activated with 5,000–10,000 molar excess of EDC and incubated with 1–20 molar excess of PrG in 50 mM Borate buffer overnight at room temperature. PrG-to-QDot excess of 10:1 typically produced sufficient degree of bioconjugation. QDot-PrG probes were purified by 5 rounds of ultrafiltration with 100 kDa MWCO concentrators (GE Healthcare) and resuspended in Borate buffer at about 1 μM stock concentration for storage at 4°C.
Transmission Electron Microscopy
TEM was performed on FEI Tecnai G2 F20 TWIN 200kV TEM using Gatan Digital Micrograph software at the University of Washington Nanotechnology User Facility. TEM sample was prepared by drop casting 10–20 μl QDots dissolved in hexane onto Formvar-coated copper grids and drying under ambient conditions.
Dynamic Light Scattering (DLS)
Nanoparticle and polymer micelle sizes were measured by light scattering analysis performed on a NanoZS nanoparticle zetasizer (Malvern Instruments). The average of the number-weighted size distributions obtained from three separate measurement runs are reported.
Characterization of Optical Properties
A UV-2450 spectrophotometer (Shimadzu) was used to obtain the QDot and PMAT light absorption profiles and estimate QDot concentration based on the method reported by Peng and coworkers.[22] A Fluoromax4 fluorometer (Horiba Jobin Yvon) was used for characterization of QDot and PMAT emission spectra. QDot quantum yield was determined in reference to Cresyl Violet Acetate (Sigma-Aldrich) in Methanol (QY=54%).
Agarose Gel Electrophoresis
Agarose gel electrophoresis was used for evaluation of QDot surface modification and bioconjugation efficiency. Additionally, gel electrophoresis was employed for purity control purposes via detection of a free PMAT band in a sample. Electrophoresis was performed on 1% agarose gel in 1x TBE buffer at 100V for 1 hour. QDot bands could be readily detected by bright fluorescence when illuminated by a UV lamp. PMAT was stained with SYBR Gold (Invitrogen) and produced characteristic green fluorescence when illuminated by a blue light source (450–500 nm range).
Cell Culture
PSMA-positive human prostate cancer LNCap cells (ATCC) were used as a model biological specimen for QDot target labeling studies. Cells were grown in glass-bottom 24-well plates (Greiner Bio-One) for 2–3 days to a density of ~60% in a humidified atmosphere at 37°C with 5% CO2. RPMI-1640 culture medium with L-Glutamine and 25mM HEPES (Lonza) supplemented with 10% Fetal Bovine Serum (PAA Laboratories) and antibiotics (60 μg/mL streptomycin and 60 U/mL penicillin) was used. For IF staining of PSMA, cells were fixed with 4% formaldehyde in TBS (prepared from methanol-free 16% stock, Thermo Scientific) for 20 min at room temperature and washed with 1x TBS. For examination of QDot non-specific binding to permeabilized specimens, cells were further treated with 2% DTAC/TBS (Dodecyltrimethylammonium chloride, Sigma-Aldrich) for 20 min and 0.25% TritonX-100/TBS (prepared from 10% stock, Thermo Scientific) for 5 min and washed with TBS. Fixed cells were stored in TBS at 4°C.
Immunofluorescence
Monoclonal mouse antibodies against prostate-specific membrane antigen (PSMA, Sigma-Aldrich) were used for specific targeting of the membrane of formalin-fixed non-permeabilized PSMA-positive prostate cancer LNCap cells. Specimen was blocked by 2% BSA-TBS (from Bovine Serum Albumin powder, Sigma-Aldrich) for 30 min and incubated with 5 μg/mL anti-PSMA IgG in 2% BSA-TBS for 1 hour. Control specimens were incubated with 2% BSA-TBS buffer alone. Following washing with TBS, cells were labeled with either 10 nM polymer-coated QDot-PrG probes pre-assembled with secondary rabbit-anti-mouse antibodies (Sigma-Aldrich) at 3:1 QDot:IgG excess or 5 nM reference QDots functionalized with secondary F(ab′)2 antibodies (Qdot goat F(ab′)2 anti-mouse IgG conjugates (H+L), Invitrogen) in 6% BSA-TBS for 1 hour. All staining steps were performed directly inside the wells of glass-bottom 24-well plate at ambient conditions. Fluorescence microscopy was performed immediately following staining.
Evaluation of QDot Non-Specific Binding to Permeabilized Specimens
Formalin-fixed LNCap cells permeabilized with cationic detergent DTAC and non-ionic detergent TritonX-100 were used for this study. Permeabilization disrupts native negatively-charged cell surface and opens access to a number of strong electrostatic interactions between the specimen and negatively-charged PMAT-coated QDots. To study the effect of PMAT modification with quaternary amine compounds as well as evaluate the behavior of an alternative zwitterionic encapsulation polymer PMAL-C8, cells were pre-blocked with 2% BSA-TBS for 30 min and incubated with 4 nM QDot-PMAT, QDot-PMAT-N+, or QDot-PMAL particles in 2% BSA-TBS for 1 hour at ambient conditions. Unbound QDots were removed by 4 rounds of washing with TBS, and non-specific labeling intensity was evaluated with fluorescence microscopy.
Fluorescence Microscopy
IX-71 inverted fluorescence microscope (Olympus) equipped with a true-color CCD (QColor5, Olympus) was used for cell imaging. Low-magnification images were obtained with 10x dry objective (NA 0.40, Olympus) and high-magnification with 100x oil-immersion objective (NA 1.40, Olympus). Wide UV filter cube (330–385 nm band-pass excitation, 420 nm long-pass emission, Olympus) was used for imaging of all QDot probes.
Supplementary Material
Acknowledgments
This work was supported in part by NIH (R01CA131797, R01CA140295), NSF (0645080), DoD-CDMRP (W81XWH0710117), the Coulter foundation, and the Department of Bioengineering at the University of Washington. X.H.G. thanks the NSF for a Faculty Early Career Development award (CAREER). P.Z. thanks the UW Center for Nanotechnology for a UIF fellowship, the National Science Foundation for Graduate Research Fellowship (DGE-0718124), and the National Cancer Institute for a T32 fellowship (T32CA138312). S.R.D. thanks the National Science Foundation for Graduate Research Fellowship (DGE-0718124). We are also grateful to UW Nanotechnology User Facility for help with TEM studies.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.a) Chan WC, Nie S. Science. 1998;281:2016. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]; b) Bruchez M, Jr, Moronne M, Gin P, Weiss S, Alivisatos AP. Science. 1998;281:2013. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- 2.a) Zrazhevskiy P, Gao X. Nat Commun. 2013;4:1619. doi: 10.1038/ncomms2635. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chattopadhyay PK, Price DA, Harper TF, Betts MR, Yu J, Gostick E, Perfetto SP, Goepfert P, Koup RA, De Rosa SC, Bruchez MP, Roederer M. Nat Med. 2006;12:972. doi: 10.1038/nm1371. [DOI] [PubMed] [Google Scholar]
- 3.Yezhelyev MV, Al-Hajj A, Morris C, Marcus AI, Liu T, Lewis M, Cohen C, Zrazhevskiy P, Simons JW, Rogatko A, Nie S, Gao X, O’Regan RM. Adv Mater. 2007;19:3146. [Google Scholar]
- 4.a) Dahan M, Levi S, Luccardini C, Rostaing P, Riveau B, Triller A. Science. 2003;302:442. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]; b) Heine M, Groc L, Frischknecht R, Beique JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Science. 2008;320:201. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cui B, Wu C, Chen L, Ramirez A, Bearer EL, Li WP, Mobley WC, Chu S. Proc Natl Acad Sci U S A. 2007;104:13666. doi: 10.1073/pnas.0706192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Akerman ME, Chan WC, Laakkonen P, Bhatia SN, Ruoslahti E. Proc Natl Acad Sci U S A. 2002;99:12617. doi: 10.1073/pnas.152463399. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cai W, Shin DW, Chen K, Gheysens O, Cao Q, Wang SX, Gambhir SS, Chen X. Nano Lett. 2006;6:669. doi: 10.1021/nl052405t. [DOI] [PubMed] [Google Scholar]; c) Tada H, Higuchi H, Wanatabe TM, Ohuchi N. Cancer Res. 2007;67:1138. doi: 10.1158/0008-5472.CAN-06-1185. [DOI] [PubMed] [Google Scholar]; d) Yang L, Mao H, Wang YA, Cao Z, Peng X, Wang X, Duan H, Ni C, Yuan Q, Adams G, Smith MQ, Wood WC, Gao X, Nie S. Small. 2009;5:235. doi: 10.1002/smll.200800714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Qi L, Gao X. ACS Nano. 2008;2:1403. doi: 10.1021/nn800280r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Probst CE, Zrazhevskiy P, Bagalkot V, Gao X. Adv Drug Deliv Rev. 2013;65:703. doi: 10.1016/j.addr.2012.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Weng KC, Noble CO, Papahadjopoulos-Sternberg B, Chen FF, Drummond DC, Kirpotin DB, Wang D, Hom YK, Hann B, Park JW. Nano Lett. 2008;8:2851. doi: 10.1021/nl801488u. [DOI] [PubMed] [Google Scholar]
- 7.Bagalkot V, Zhang L, Levy-Nissenbaum E, Jon S, Kantoff PW, Langer R, Farokhzad OC. Nano Lett. 2007;7:3065. doi: 10.1021/nl071546n. [DOI] [PubMed] [Google Scholar]
- 8.a) Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Nat Meth. 2008;5:763. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]; b) Zrazhevskiy P, Gao X. Nano Today. 2009;4:414. doi: 10.1016/j.nantod.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Smith AM, Dave S, Nie S, True L, Gao X. Expert Rev Mol Diagn. 2006;6:231. doi: 10.1586/14737159.6.2.231. [DOI] [PubMed] [Google Scholar]; d) Alivisatos P. Nat Biotechnol. 2004;22:47. doi: 10.1038/nbt927. [DOI] [PubMed] [Google Scholar]; e) Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Science. 2005;307:538. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zrazhevskiy P, Sena M, Gao XH. Chem Soc Rev. 2010;39:4326. doi: 10.1039/b915139g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Murray CB, Norris DJ, Bawendi MG. J Am Chem Soc. 1993;115:8706. [Google Scholar]; b) Peng ZA, Peng XG. J Am Chem Soc. 2001;123:183. doi: 10.1021/ja003633m. [DOI] [PubMed] [Google Scholar]; c) Peng ZA, Peng XG. J Am Chem Soc. 2002;124:3343. doi: 10.1021/ja0173167. [DOI] [PubMed] [Google Scholar]; d) Talapin DV, Rogach AL, Kornowski A, Haase M, Weller H. Nano Lett. 2001;1:207. doi: 10.1021/nl0155126. [DOI] [PubMed] [Google Scholar]; e) Li JJ, Wang YA, Guo WZ, Keay JC, Mishima TD, Johnson MB, Peng XG. J Am Chem Soc. 2003;125:12567. doi: 10.1021/ja0363563. [DOI] [PubMed] [Google Scholar]
- 10.a) Wu XY, Liu HJ, Liu JQ, Haley KN, Treadway JA, Larson JP, Ge NF, Peale F, Bruchez MP. Nat Biotechnol. 2003;21:41. doi: 10.1038/nbt764. [DOI] [PubMed] [Google Scholar]; b) Yu WW, Chang E, Falkner JC, Zhang J, Al-Somali AM, Sayes CM, Johns J, Drezek R, Colvin VL. J Am Chem Soc. 2007;129:2871. doi: 10.1021/ja067184n. [DOI] [PubMed] [Google Scholar]; c) Gao X, Cui Y, Levenson RM, Chung LW, Nie S. Nat Biotechnol. 2004;22:969. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 11.Pellegrino T, Manna L, Kudera S, Liedl T, Koktysh D, Rogach AL, Keller S, Radler J, Natile G, Parak WJ. Nano Lett. 2004;4:703. [Google Scholar]
- 12.a) Bryant GW, Jaskolski W. J Phys Chem B. 2005;109:19650. doi: 10.1021/jp0535543. [DOI] [PubMed] [Google Scholar]; b) Gomez DE, Califano M, Mulvaney P. Phys Chem Chem Phys. 2006;8:4989. doi: 10.1039/b607661k. [DOI] [PubMed] [Google Scholar]
- 13.a) Morris-Cohen AJ, Donakowski MD, Knowles KE, Weiss EA. J Phys Chem C. 2009;114:897. [Google Scholar]; b) Zorn G, Dave SR, Gao X, Castner DG. Anal Chem. 2011;83:866. doi: 10.1021/ac102516n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frantsuzov P, Kuno M, Janko B, Marcus RA. Nat Phys. 2008;4:519. [Google Scholar]
- 15.a) Bentzen EL, Tomlinson ID, Mason J, Gresch P, Warnement MR, Wright D, Sanders-Bush E, Blakely R, Rosenthal SJ. Bioconjug Chem. 2005;16:1488. doi: 10.1021/bc0502006. [DOI] [PubMed] [Google Scholar]; b) Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanomedicine (Lond) 2011;6:715. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu W, Howarth M, Greytak AB, Zheng Y, Nocera DG, Ting AY, Bawendi MG. J Am Chem Soc. 2008;130:1274. doi: 10.1021/ja076069p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Muro E, Pons T, Lequeux N, Fragola A, Sanson N, Lenkei Z, Dubertret B. J Am Chem Soc. 2010;132:4556. doi: 10.1021/ja1005493. [DOI] [PubMed] [Google Scholar]; b) Breus VV, Heyes CD, Tron K, Nienhaus GU. ACS Nano. 2009;3:2573. doi: 10.1021/nn900600w. [DOI] [PubMed] [Google Scholar]; c) Park J, Nam J, Won N, Jin H, Jung S, Jung S, Cho SH, Kim S. Adv Funct Mater. 2011;21:1558. [Google Scholar]; d) Cao Z, Jiang S. Nano Today. 2012;7:404. [Google Scholar]; e) Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG, Frangioni JV. Nat Nanotechnol. 2009;5:42. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Goldman ER, Anderson GP, Tran PT, Mattoussi H, Charles PT, Mauro JM. Anal Chem. 2002;74:841. doi: 10.1021/ac010662m. [DOI] [PubMed] [Google Scholar]; b) Bakalova R, Zhelev Z, Ohba H, Baba Y. J Am Chem Soc. 2005;127:9328. doi: 10.1021/ja0510055. [DOI] [PubMed] [Google Scholar]
- 18.a) Bakalova R, Zhelev Z, Ohba H, Baba Y. J Am Chem Soc. 2005;127:11328. doi: 10.1021/ja051089h. [DOI] [PubMed] [Google Scholar]; b) Medintz IL, Clapp AR, Mattoussi H, Goldman ER, Fisher B, Mauro JM. Nat Mater. 2003;2:630. doi: 10.1038/nmat961. [DOI] [PubMed] [Google Scholar]; c) Medintz IL, Goldman ER, Lassman ME, Mauro JM. Bioconjug Chem. 2003;14:909. doi: 10.1021/bc020062+. [DOI] [PubMed] [Google Scholar]
- 19.a) Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Nat Biotechnol. 2003;21:47. doi: 10.1038/nbt767. [DOI] [PubMed] [Google Scholar]; b) Jin T, Tiwari DK, Tanaka S, Inouye Y, Yoshizawa K, Watanabe TM. Mol BioSyst. 2010;6:2325. doi: 10.1039/c0mb00056f. [DOI] [PubMed] [Google Scholar]
- 20.Xing Y, Chaudry Q, Shen C, Kong KY, Zhau HE, Chung LW, Petros JA, O’Regan RM, Yezhelyev MV, Simons JW, Wang MD, Nie S. Nat Protoc. 2007;2:1152. doi: 10.1038/nprot.2007.107. [DOI] [PubMed] [Google Scholar]
- 21.Saha K, Bender F, Gizeli E. Anal Chem. 2003;75:835. doi: 10.1021/ac0204911. [DOI] [PubMed] [Google Scholar]
- 22.Yu WW, Qu L, Guo W, Peng X. Chem Mater. 2003;15:2854. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.