

Abstract
Salmonella enterica serovar Typhimurium is the most common Salmonella serovar causing foodborne infections in Australia and many other countries. Twenty-one S. Typhimurium strains from Salmonella reference collection A (SARA) were analyzed using Illumina high-throughput genome sequencing. Single nucleotide polymorphisms (SNPs) in 21 SARA strains ranged from 46 to 11,916 SNPs, with an average of 1,577 SNPs per strain. Together with 47 strains selected from publicly available S. Typhimurium genomes, the S. Typhimurium core genes (STCG) were determined. The STCG consist of 3,846 genes, a set that is much larger than that of the 2,882 Salmonella core genes (SCG) found previously. The STCG together with 1,576 core intergenic regions (IGRs) were defined as the S. Typhimurium core genome. Using 93 S. Typhimurium genomes from 13 epidemiologically confirmed community outbreaks, we demonstrated that typing based on the S. Typhimurium core genome (STCG plus core IGRs) provides superior resolution and higher discriminatory power than that based on SCG for outbreak investigation and molecular epidemiology of S. Typhimurium. STCG and STCG plus core IGR typing achieved 100% separation of all outbreaks compared to that of SCG typing, which failed to separate isolates from two outbreaks from background isolates. Defining the S. Typhimurium core genome allows standardization of genes/regions to be used for high-resolution epidemiological typing and genomic surveillance of S. Typhimurium.
INTRODUCTION
Salmonella enterica serovar Typhimurium is one of the leading causes of Salmonella-related gastroenteritis in humans (1). Rapid and accurate identification and characterization of S. Typhimurium isolates is one of the most important tasks of public health laboratory surveillance for outbreak investigation and long-term epidemiology, especially when S. Typhimurium outbreaks pose threats of national/international spread (2). Currently, a variety of techniques have been employed to subtype S. Typhimurium for epidemiological purposes (3–6). Traditionally, phage typing was used in Australia and Europe (7, 8). However, our studies showed that phage types may arise independently through mutation, and thus isolates of the same phage type may not share the same evolutionary history (9). Multilocus sequence typing (MLST) has a low resolution to type S. Typhimurium, as the majority of isolates belong to sequence type 19 (ST19) (10). Multilocus variable-number tandem-repeat analysis (MLVA) has been adopted as a standardized method in Australia (9, 11) and Europe (12). However, the numerical dominance of endemic MLVA types reduces the power of MLVA for outbreak investigations. In addition, MLVA has limited value for long-term epidemiology (13).
Next-generation sequencing (NGS) has been increasingly employed to prospectively identify and track outbreaks (14). NGS has major advantages over other pathogen-typing methods, as it promises a standardized universal solution for high-resolution typing. We recently demonstrated the utility and resolution of NGS for outbreak investigation of S. Typhimurium by sequencing 57 isolates from five distinct and epidemiologically confirmed point-source S. Typhimurium DT170 outbreaks in Australia (15). Our study utilized all single nucleotide polymorphisms (SNPs) identified from the genomes of the outbreak isolates. In contrast, Leekitcharoenphon et al. (16) utilized SNPs identified from 2,882 Salmonella core genes for outbreak investigation of S. Typhimurium. The Salmonella core genes were defined by analyzing 73 representative and publicly available genomes from 23 serovars and proposed a core genome typing scheme for S. enterica (16). The use of the core genome for typing allows standardization of genome-based typing. However, since Salmonella is a diverse species with 7 subspecies and more than 2,400 serovars, the species core genome may be substantially smaller than the core genome of a serovar, and thus the use of the species core genome for typing may lead to substantial loss of resolution at the serovar level.
In this study, we defined the core genome of S. Typhimurium using 21 S. Typhimurium reference strains from Salmonella reference collection A (SARA) and 47 publicly available genomes. To obtain the core genome of the serovar, it is critical to select strains to represent the spectrum of diversity. SARA is a collection of 21 diverse S. Typhimurium strains representing 17 electrophoretic types and four sequence types (STs)and 51 strains from four closely related serovars, which are referred to as the S. Typhimurium complex (4). The strains were collected internationally in the 1980s or earlier from various host sources and have been characterized using a range of molecular methods, which showed considerable diversity (5, 6). We further evaluated the performance of epidemiological typing based on Salmonella core genes (SCG), S. Typhimurium core genes (STCG), and STCG plus core intergenic regions (IGRs) using published genomic data from epidemiologically confirmed outbreaks. To avoid confusion over the terminologies used, the S. Typhimurium core genome includes STCG and core IGRs while STCG denotes S. Typhimurium core genes only. S. Typhimurium core genome typing refers to typing using either STCG or STCG plus core IGRs as a generic term if not specified.
MATERIALS AND METHODS
Bacterial strains and genomic DNA isolation.
Twenty-one S. Typhimurium strains from SARA were used (4) (Table 1). These strains were isolated from various sources, four of which came from human sources and eight of which came from animal sources while the remaining nine came from unknown sources. The phenol-chloroform method was used to extract genomic DNA from each strain as described previously (17).
TABLE 1.
Strains sequenced in this study
| Strain | ETa | Original name | Source | Locality | STb | Year |
|---|---|---|---|---|---|---|
| SARA1 | Tm 1 | INSP 24 | Human | Mexico | ST19 | |
| SARA2 | Tm 1 | LT2 | Lab strain | ST19 | ||
| SARA3 | Tm 1 | NVSL 7095 | Horse | Rhode Island | ST19 | 1987 |
| SARA4 | Tm 1 | NVSL 5820 | Rabbit | Indiana | ST19 | 1986 |
| SARA5 | Tm 1 | IVB 232 | Mongolia | ST19 | ||
| SARA6 | Tm 2 | CDC B1213 | Human | Ohio | ST19 | |
| SARA7 | Tm 3 | IVB 665/81 | Norway | ST36 | ||
| SARA8 | Tm 5 | IVB 5560 | Finland | ST36 | ||
| SARA9 | Tm 7 | NVSL 2816 | Parrot | California | ST98 | 1987 |
| SARA10 | Tm 9 | NVSL 6814 | Opposum | California | ST19 | 1987 |
| SARA11 | Tm 10 | IVB 276/25 | Thailand | ST19 | ||
| SARA12 | Tm 11 | NVSL 6993 | Horse | Louisiana | ST19 | 1987 |
| SARA13 | Tm 12 | IVB 1430 | France | ST19 | ||
| SARA14 | Tm 13 | IVB 75/67 | Panama | ST19 | ||
| SARA15 | Tm 14 | NVSL 6968 | Dog | Texas | ST19 | 1987 |
| SARA16 | Tm 15 | CDC B1236 | Human | North Carolina | ST19 | |
| SARA17 | Tm 16 | IVB 48/81 | Yugoslavia | ST19 | ||
| SARA18 | Tm 17 | NVSL 6938 | Horse | Iowa | ST19 | 1987 |
| SARA19 | Tm 21 | INSP 85 | Human | Mexico | ST19 | |
| SARA20 | Tm 22 | IVB 1544 | France | ST19 | ||
| SARA21 | Tm 23 | USFW 318 | Heron | Oregon | ST99 |
Genome sequencing, de novo assembly, and core genome analysis.
Genomic DNA was sequenced using the MiSeq genome analyzer (Illumina). We used 250-bp paired-end sequencing. Contigs were assembled de novo using the software package Velvet version 1.0.8 and VelvetOptimiser (18). Large scaffolds and short contigs generated by Velvet were aligned to the S. Typhimurium LT2 genome (GenBank accession number NC_003197) using progressiveMauve version 2.3.1 (19). RAST was used to annotate the sequences from each NGS genome (20). These annotated genes were also grouped into functional categories.
Twenty-one strains from SARA and 47 S. Typhimurium genomes from GenBank were used for the identification of the core genome content (Table 1; see also Table S1 in the supplemental material). The assembled genomes were aligned to the reference S. Typhimurium strain LT2 in progressiveMauve (19). Genes and intergenic regions (IGRs) (>100 bp) that appeared in all isolates were considered core genes and core IGRs, respectively. Mobile genes and repetitive elements were excluded from the core genome. In order to find the core genome of the Typhimurium complex, 9 Salmonella enterica serovar Heidelberg genomes, 2 Salmonella enterica serovar Saintpaul genomes, 3 Salmonella enterica serovar Muenchen genomes, and 2 Salmonella enterica serovar Paratyphi B genomes were used in addition to the 68 S. Typhimurium genomes (see Table S1 in the supplemental material).
To determine whether a stable core genome was obtained, a regression analysis of the number of isolates against their shared genes was performed by fitting a double exponential decay function (21), Nc = θ + k1 × exp(m1 × Ng) + k2 × exp(m2 × Ng), where θ is a constant value representing the predicted minimum number of core genes, Nc is the number of core genes, Ng is the number of genomes, and k1, m1, k2, and m2 are parameters where the model was determined by a weighted least-squares regression analysis.
Identification of single nucleotide polymorphisms.
For SNP detection in the draft genomes, SNP calling was conducted as described previously (17). The Burrows-Wheeler alignment (BWA) tool (version 0.7.5) was used to map the reads against the S. Typhimurium LT2 genome (22). SAMtools version 0.1.19 was used to further filter the SNPs identified from BWA mapping by SNP quality (23). SNP calling followed the previously described criteria (17). SNPs with quality score of less than 30 were removed. A custom script was used to determine whether an SNP in the genic region was a synonymous SNP (sSNP) or a nonsynonymous SNP (nsSNP). For the complete genomes, SNPs were determined by using the NUCmer program “show-snps” (with options “-CIlrT”) in the MUMmer package version 3.0 (24).
Phylogenetic analysis.
To evaluate the performance of the S. Typhimurium core genome (STCG with and without core IGRs) typing, 21 strains from SARA sequenced in this study and 94 isolates from 13 different outbreaks were used (see Table S2 in the supplemental material) (14, 15, 25, 26). The selection of outbreaks was either based on representative phage type or ST, including DT135, DT135a, U292, DT3, DT104, DT12, DT120, and ST313. Phylogenetic trees based on SNPs were constructed using the minimum evolution algorithm in MEGA 6.0 (27). Bootstrap analysis was performed with 1,000 replicates. The percentage of concordance was calculated by Ridom EpiCompare (version 1.0) (28). The discriminatory power was assessed by calculating Simpson's index of diversity (D value) (29).
Sequence divergence in coding genes and IGR.
Sequence divergence was estimated using the Kimura 2-parameter (K2P) model implemented in MEGA 6.0 (27). The Student t test was performed with SPSS for Windows version 11.5 (SPSS Inc., Chicago, IL, USA).
Nucleotide sequence accession numbers.
The annotated S. Typhimurium whole-genome sequences were deposited in the Sequence Read Archive under accession numbers SAMN03470046 to SAMN03470066.
RESULTS
General statistics of the SARA genome sequencing.
The 21 SARA S. Typhimurium strains were sequenced using MiSeq 250-bp paired-end sequencing in a multiplexed format. The average number of reads generated per genome was ∼2,651,000. The average coverage depth for all genomes was ∼42×, with a lowest coverage depth of 24× (Table 2). Percentage match of reads to reference strain LT2 chromosome ranged from 96.5% to 99.0%. The reads were assembled de novo. The number of contigs ranged from 228 to 619, with an average of 330. SARA10 had the largest genome size of 5.0 Mb, while SARA16 had the smallest (4.7 Mb).
TABLE 2.
General features of the 21 S. Typhimurium genomes sequenced in this study
| Strain | Total no. of reads | N50 | No. of contigs | Total length (bp) | Coverage (×) | Match to LT2 chromosome (%) | No. (%) of nonsynonymous SNPs | No. (%) of synonymous SNPs | No. (%) of intergenic SNPs | No. (%) of indels | Total no. of SNPs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SARA1 | 1,581,754 | 194,269 | 283 | 4,900,132 | 46 | 97.8 | 88 (49.42) | 32 (18.00) | 50 (28.09) | 8 (4.49) | 178 |
| SARA2 | 1,136,806 | 324,050 | 307 | 4,892,963 | 35 | 99.0 | 22 (47.83) | 24 (52.17) | 0 (0.00) | 0 (0) | 46 |
| SARA3 | 1,003,706 | 172,050 | 280 | 4,789,145 | 34 | 96.5 | 189 (42.76) | 150 (33.94) | 95 (21.49) | 8 (1.36) | 442 |
| SARA4 | 1,500,444 | 222,200 | 363 | 4,967,277 | 39 | 97.2 | 363 (42.52) | 319 (34.81) | 188 (21.32) | 12 (1.07) | 882 |
| SARA5 | 3,212,530 | 168,092 | 479 | 4,965,739 | 46 | 97.5 | 6 (21.88) | 34 (40.63) | 24 (37.50) | 0 (0.00) | 64 |
| SARA6 | 2,683,486 | 225,480 | 372 | 4,937,837 | 64 | 98.6 | 204 (43.68) | 143 (30.62) | 115 (24.63) | 5 (1.07) | 467 |
| SARA7 | 2,700,978 | 240,373 | 327 | 4,918,149 | 63 | 97.9 | 1,913 (16.05) | 8,509 (71.41) | 1,372 (11.51) | 122(1.02) | 11,916 |
| SARA8 | 1,291,676 | 219,093 | 289 | 4,807,626 | 37 | 97.1 | 1,844 (16.32) | 8,013 (71.71) | 1,258 (11.13) | 184(1.63) | 11,299 |
| SARA9 | 1,187,376 | 214,955 | 288 | 4,919,127 | 34 | 97.5 | 280 (36.61) | 265 (37.48) | 154 (21.78) | 8 (1.13) | 707 |
| SARA10 | 1,624,246 | 194,273 | 273 | 5,025,508 | 45 | 97.0 | 296 (41.69) | 255 (35.92) | 151 (21.27) | 8 (1.13) | 710 |
| SARA11 | 1,207,350 | 277,613 | 307 | 4,921,121 | 37 | 97.2 | 302 (39.27) | 284 (36.93) | 165 (21.46) | 21 (2.73) | 769 |
| SARA12 | 1,193,274 | 257,533 | 265 | 4,899,590 | 40 | 97.2 | 454 (41.35) | 341 (30.05) | 260 (23.68) | 43 (3.91) | 1,098 |
| SARA13 | 1,172,002 | 278,182 | 274 | 4,856,144 | 34 | 97.0 | 437 (44.26) | 310 (29.70) | 240 (23.76) | 23 (2.28) | 1,010 |
| SARA14 | 1,789,596 | 356,416 | 228 | 4,822,760 | 69 | 97.9 | 233 (44.13) | 153 (28.98) | 107(20.27) | 35 (6.63) | 528 |
| SARA15 | 2,668,118 | 270,525 | 242 | 4,862,737 | 49 | 97.5 | 20 (7.75) | 159 (61.63) | 74 (28.68) | 5 (1.94) | 258 |
| SARA16 | 2,930,600 | 144,590 | 619 | 4,727,259 | 37 | 96.5 | 77 (31.40) | 218 (47.67) | 99 (19.77) | 0 (0.00) | 394 |
| SARA17 | 545,298 | 118,536 | 433 | 4,905,073 | 50 | 97.1 | 194 (43.69) | 156 (35.14) | 90 (20.27) | 4 (0.90) | 444 |
| SARA18 | 1,284,792 | 149,251 | 387 | 4,769,756 | 37 | 97.6 | 219 (41.17) | 192 (36.09) | 117 (21.99) | 4 (0.75) | 532 |
| SARA19 | 2,430,606 | 223,024 | 313 | 4,940,693 | 35 | 97.3 | 104 (45.61) | 61 (26.75) | 57 (25.00) | 4 (1.75) | 228 |
| SARA20 | 819,486 | 272,276 | 332 | 4,930,836 | 24 | 97.3 | 173 (45.53) | 115 (30.27) | 86 (22.63) | 4 (1.05) | 380 |
| SARA21 | 1,073,366 | 306,796 | 250 | 4,853,595 | 34 | 97.0 | 360 (47.57) | 231 (30.52) | 162 (21.40) | 4 (0.53) | 757 |
Identification of the S. Typhimurium core genes.
Overall, the pan genome that includes all genes present in one or more of the 21 strains contained 4,955 genes, of which 4,177 were present in all strains. We screened the 4,177 genes against 47 genomes selected from available S. Typhimurium genomes in GenBank, from which we excluded genomes identical or closely related to outbreak strains. We removed remnant prophage and mobile genes from the core genes even if they were present in all strains but kept virulence genes and genes on genomic islands (GIs). In the final set, 3,846 genes (see Table S3 in the supplemental material) were defined as the S. Typhimurium core genes (STCG). These core genes were present as 64 conserved segments with an average of 60 genes per segment, the longest segment having 249 genes (see Table S3 in the supplemental material). The 3,846 core genes were unevenly distributed across the functional categories (Fig. 1A). Core genes account for 98.87% and 97.19% in the functional categories of protein metabolism and respiration, respectively.
FIG 1.
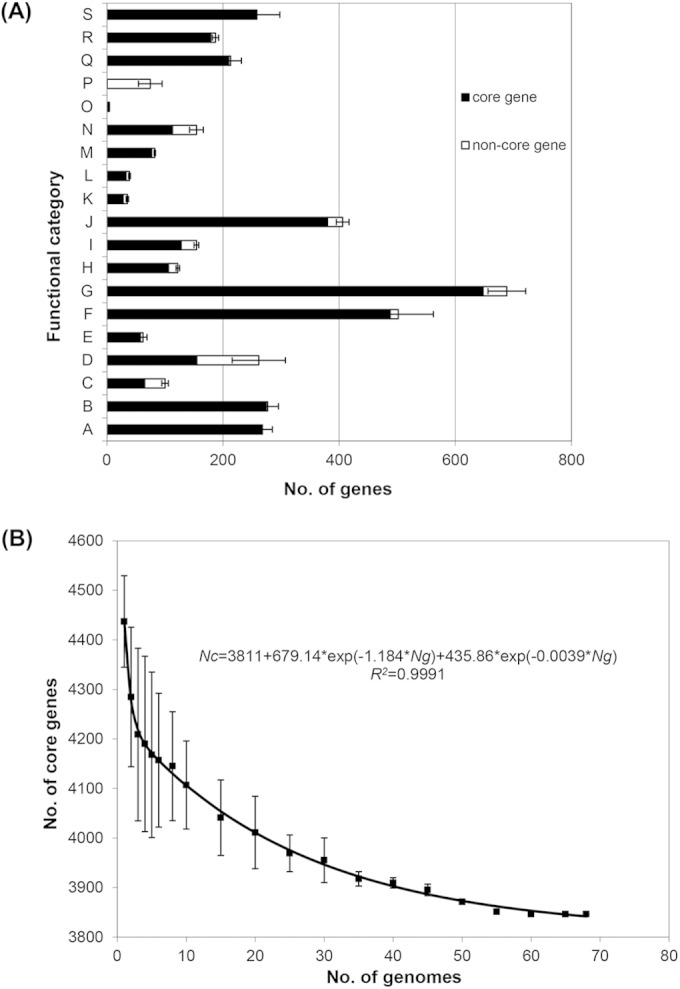
Core genome variation of S. Typhimurium. (A) Functional catalogues of core and noncore genes in the LT2 genome. A, Cofactors, vitamins transport, and metabolism; B, cell wall and capsule; C, virulence, disease and defense; D, membrane transport; E, iron acquisition and metabolism; F, nucleotide transport and metabolism; G, carbohydrates metabolism; H, fatty acids, lipids, and isoprenoids metabolism; I, nitrogen, sulfur, and phosphorus metabolism; J, amino acid transport and metabolism; K, metabolism of aromatic compounds; L, cell division and cell cycle; M, motility and chemotaxis; N, regulation and cell signaling; O, secondary metabolism; P, phages, prophages, transposable elements, plasmids; Q, respiration; R, stress response; S, protein metabolism; T, general prediction only or unknown function. The catalogues of gene functions were based on the subsystem of RAST. Error bars indicate variations in the number of accessory genes among the 21 SARA strains. (B) Estimation of core genome size. The graph shows the descending trend of the core genome size of S. Typhimurium with the increasing number of genomes. The number of shared genes is plotted against the number of genomes that were sequentially added. Black squares are the averages of such values. Error bars indicate variations in the number of core genes among different strains.
To determine whether the diversity and number of strains used were sufficiently large to derive a stable core genome, we used the statistical model developed by Bottacini et al. (21) by random sampling of the 68 genomes (see Table S1 in the supplemental material) to fit a double exponential decay function. The regression function is expressed as Nc = 3,811 + 679.14 × exp(−1.184 × Ng) + 345.86 × exp(−0.0039 × Ng) (R2 = 0.9991) (Fig. 1B), and the predicted minimum number of genes was 3,811. The number of core genes (3,846) for the 68 genomes derived above is close to the predicted value, which suggests that we derived a stable core genome. The fitted curve reached a plateau at 55 genomes (see Fig. S1A in the supplemental material), which is the expected minimum number of genomes that would be required to obtain the core genome content.
The S. Typhimurium complex includes four serovars closely related to S. Typhimurium (6). For the 3,846 S. Typhimurium core genes identified, 3,809 genes were shared by all strains in the S. Typhimurium complex while 37 genes were present only in S. Typhimurium (see Table S4 in the supplemental material). Interestingly, most of the S. Typhimurium unique core genes are located between 3.7 Mb and 4.7 Mb in LT2 (see Fig. S1B in the supplemental material), including a part of the rfb gene cluster, sugR, rhuM, oadA, oadB, ilvA, yjdC, mgtA, and other nonessential genes. These genes were variably present in S. Paratyphi B SPB7, S. Heidelberg SL486, S. Muenchen baa1594, and S. Saintpaul SARA26 (see Table S4 in the supplemental material). Most of these genes encode proteins that are associated with extracellular components or functions, including putative transporters, putative cytoplasmic proteins, and putative membrane proteins. In general, genes encoding proteins located outside the cell are known to be more variable (30). Five genes from rfb gene clusters are absent in S. Muenchen. This may reflect the difference between S. Muenchen (a serovar of the O:8 [C2-C3] serogroup) and the other four serovars (O:4 [B] serogroup) in the O-antigen gene cluster.
Characterization of core IGRs.
We identified the S. Typhimurium core IGRs using the procedure similar to that used to identify the core genes. Noncoding IGRs greater than 100 bp were extracted according to the coordinates of their adjacent consecutive coding genes for LT2. There are 1,857 IGRs, of which 281 were variably present in the 68 S. Typhimurium strains, and 1,576 were core IGRs (see Table S5 in the supplemental material). The size of core IGRs ranged from 101 bp to 12,151 bp, with an average length of 285 bp. The largest IGR has a length of 12,151 bp, which is a large repetitive region in S. Typhimurium.
To determine whether the core IGRs evolve at a similar rate to the core genes, we performed a pairwise comparison of core genes and core IGRs between LT2 and D23580, which are the only complete S. Typhimurium genomes. The divergence was 2.12 × 10−4 substitutions per site and 2.15 × 10−4 substitutions per site for core genes and core IGRs, respectively, indicating that the core IGRs and core coding regions evolve at a similar rate (t test, P = 0.31; n = 1576) (see Fig. S2 in the supplemental material).
Identification of genome SNPs in the SARA strains.
SNP calling was performed for the 21 SARA strains sequenced in this study. The total SNPs ranged from 46 to 11,916 (Table 2). Note that SARA2 is also a LT2 strain. To examine whether the SNPs observed in SARA2 are due to sequencing errors of the reference sequence, we investigated three other LT2 genomes in the public database (accession numbers ERS007491, ERS007500, and AHUZ00000000). The number of SNPs ranged from 56 to 68. Moreover, all of the 44 SNPs observed in SARA2 also existed in these genomes, indicating that they are probably not due to sequencing errors.
We further analyzed the nature of the SNPs of the SARA genomes. The SNPs were divided into four categories: nonsynonymous (nsSNP), synonymous (sSNP), intergenic (IG), and single base indels. IG SNPs on average accounted for approximately 21% of the total number of SNPs in each strain with the exception of SARA2, which had no IG SNP (0%). The average proportion of nsSNP for each strain was 37.6% ranging from 7.8% to 49.4% while the sSNPs ranged from 18.0% to 71.7% with an average of 39.1%. Single base indels accounted for only 1.8% of the SNPs. SARA8 had the highest number of indels (184 indels) representing 1.63% of the SNPs. Interestingly, SARA7 and SARA8 had 71.4% and 71.7% SNPs belonging to sSNP, many of which were shared between the two strains. Thirty-two genes among 21 SARA strains terminated earlier due to a stop codon and led to proteins that were >20% shorter compared to strain LT2 (see Table S6 in the supplemental material). Thus, these genes were considered pseudogenes. We then extracted the S. Typhimurium core genome SNPs and SCG SNPs. The average number of S. Typhimurium core genome SNPs and SCG SNPs for 21 SARA strains were 1,267 and 825, respectively, indicating that the S. Typhimurium core genome covers 34.8% more SNP sites than SCG (Table 3).
TABLE 3.
Average number of SNPs found in three typing schemes
| Group | Total no. of SNPs | No. of STCG plus core IGR SNPs | No. of STCG SNPs | No. of SCG SNPs |
|---|---|---|---|---|
| SARA | 1,577 | 1,267 | 957 | 825 |
| Outbreak 1 | 869 | 719 | 630 | 560 |
| Outbreak 2 | 717 | 582 | 454 | 398 |
| Outbreak 3 | 670 | 422 | 302 | 261 |
| Outbreak 4 | 815 | 625 | 482 | 417 |
| Outbreak 5 | 629 | 485 | 381 | 332 |
| Outbreak 6 | 649 | 545 | 415 | 345 |
| Outbreak 7 | 643 | 510 | 420 | 360 |
| Outbreak 8 | 694 | 584 | 457 | 400 |
| Outbreak 9 | 788 | 678 | 599 | 249 |
| Outbreak 10 | 711 | 620 | 554 | 230 |
| Outbreak 11 | 675 | 605 | 539 | 240 |
| Outbreak 12 | 672 | 604 | 533 | 233 |
| Outbreak 13 | 708 | 623 | 540 | 231 |
Application of S. Typhimurium core genome for epidemiological typing.
With the STCG plus core IGRs defined above, we compared the resolution of the use of STCG, STCG plus core IGRs, and SCG for epidemiological typing of S. Typhimurium. The SCG of 2,882 genes was previously defined by Leekitcharoenphon et al. (16). The publicly available genome sequences of S. Typhimurium strains were used as test samples, including strains from 8 prolonged outbreaks (over 1 year) and 5 short duration outbreaks (ranging from 9 to 46 days) (see Table S2 in the supplemental material). SNPs were called by comparison with LT2 and were used to compare the typing methods.
For the isolates from the 13 outbreaks, the number of SNPs ranged from 629 to 869 (Table 3). The SNPs for STCG plus core IGRs for the 13 outbreaks were between 422 and 719, which were only slightly lower than the total number of SNPs, while the STCG SNPs ranged from 302 to 630. SCG SNPs ranged from 230 to 560, which is 43.5% lower on average than STCG plus core IGRs.
We used several parameters to measure the difference in resolving power between S. Typhimurium core genome typing and that of SCG typing. Phylogenetic trees constructed using the minimum evolution method (31) were used to determine whether STCG and SCG resolve the strain relationships differently. Simpson's index of diversity (D value) was used to measure differences in discriminatory power within an outbreak, while SNP difference was used to measure the separation of outbreak isolates from background isolates or the nearest isolates unrelated to a given outbreak.
As shown in Fig. 2, STCG typing achieved 100% separation of the outbreaks (Fig. 2A). However, the performance of the SCG typing was much poorer (Fig. 2B). Two isolates from outbreak 2 were assigned to different branches. Moreover, it is notable that the SCG tree failed to distinguish some background isolates from outbreak isolates. Strain 0909R12120 (231) was not related to any of the outbreaks but was clustered with isolates from outbreak 6. The epidemiological concordance of SCG typing decreased to 97.9%.
FIG 2.

Phylogenetic relationships of S. Typhimurium isolates based on S. Typhimurium core genes (A) and Salmonella core genes (B). The minimum evolution method was used to infer evolutionary relationships of the isolates. The numbers in brackets represent outbreak numbers. Bootstrap was performed with 1,000 replicates. The bootstrap values (1,000 replicates; >50%) are shown next to the branches. The unit of the scale bar indicates the evolutionary distance in substitutions per nucleotide. The solid circles and squares indicate the wrongly placed isolates from outbreaks 2 and 6, respectively, by SCG typing.
We further examined the level of differentiation to the genotype level and calculated Simpson's index of diversity (D value) within the 13 outbreaks. Overall, compared to those of STCG typing, the number of genotypes and the D values were decreased in SCG typing (Fig. 3A and B).
FIG 3.

Comparison of core genome typing. (A) Number of genotypes among 13 outbreaks based on STCG plus core IGRs (blue bars), STCG (red bars), and SCG (green bars). (B) Simpson's index of diversity (D value) among 13 outbreaks based on STCG plus core IGRs (blue bars), STCG (red bars), and SCG (green bars). (C) The average number of SNP difference between outbreak isolates and background strains/closest outbreak-unrelated isolates based on STCG plus core IGRs (blue bars), STCG (red bars), and SCG (green bars).
The SNP differences between outbreak-related strains and background strains in STCG and SCG typing as measured by the average number of SNP difference are shown in Fig. 3C. The SNP differences in STCG typing averaged 39 SNPs with a smallest difference of 7 SNPs. In contrast, SCG typing separated background isolates from outbreak isolates by as few as 3 SNPs (outbreak 13) with an average of 27 SNPs.
We further examined STCG plus core IGRs. It separated all outbreaks from each other and from background isolates (see Fig. S3 in the supplemental material). The discriminatory power of STCG plus core IGRs for typing was slightly higher than STCG typing (Fig. 3). For 3 of the 13 outbreaks, adding core IGRs increased the resolution, while for the remaining 10 outbreaks the resolution was the same as that for STCG typing.
DISCUSSION
In this study, we sequenced 21 SARA strains to determine the core genome of S. Typhimurium, which consisted of 3,846 core genes and 1,576 core IGRs. The S. Typhimurium core genome contained 956 (33.45%) more genes than the Salmonella core genome, which contains only 2,882 genes as defined previously (16). We showed that the use of the STCG or the STCG plus core IGRs increases the resolution of the genome sequencing for epidemiological typing using published and epidemiologically investigated outbreaks (14, 15, 25, 26).
The number of core genes in a serovar depends on the sampled diversity of the given serovar. We used SARA strains that were selected previously based on multilocus enzyme electrophoresis (MLEE) diversity (5, 6) and also cover most of the continents and seven major STs. The 21 SARA strains were obtained from nine different countries with different prevalence profiles of S. Typhimurium infections, including four European countries (Norway, France, Yugoslavia, and Finland), three countries of the American continent (United States, Panama, and Mexico), and two Asian countries (Mongolia and Thailand). Other isolates from public databases include 13 isolates from Denmark, nine isolates from Africa (Malawi), nine isolates from Australia, six isolates each from the United Kingdom and the United States, three isolates from China, and one isolate from Japan. We used a statistical model to estimate the underlying number of core genes by fitting a double exponential decay function and found that the core genome content reached a plateau in the sampling, suggesting that the core genome has reached its stable stage from the strains analyzed and adding more strains would not substantially reduce the core genome size.
We also analyzed the core intergenic regions for consideration as a source of SNPs to increase the power of genome-based typing. Intergenic regions are generally considered to have less or no functional constraints, and thus mutational changes in IGRs are thought to be generally neutral and have little or no selective cost (32). The substitution rates for IGRs are, therefore, expected to be higher than those for housekeeping genes, where there are major functional constraints on mutation as many will be deleterious. Interestingly, the overall substitution rate was similar between core genes and core IGRs. Core IGRs may contain regulatory elements such as small RNAs (sRNAs), which are highly conserved. Hu et al. used IG SNPs for typing different phage types of S. Typhimurium previously (32). Thus, IGRs may increase the resolution of genome-based typing. We found 1,576 core IGRs with an average size of 285 bp and a total size of 448,581 bp, contributing to 11.5% of the S. Typhimurium core genome. For the 68 genomes used for determining core IGRs, we found that 316 had variations, including SNP changes in 296 IGRs. However, no variation was observed in 1,260 core IGRs. The Salmonella core IGRs were not defined previously (16), and a comparison cannot be made. We expect that conservation of Salmonella core IGRs will be similar in proportion to that observed for the core genes at the species level.
Using the isolates from 13 outbreaks, we demonstrated that the use of STCG and S. Typhimurium core genome (STCG ± core IGRs) increased the resolution of typing. SCG typing offered the poorest resolution, as some outbreak isolates cannot be differentiated from background isolates (outbreak 6). By total number of bases, SCG covers 2.9 Mb while STCG covers 3.5 Mb and STCG plus core IGRs covers 3.9 Mb. The resolution of the typing scheme increases with the total number of bases under consideration. This was reflected in the number of SNP sites observed in the 21 SARA strains with 957 and 1,267 SNP sites in total for STCG and STCG plus core IGRs, an increase of 16% and 54%, respectively, compared to the 825 SNP sites covered by SCG. Additionally, typing using STCG and STCG plus core IGRs increased the differentiation of isolates within an outbreak. For 3 outbreaks, the inclusion of core IGR increased the differentiation compared to that of STCG typing. Compared to SCG typing, the use of STCG or STCG plus core IGRs increased the differentiation of isolates within an outbreak for 7 and 8 outbreaks, respectively. Although the discriminatory power of typing using STCG ± core IGRs is no better than that of SCG typing to subtype isolates for some outbreaks, this situation is likely to change when there is an increased number of background isolates sequenced from the same region and/or time period (15). There will be an increased need for higher resolution to differentiate outbreak isolates from closely related and/or endemic isolates.
NGS data may be used to trigger an investigation of an outbreak either locally or globally. However, considering that there are variations between isolates within an outbreak and background isolates may be very closely related to isolates from a potential outbreak, genome sequence alone may not be adequate for the determination of an epidemiological link. Our previous study showed that to rule in or rule out whether an isolate is part of an outbreak requires the determination of a cutoff of the number of SNP differences between isolates being analyzed (15). We found a cutoff of a maximum of 4 SNP differences for an ex vivo/in vivo evolution time of up to 40 days using the fastest mutation rate known (15). However, for prolonged outbreaks such as outbreak 1, which lasted more than 2 years, one may need to set a larger cutoff as the expected number of SNPs is proportional to the ex vivo/in vivo evolution time. Therefore, for prolonged outbreaks, differentiation of outbreak isolates from background isolates becomes more challenging, as larger cutoffs may falsely rule in background isolates as outbreak isolates. Defining the S. Typhimurium core genome will help to further determine appropriate cutoff values for outbreak investigations.
Our study highlights the importance of defining the core genome at an appropriate level for epidemiological typing to retain a sufficient number of core genes/regions to achieve the resolution required for short- and long-term epidemiology. For Salmonella, there is a clear subspecies structure and good separation of serotypes; defining the core genome at serovar level offers the best resolution for epidemiological typing of a serovar. For species that contain high diversity and with less distinctive subspecies structure, the core genome at species level is likely to become progressively smaller as the number of genomes sequenced increases, which clearly would lead to a substantial loss of discriminatory power. The core genome of Escherichia coli is estimated to be only around 1,500 to 2,000 genes (33, 34). This led to the proposal of a soft core genome by Kaas et al. (34), which is defined as core genes found in 95% of all the analyzed genomes in contrast to the usual 100%. The E. coli soft core genome was found to consist of 3,051 genes, nearly twice the core genome defined traditionally (33, 34). We did not determine whether the soft core genome approach would alleviate the need to define the core genome at serovar level in Salmonella. Serovar-level core genome typing offers the best resolution to meet the needs of outbreak investigation, as only a few serovars predominate in human infections (35, 36).
The SARA genomes also contained rich genome content variation, including plasmids, prophages, and antibiotic resistance genes (see Text S1 in the supplemental material). An alternative approach to core genome typing is pan-genome typing, including all accessory genes to distinguish isolates based on the presence or absence of genes (14). The study by Leekitcharoenphon et al. (14) compared pan-genome typing and core genome typing and found that pan-genome typing had only 64% to 65% concordance with core genome typing based on the phylogenetic trees derived, and four outbreaks cannot be separated by pan-genome typing. Clearly accessory genome variation within and between outbreaks is not sufficient to support a high-resolution epidemiological typing.
In conclusion, our analysis of 21 SARA strains and 47 publicly available genomes found that the core genome of S. Typhimurium is significantly larger than the Salmonella core genome. We show that the S. Typhimurium core genome offers higher resolution for epidemiological typing of outbreaks. In this study, we employed a core genome SNP-based approach for epidemiological typing for the comparison of discriminatory power. Genome typing can be based on either SNP comparison or gene by gene comparison (37). The S. Typhimurium core genome defined in this study allows standardization of the genome regions to be used for high-resolution typing for genomic surveillance and public health investigation of outbreaks.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Health and Medical Research Council of Australia.
We thank Ken Sanderson for the SARA strains.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.03407-14.
REFERENCES
- 1.Hohmann EL. 2001. Nontyphoidal salmonellosis. Clin Infect Dis 32:263–269. doi: 10.1086/318457. [DOI] [PubMed] [Google Scholar]
- 2.Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. 2012. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 13:601–612. doi: 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porwollik S, Wong RM, McClelland M. 2002. Evolutionary genomics of Salmonella: gene acquisitions revealed by microarray analysis. Proc Natl Acad Sci U S A 99:8956–8961. doi: 10.1073/pnas.122153699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beltran P, Plock SA, Smith NH, Whittam TS, Old DC, Selander RK. 1991. Reference collection of strains of the Salmonella Typhimurium complex from natural populations. J Gen Microbial 137:601–606. doi: 10.1099/00221287-137-3-601. [DOI] [PubMed] [Google Scholar]
- 5.Bell RL, Gonzalez-Escalona N, Stones R, Brown EW. 2011. Phylogenetic evaluation of the ‘Typhimurium’ complex of Salmonella strains using a seven-gene multi-locus sequence analysis. Infect Genet Evol 11:83–91. doi: 10.1016/j.meegid.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Achtman M, Hale J, Murphy RA, Boyd EF, Porwollik S. 2013. Population structures in the SARA and SARB reference collections of Salmonella enterica according to MLST, MLEE and microarray hybridization. Infect Genet Evol 16:314–325. doi: 10.1016/j.meegid.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Anderson ES, Ward LR, Saxe MJ, de Sa JD. 1977. Bacteriophage-typing designations of Salmonella Typhimurium. J Hyg (Lond) 78:297–300. doi: 10.1017/S0022172400056187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsen JE, Skov MN, Angen O, Threlfall EJ, Bisgaard M. 1997. Genomic relationships between selected phage types of Salmonella enterica subsp. enterica serotype Typhimurium defined by ribotyping, IS200 typing and PFGE. Microbiology 143:1471–1479. doi: 10.1099/00221287-143-4-1471. [DOI] [PubMed] [Google Scholar]
- 9.Pang S, Octavia S, Reeves PR, Wang Q, Gilbert GL, Sintchenko V, Lan R. 2012. Genetic relationships of phage types and single nucleotide polymorphism typing of Salmonella enterica serovar Typhimurium. J Clin Microbiol 50:727–734. doi: 10.1128/JCM.01284-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sangal V, Harbottle H, Mazzoni CJ, Helmuth R, Guerra B, Didelot X, Paglietti B, Rabsch W, Brisse S, Weill FX, Roumagnac P, Achtman M. 2010. Evolution and population structure of Salmonella enterica serovar Newport. J Bacteriol 192:6465–6476. doi: 10.1128/JB.00969-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sintchenko V, Wang Q, Howard P, Ha CW, Kardamanidis K, Musto J, Gilbert GL. 2012. Improving resolution of public health surveillance for human Salmonella enterica serovar Typhimurium infection: 3 years of prospective multiple-locus variable-number tandem-repeat analysis (MLVA). BMC Infect Dis 12:78. doi: 10.1186/1471-2334-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wuyts V, Mattheus W, De Laminne de Bex G, Wildemauwe C, Roosens NH, Marchal K, De Keersmaecker SC, Bertrand S. 2013. MLVA as a tool for public health surveillance of human Salmonella Typhimurium: prospective study in Belgium and evaluation of MLVA loci stability. PLoS One 8:e84055. doi: 10.1371/journal.pone.0084055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenke C, Lindstedt BA, Harmsen D, Karch H, Brandal LT, Mellmann A. 2011. Comparison of multilocus variable-number tandem-repeat analysis and multilocus sequence typing for differentiation of hemolytic-uremic syndrome-associated Escherichia coli (HUSEC) collection strains. J Clin Microbiol 49:3644–3646. doi: 10.1128/JCM.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leekitcharoenphon P, Nielsen EM, Kaas RS, Lund O, Aarestrup FM. 2014. Evaluation of whole genome sequencing for outbreak detection of Salmonella enterica. PLoS One 9:e87991. doi: 10.1371/journal.pone.0087991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Octavia S, Wang Q, Tanaka MM, Kaur S, Sintchenko V, Lan R. 2015. Delineating community outbreaks of Salmonella enterica serovar Typhimurium by use of whole-genome sequencing: insights into genomic variability within an outbreak. J Clin Microbiol 53:1063–1071. doi: 10.1128/JCM.03235-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leekitcharoenphon P, Lukjancenko O, Friis C, Aarestrup FM, Ussery DW. 2012. Genomic variation in Salmonella enterica core genes for epidemiological typing. BMC Genomics 13:88. doi: 10.1186/1471-2164-13-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pang S, Octavia S, Feng L, Liu B, Reeves PR, Lan R, Wang L. 2013. Genomic diversity and adaptation of Salmonella enterica serovar Typhimurium from analysis of six genomes of different phage types. BMC Genomics 14:718. doi: 10.1186/1471-2164-14-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Osterman AL, Overbeek RA, McNeil LK, Paczian T, Parrello B, Pusch GD, Reich C, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bottacini F, Medini D, Pavesi A, Turroni F, Foroni E, Riley D, Giubellini V, Tettelin H, van Sinderen D, Ventura M. 2010. Comparative genomics of the genus Bifidobacterium. Microbiology 156:3243–3254. doi: 10.1099/mic.0.039545-0. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delcher AL, Salzberg SL, Phillippy AM. 2003. Using MUMmer to identify similar regions in large sequence sets. Curr Protoc Bioinformatics Chapter 10:Unit 10.3. [DOI] [PubMed] [Google Scholar]
- 25.Kingsley RA, Msefula CL, Thomson NR, Kariuki S, Holt KE, Gordon MA, Harris D, Clarke L, Whitehead S, Sangal V, Marsh K, Achtman M, Molyneux ME, Cormican M, Parkhill J, MacLennan CA, Heyderman RS, Dougan G. 2009. Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res 19:2279–2287. doi: 10.1101/gr.091017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawkey J, Edwards DJ, Dimovski K, Hiley L, Billman-Jacobe H, Hogg G, Holt KE. 2013. Evidence of microevolution of Salmonella Typhimurium during a series of egg-associated outbreaks linked to a single chicken farm. BMC Genomics 14:800. doi: 10.1186/1471-2164-14-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Bio Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellmann A, Mosters J, Bartelt E, Roggentin P, Ammon A, Friedrich A, Karch H, Harmsen D. 2004. Sequence-based typing of flaB is a more stable screening tool than typing of flaA for monitoring of Campylobacter populations. J Clin Microbiol 42:4840–4842. doi: 10.1128/JCM.42.10.4840-4842.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunter PR, Gaston MA. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J Clin Microbiol 26:2465–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Julenius K, Pedersen AG. 2006. Protein evolution is faster outside the cell. Mol Biol Evol 23:2039–2048. doi: 10.1093/molbev/msl081. [DOI] [PubMed] [Google Scholar]
- 31.Rzhetsky A, Nei M. 1992. Statistical properties of the ordinary least-squares, generalized least-squares, and minimum-evolution methods of phylogenetic inference. J Mol Evol 35:367–375. doi: 10.1007/BF00161174. [DOI] [PubMed] [Google Scholar]
- 32.Hu H, Lan R, Reeves PR. 2006. Adaptation of multilocus sequencing for studying variation within a major clone: evolutionary relationships of Salmonella enterica serovar Typhimurium. Genetics 172:743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukjancenko O, Wassenaar TM, Ussery DW. 2010. Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol 60:708–720. doi: 10.1007/s00248-010-9717-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaas RS, Friis C, Ussery DW, Aarestrup FM. 2012. Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics 13:577. doi: 10.1186/1471-2164-13-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffmann M, Zhao S, Luo Y, Li C, Folster JP, Whichard J, Allard MW, Brown EW, McDermott PF. 2012. Genome sequences of five Salmonella enterica serovar Heidelberg isolates associated with a 2011 multistate outbreak in the United States. J Bacteriol 194:3274–3275. doi: 10.1128/JB.00419-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galanis E, Lo Fo Wong DM, Patrick ME, Binsztein N, Cieslik A, Chalermchikit T, Aidara-Kane A, Ellis A, Angulo FJ, Wegener HC; World Health Organization Global Salm-Surv. 2006. Web-based surveillance and global Salmonella distribution, 2000-2002. Emerg Infect Dis 12:381–388. doi: 10.3201/eid1203.050854, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, McCarthy ND. 2013. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol 11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.