

Abstract
Background
Defence mechanisms of organisms are shaped by their lifestyle, environment and pathogen pressure. Carpenter ants are social insects which live in huge colonies comprising genetically closely related individuals in high densities within nests. This lifestyle potentially facilitates the rapid spread of pathogens between individuals. In concert with their innate immune system, social insects may apply external immune defences to manipulate the microbial community among individuals and within nests. Additionally, carpenter ants carry a mutualistic intracellular and obligate endosymbiotic bacterium, possibly maintained and regulated by the innate immune system. Thus, different selective forces could shape internal immune defences of Camponotus floridanus.
Results
The immune gene repertoire of C. floridanus was investigated by re-evaluating its genome sequence combined with a full transcriptome analysis of immune challenged and control animals using Illumina sequencing. The genome was re-annotated by mapping transcriptome reads and masking repeats. A total of 978 protein sequences were characterised further by annotating functional domains, leading to a change in their original annotation regarding function and domain composition in about 8 % of all proteins. Based on homology analysis with key components of major immune pathways of insects, the C. floridanus immune-related genes were compared to those of Drosophila melanogaster, Apis mellifera, and other hymenoptera. This analysis revealed that overall the immune system of carpenter ants comprises many components found in these insects. In addition, several C. floridanus specific genes of yet unknown functions but which are strongly induced after immune challenge were discovered. In contrast to solitary insects like Drosophila or the hymenopteran Nasonia vitripennis, the number of genes encoding pattern recognition receptors specific for bacterial peptidoglycan (PGN) and a variety of known antimicrobial peptide (AMP) genes is lower in C. floridanus. The comparative analysis of gene expression post immune-challenge in different developmental stages of C. floridanus suggests a stronger induction of immune gene expression in larvae in comparison to adults.
Conclusions
The comparison of the immune system of C. floridanus with that of other insects revealed the presence of a broad immune repertoire. However, the relatively low number of PGN recognition proteins and AMPs, the identification of Camponotus specific putative immune genes, and stage specific differences in immune gene regulation reflects Camponotus specific evolution including adaptations to its lifestyle.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1748-1) contains supplementary material, which is available to authorized users.
Keywords: Camponotus floridanus, Carpenter ant, Transcriptome, Re-annotation, Immune system
Background
Insects are among the most successful animal life forms on Earth in terms of species richness and abundance. Like all other living organisms they are under permanent threat of infection by harmful microorganisms.
Insects are not endowed with an adaptive immune system and must rely entirely on innate immune mechanisms or externally applied immune defences [1, 2]. Invading microorganisms that break the primary passive protective barriers such as the cuticle or the peritrophic membrane in the gut encounter immediate-acting defence strategies such as phagocytic cells, phenoloxidase activity and reactive oxygen species. As a second line of defence a powerful antimicrobial immune response is mounted, mainly based on AMPs but also including serine proteases, stress factors and factors involved in opsonisation and clotting [3, 4]. Based on several structural features, the AMPs can be classified into several groups such as α-helical peptides, glycine-rich peptides, cysteine-rich peptides or proline-rich peptides [5].
Detection of microbial invaders is achieved by pattern recognition receptors (PRRs) which recognise conserved structural motifs of the microorganisms such as DAP- or Lys-containing PGN of respectively Gram-negative or Gram-positive bacteria [4]. The conserved molecular patterns of the microbes they recognise are called microbe associated molecular patterns (MAMPs) [6]. These PRRs then interact with cellular signalling systems such as Toll, IMD, Jak-Stat, and JNK pathways, which ultimately lead to the activation of an immune response [7, 8]. Early work on the Drosophila melanogaster immune system already revealed striking similarities of these signal transduction pathways with those of vertebrates. For instance, the identification of the Drosophila Toll receptor was a milestone discovery, since later it was found that related PRRs of vertebrates, the so-called Toll-like receptors, also play a dominant role in the innate immune system of vertebrates [8].
Here we characterise the immune system of the carpenter ant C. floridanus. These ants live in huge colonies of genetically highly related animals. The high density of closely related individuals within the nest may pose specific hygiene problems since pathogen transfer may be facilitated by the close contact of colony members. On the other hand, social insects have evolved many additional hygienic measures on the colony level which may improve health of the individuals, such as cleaning behaviours or the use of other external immune defences such as the application of antimicrobial secretions, a phenomenon termed ‘social immunity’ [2, 9, 10]. Thus, not only the canonical genes encoding AMPs or factors involved in signalling pathways but also genes encoding traits involved in external or social immune defences should be viewed as part of the immune system of a social insect [2]. For example, Le Conte and co-workers discovered several genes which might contribute to social immunity in honey bees [11]. A striking new finding was reported recently for the closely related ant C. pennsylvanicus, in which a Cathepsin D like protease was found to be transmitted to other ants by trophallaxis, leading to an increased infection resistance in the recipients [12]. The first genome sequence of a social insect, the honey bee, revealed an apparently low number of genes with immune-related functions as compared to solitary insects [13]. Accordingly, it was suggested that external immune defences including social immunity may have alleviated selection pressure from the canonical innate immune factors as internal and external immune defences may trade off against each other. However, the recent honey bee genome upgrade identified about 5000 more protein encoding genes than previously reported, which need to be analysed carefully in the future [14]. In addition, the genome sequences of several other hymenopteran and dipteran species reveal that dipterans appear to have an unusually high number of immune genes and that a social lifestyle may not directly correlate with this number [15].
A specific feature of C. floridanus is its obligate interaction with an intracellular mutualistic y-Proteobacterium, Blochmannia floridanus, which resides in midgut cells and in the ovaries and supplements nutrients to its host [16]. The host recognises the endosymbiont as non-self and the immune system can therefore play a role in maintenance and regulation of the chronic infection by the endosymbiont [17], while on the other side it has to defeat pathogenic microorganisms, thus possibly requiring specific adaptations of its immune system [18]. For instance, previous work revealed a localised down-modulation of the immune response that is restricted to the midgut tissue of the ants and correlates with massive replication of the endosymbiont in this tissue [18].
To get insight into the immune system of the carpenter ant and to unravel possible specific adaptations to its lifestyle as a social insect living in an obligate mutualistic interaction with a bacterial endosymbiont, we re-evaluated the data available from the recently published genomic sequence of C. floridanus [19] and extended this dataset with genome-wide transcriptome data generated from animals with or without previous immune challenge. To identify and functionally annotate the key players of the C. floridanus immune response and to determine the interactions among these players, we performed a detailed analysis including sequence and domain analysis and pathway annotation (including signalogs [20]). Transferring annotation of proteins at a domain level allows more accurate functional inference [21] and is useful for predicting the function of multi-domain proteins [22] and novel domain combinations that possibly give rise to new protein functions [23]. Furthermore, we compared the C. floridanus immune genes and pathways to other recently sequenced ant genomes including Acromyrmex echinatior, Atta cephalotes, Cerapachys biroi, Harpegnathos saltator, Linipithema humile, Pogonomyrmex barbatus, Solenopsis invicta [19, 24–29]. We used clusters of orthologous genes and protein families (e.g. according to PFAM) for many of the above-mentioned comparisons and thus the comparisons based on these entities are quite broad. Nevertheless, regarding the number of complete genomes available there are limitations for drawing general conclusions. Moreover, other hymenoptera such as the honey bee A. mellifera [13], the solitary parasitic wasp Nasonia vitripennis [30], and, as a model organism, D. melanogaster [31], served to investigate possible differences in the immune system of endosymbiont-bearing, eu-social and solitary insects.
Results and discussion
C. floridanus genome structure and re-annotation
To establish the immune repertoire of C. floridanus the full transcriptome was analysed by Illumina sequencing, comparing challenged and unchallenged animals (pools of whole larvae L2 and workers W2 for each sample). Sequencing resulted in 125,873,897 reads for immune-challenged and 118,142,837 reads for untreated animals. Sequence statistics of Illumina sequencing are listed in Table 1. The first C. floridanus genome annotation (v3.3) contains 17,064 protein-coding genes [19]. The updated version of the previous annotation is labelled as cflo_OGSv3.3 (available at http://hymenopteragenome.org/camponotus/). Compared with the previously published annotation of the C. floridanus genome, the optimised Augustus software with species-specific parameters applied here predicted 15,631 protein-coding genes based on data from C. floridanus expressed sequence tags (ESTs) and the large-scale Illumina sequencing data (raw reads and assembled reads) presented here. Our revised annotation counts fewer genes and an increase in the number of multi-exon genes as compared to the published version v3.3. The accuracy of annotation for both was improved using the latest version of the gene prediction tool Augustus v2.7. Furthermore, over-prediction (false positives) of single exon genes was reduced by this software. The improved quality of the annotation was further validated as 14,956 (81.41 %) predicted transcripts could now be supported by extrinsic evidence such as introns and exons hints generated by sources such as Illumina sequence data and ESTs. The new annotation predicts a total of 18,369 proteins as compared to 17,064 proteins in the previous version v3.3. We found 1928 genes with two or more alternative transcripts. The optimised parameters with 80.8 % exon level accuracy allowed the improvement of annotation of the C. floridanus genome (http://bioinf.uni-greifswald.de/augustus/binaries/species/), e.g. with regard to splicing events. The prediction accuracy of Augustus with the new optimised parameters is tabulated in Additional file 1: Table S1. The optimised parameters are provided as part of the latest Augustus package (http://bioinf.uni-greifswald.de/augustus/submission/). Bonasio and co-workers (2011) detected 7583 alternative splicing events in 2538 genes (cflo_OGSv3.3) overall, while our analysis revealed 1928 genes affected by alternative splicing events coding for 4666 alternative transcripts (Additional file 2: Table S2). However, the OGSv3.3 data available for C. floridanus contain 17,064 transcripts and 17,064 proteins without distinction between alternative isoforms and thus cannot be used for further analysis. The new data reported here can be accessed at our web repository (http://camponotus.bioapps.biozentrum.uni-wuerzburg.de) and distinguish the alternative splicing products of the genes with the suffix in the accession number as t1, t2, t3 etc. The exact distribution of alternative transcripts over the annotated 15,631 genes is listed in Additional file 1: Table S1.
Table 1.
Quantitative overview on the transcriptome sequencing data
| (A) Illumina sequencing – quantitative overview | |||
| Immune challenged | Non-immune challenged | ||
| Sequencing | Paired end 2 x 50 bp | ||
| Total number of reads | 125,873,897 | 118,142,837 | |
| Median insert length (bp) | 176 bp | 163 bp | |
| Overall mapping rate | 87.4 % | 88.6 % | |
| Mapping to Camponotus genome | 99.45 % | 99.37 % | |
| Mapping to Blochmannia genome | 0.55 % | 0.63 % | |
| (B) Repeats distribution | |||
| Number of elements | Length occupied | Percentage of sequence | |
| LTR elements | 662 | 445,584 bp | 0.19 % |
| DNA elements | 163 | 44,183 bp | 0.02 % |
| Unclassified | 41,722 | 14,401,396 bp | 6.13 % |
| Total interspersed repeats | - | 14,891,163 bp | 6.34 % |
| Small RNA | 26 | 13,938 bp | 0.01 % |
| Satellites | 50 | 9914 bp | 0.00 % |
| Simple repeats | 67,608 | 3,996,593 bp | 1.70 % |
| Low complexity | 284,104 | 15,360,412 bp | 6.54 % |
We compared the improved genome annotation of C. floridanus to published data on other genomes, showing that C. floridanus has fewer repetitive elements (15.05 %) than D. melanogaster (27.38 %) or N. vitripennis (24.31 %), but more than A. mellifera (6.86 %). Using the major database of repetitive elements, Repbase [32], we list 12 specific repetitive elements for the C. floridanus genome and we give a summary of these elements in Table 1. Based on a de novo repeat library constructed with the RepeatModeler programme we detected 62 repetitive elements in the C. floridanus genome. With the assembled repeat library, 14.57 % of the C. floridanus genome was identified to contain repetitive sequences, consisting of 6.34 % of interspersed repeat elements, 1.70 % of simple repeats, 6.54 % low complexity stretches, and 0.01 % of small RNAs and satellites (Table 1).
Functional annotation and classification
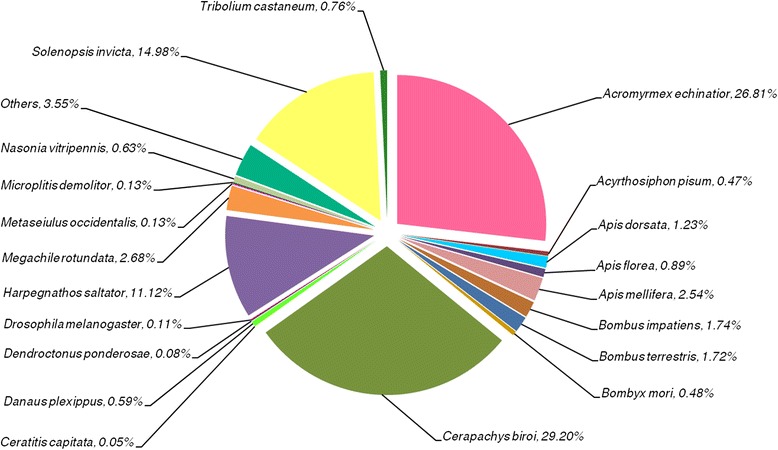
In comparison to the previous C. floridanus genome annotation (v3.3) we found 978 more proteins with functional domains (Additional file 3: Figure S1; new annotation Cflo-New). These protein sequences were further analysed with regards to their function. The total number of additional proteins identified was higher but not all of them contained functional domains. We further compared the changes in level-2 GO annotations in all three GO categories, i.e. biological process, molecular function and cellular component. As expected, based on best hits of sequence-based similarity searches we found that the C. floridanus sequences are generally most closely related to recently sequenced ant species (Fig. 1). Furthermore, we examined their overall functions. Among the 18,369 proteins, the software Blast2GO assigned level 2 GO terms to 8490 proteins and stressed important functions (Fig. 2). Regarding the term “biological process” (Fig. 2a), various “cellular processes” (green) were most abundant (19.98 %), regarding “molecular function” (Fig. 2b), enzymes presented the highest fraction among the proteins (“catalytic activity”, 40.07 %, green), directly followed by “binding” with 39 %. Regarding “cellular compartment” the subcategory “Cell” with 33.74 % was found to be most abundant (Fig. 2c). However, consider that two thirds of the proteins are located in membranes, organelles or part of macromolecular complexes (Fig. 2 shows in detail all other functional categories). In total 7143 proteins of Cflo(v3.3) could be annotated by Blast2GO. The proportion of Cflo(v3.3) proteins annotated previously falling into each functional category is shown in Additional file 4: Figure S2. Comparing our functional annotation with Cflo(v3.3) in terms of GO classification, we note an increase of 18 to 23 % regarding successful subcategory assignments covering all three major GO categories (13,354 terms in comparison to 10,819 terms in Cflo(v3.3) for “biological process”; 6088 terms versus 5141 in “molecular function”, and 5441 terms versus 4568 terms respectively for the GO category “cellular component”; Additional file 5: Table S3). In conclusion, by refining the accuracy of prediction regarding single and multiple transcript genes and evidence for their expression the transcriptome sequencing improved the annotation of the C. floridanus proteome, the identification of repetitive elements as well as alternative splicing predictions.
Fig. 1.
Distribution of closest related sequences to C. floridanus proteins. All proteins within the NCBI nr database (without C. floridanus) were analysed using the sequence comparison tool BLAST [118] with an e-value cut-off of 1e-5. The best sequence-similar hit of each re-annotated C. floridanus sequence was used for the analysis
Fig. 2.
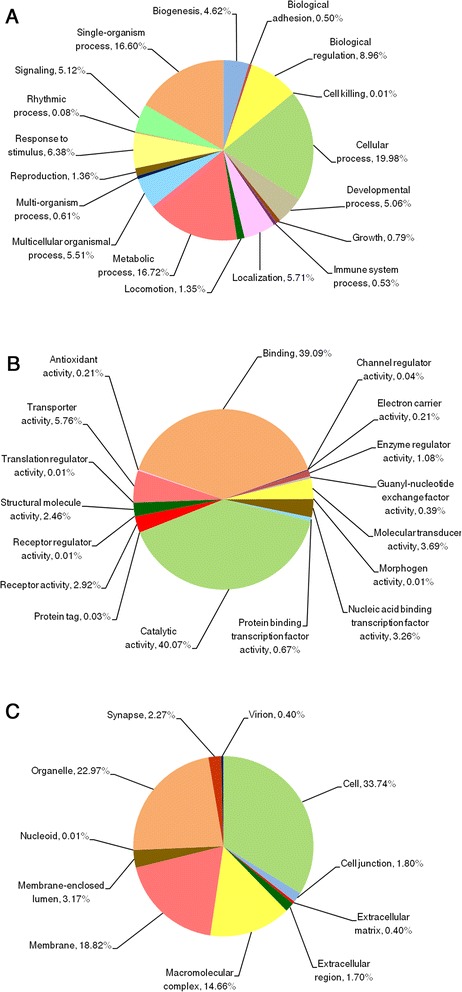
Distribution of functions in C. floridanus proteins. Categorisation of 8490 proteins of C. floridanus in GO terms (level two) for a biological process, b molecular function, and c cellular component with a filter score e-value cut-off of 1e-5
Comparison of the immune gene repertoire of C.floridanus with different insect species
We present the C. floridanus immunome using sequence-based protein orthology with the previously published data of D. melanogaster, A. mellifera and N. vitripennis assuming their functions and modes of action are conserved (Fig. 3). C. floridanus shares 307 orthologs with N. vitripennis, 271 orthologs with D. melanogaster, and 221 with A. mellifera. All four species share 65 immune protein orthologs which mostly comprise proteins involved in core immune signalling pathways, PRRs or serine proteases (Additional file 6: Table S4). Overall, in accordance with previous findings [33] signalling pathways seem to be more conserved in comparison to effector molecules that show higher levels of taxon specificity. We classified the 474 immune related genes of C. floridanus identified here into several categories including microbial recognition, signalling pathways (Toll, Jak-Stat, IMD and JNK), AMPs, phagocytosis, melanisation, encapsulation, cytoskeleton immune proteins, antiviral defence, coagulation, haematopoiesis and other immune responses (Fig. 4). In the Additional file 7: Table S5 the accession numbers and annotations of all categorised immune proteins of C. floridanus are listed.
Fig. 3.
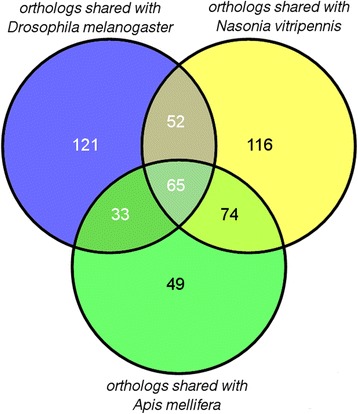
Immune gene repertoire of C. floridanus shared with different insect species. The Venn diagram shows the number of immune protein orthologs (including alternative splice isoforms) of C. floridanus shared with D. melanogaster, N. vitripennis and A. mellifera
Fig. 4.
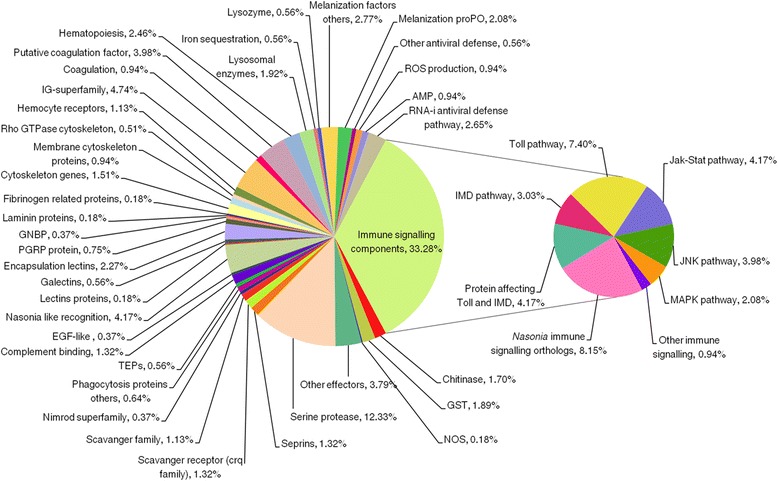
Functions of immune-related genes of C. floridanus. The pie chart shows the percentage of immune genes (474 genes in total) in each functional sub-category relative to the entire set of immune genes. The category ’immune signalling components’ is further divided into different pathways
Signal transduction via major immune signalling pathways
Recognition of a pathogen is the first step in promoting an efficient immune response. Therefore, pattern recognition receptors (PRRs) recognise the so-called MAMPs such as bacterial PGN or fungal beta-1,3-glucans [4, 6]. Upon binding to microbial components they trigger the activation of signal transduction systems either directly or after a series of proteolytic events mediated by serine proteases, ultimately resulting in the activation of antimicrobial defence mechanisms including the expression of AMPs [4]. Due to the in-depth knowledge and functional assays of its major immune signalling pathways [34, 35] we used D. melanogaster as a template to identify components of the Toll, IMD, Jak-Stat, and JNK signal transduction pathways in C. floridanus. The reconstructed immune related signalling repertoire of C. floridanus is highly conserved and largely similar to the signalling components identified by experiments mainly conducted with Drosophila [36].
Toll signalling pathway
The Toll pathway of insects is mainly activated by fungal pathogens and Gram-positive bacteria. The Toll pathway not only regulates the antimicrobial response but is also required for proper haemocyte proliferation [4, 37]. Therefore, Toll activation leads to a coordinated immune response that comprises both cellular and humoral immunity [38]. The Toll signalling pathway was found to be highly conserved in terms of the presence of homologs in C. floridanus (Fig. 5). Recognition of Lys-type PGN characteristic for most Gram-positive bacteria or of fungal beta-1,3-glucans by specific PRRs leads to the activation of proteolytic cascades which finally activate the Toll-dependent signalling cascade. In C. floridanus three PRRs likely feeding into the Toll pathway are found: a PGRP-SA (Cflo_N_g8526t1) which according to sequence homology probably recognises Lys-type PGN, and two proteins annotated as beta-1,3-glucan binding proteins (Cflo_N_g15215t1 and Cflo_N_g5742t1) with high homologies to both GNBP1 and GNBP3 of D. melanogaster. GNBP1 is known to perceive Lys-type PGN, while GNBP3 recognises fungal cell wall components [39]. The homology data of the C. floridanus proteins do not allow a clear identification of the signal sensed by the two GNBPs. However, the previous expression data acquired post immune challenge of C. floridanus with Gram-negative or Gram-positive bacteria revealed a long-lasting up-regulation of the gene encoding one of the GNBPs (EFN66519.1; Cflo_N_g5742t1) only after infection with Gram-positive bacteria [17]. This suggests that this Camponotus protein may be able to recognise Lys-type PGN and thus may be a functional homolog of GNBP1 of D. melanogaster (Fig. 5). In addition, C. floridanus encodes a homolog of the protease Persephone which was previously shown to be involved in the detection of danger signals indicative for infection with Gram-positive bacteria and fungi [40]. In D. melanogaster the Toll pathway is triggered upon microbially induced proteolytic cleavage of the circulating cytokine-like ligand molecule Spätzle that binds to the Toll receptor, thus finally leading to the nuclear translocation of the NF-κB-like transcription factors Dorsal and DIF (Dorsal-related immunity factor). A single gene encoding Dorsal is present in C. floridanus, but similar to A.mellifera no ortholog of DIF was found. This is in agreement with the recent suggestion that DIF belongs to a highly derived branch possibly found only in brachyceran flies [13]. Therefore, in C. floridanus Dorsal appears to be the unique transcription factor required for induction of AMPs during the Toll mediated immune response.
Fig. 5.
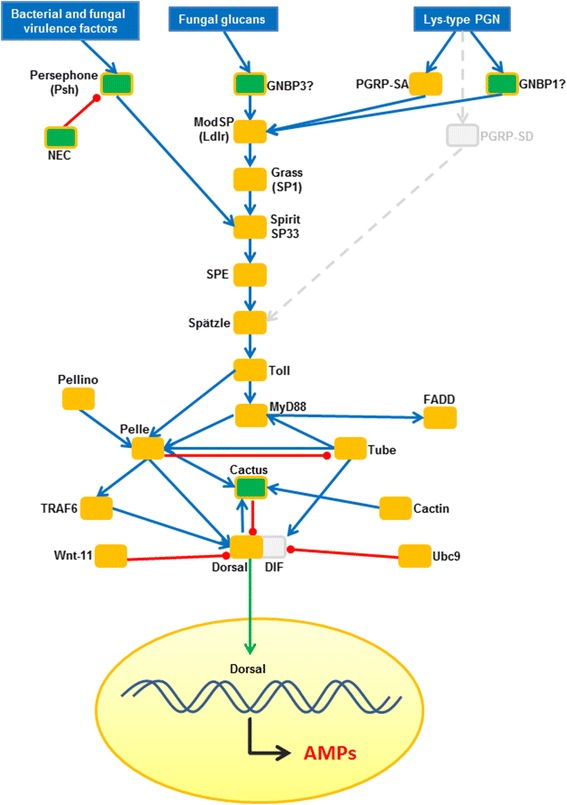
The Toll signalling pathway of C. floridanus. All identified signalling components are mapped on the comprehensive immune network of D. melanogaster. The names of the factors correspond to the Drosophila designations. Connectivity among nodes is based either on positive attribute (blue arrow) or negative attribute (red arrow). Missing components are shown in grey colour. Nuclear translocation is shown by a green arrow. Factors significantly upregulated on the transcriptional level upon immune-challenge are shown by green boxes
IMD and JNK signalling pathways
The activation of the IMD pathway of insects is triggered after infection predominantly by Gram-negative bacteria and is also involved in the induction of expression of AMPs [4, 41]. C. floridanus harbours the Gram-negative obligate intracellular endosymbiont B. floridanus and it was suggested that the IMD pathway may also contribute to control and tolerance of the endosymbiont [17, 18]. The present data show that most components of the IMD pathway are present in C. floridanus (Fig. 6). However, in C. floridanus DAP-type PGN mainly characteristic for Gram-negative bacteria is recognised via a single signal-transducing PRR, originally annotated as PGRP-LE, while the organism appeared to lack PGRP-LC-like receptors. The careful re-evaluation of the genomic data revealed, however, that this protein has a much longer N-terminal sequence and comprises a transmembrane domain at sequence position 264 to 287. In fact, a cluster analysis revealed that the protein is more related to PGRP-LC sequences of D. melanogaster and A. mellifera than to PGRP-LE of D. melanogaster. Thus, the C. floridanus protein is likely to be a PGRP-LC homolog (Cflo_N_g10272t1) and appears to be the only receptor for DAP-type PGN that activates the IMD pathway. In addition, two PGN recognition proteins with regulatory function (PGRP-SC and PGRP-LB) were previously found [18]. Due to the presence of an amidase domain both of them may down-modulate the signal transduction pathways by cleavage of PGN [6, 42]. In fact, PGRP-LB was recently found to be implicated in the tolerance towards the obligate intracellular endosymbiont B. floridanus in the midgut tissue during pupation of the animals. PRGP-LB is highly up-regulated only in the midgut tissue and not in other parts of the pupa body cavity, coinciding with a massive multiplication of the endosymbiont and a reduction of the immune competence in this tissue [18].
Fig. 6.
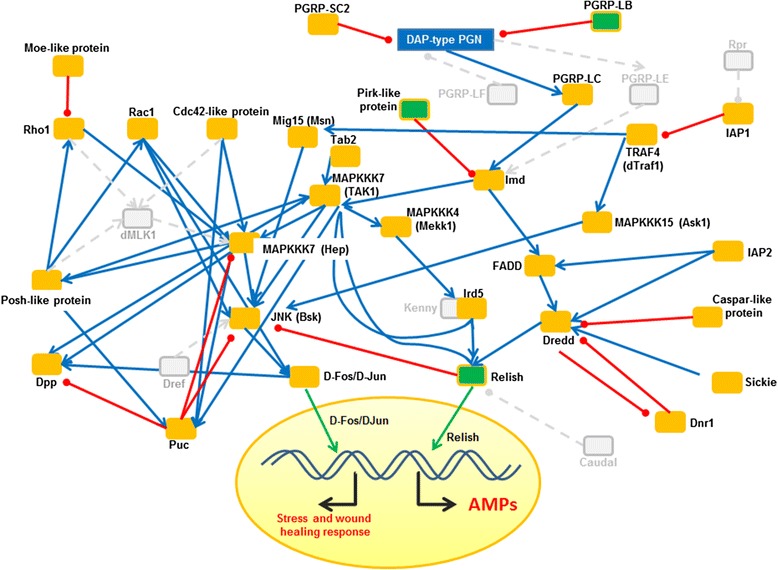
The Imd and JNK pathways of C. floridanus. All identified signalling components are mapped on the comprehensive immune network of D. melanogaster. The names of the factors correspond to the Drosophila designations. Connectivity among nodes is based either on positive attribute (blue arrow) or negative attribute (red arrow). Missing components are shown in grey colour. Nuclear translocation is shown by a green arrow. Factors significantly upregulated on the transcriptional level upon immune-challenge are shown by green boxes
A major difference with regard to D. melanogaster in the cytosolic C. floridanus IMD signalling cascade concerns the so-called IKK complex which activates the NF-κB-like transcription factor Relish by phosphorylation [43]. In Drosophila the IKK complex is composed of the enzymatically active Ird5 subunit and the regulatory subunit Kenny. However, in C. floridanus the regulatory subunit is missing. It has been published that a Kenny mutant of D. melanogaster is highly susceptible to bacterial infections [44]. Iterative sequence analyses also verified the lack of the Kenny subunit in A. mellifera, N. vitripennis and other ant species, suggesting a common character of the IKK complex in hymenoptera. Whether the lack of the Kenny subunit may reflect a reduction in the immune potential of hymenoptera or whether so far unknown factors may be involved in building a functional IKK complex is not yet known.
The IMD pathway also leads to TAK1 (transforming growth factor β-activated kinase 1) mediated activation of the JNK signalling cascade [45]. JNK signalling contributes to regulation of many developmental processes [46], wound healing [47], activation of stress-protective proteins [48], inflammatory [49] and cellular immune responses [50]. The data reported here indicate that the JNK pathway in C. floridanus is quite similar to the pathway in D. melanogaster, since most of the core components of the JNK pathway of D. melanogaster have homologs in C. floridanus (Fig. 6).
Additional receptor proteins and Jak-Stat signalling pathway
Besides the above mentioned PRRs, we also identified proteins known to be involved in pathogen recognition and/or promotion of phagocytosis including scavenger receptors, croquemort family members, nimrod and draper orthologs, vitellogenin, galectins, c-type lectins, brain angiogenesis inhibitor 1 (BAI1), fibrinogen-related protein, down syndrome cell adhesion molecular (Dscam) and thioester containing proteins (TEPs). The latter are known to play a role in the Jak-Stat pathway, which contributes to stem cell regulation in the intestine of Drosophila and thus to midgut homeostasis [51, 52]. In C. floridanus all core components of the Jak-Stat pathway are present (Fig. 7) except extracellular ligand proteins identified in Drosophila, which activate the pathway and which are also not found in other insects, including the honey bee [13]. While TEPs and Turandot proteins are among the downstream effectors of the Jak-Stat pathway in D. melanogaster [53, 54], similar to A. mellifera no homologs of Turandot proteins were identified by BlastP searches in C. floridanus, but several TEPs were found. TEPs may play a role in insect immunity by promoting phagocytosis of bacteria [53, 55]. Most TEPs share the common CGEQ motif defining the thioester site, which allows the formation of a covalent bond to microbial surfaces [55]. However, several TEPs in insects lack the thioester motif, but these TEPs may act as adaptors for the initiation of the membrane attack complex as is found in vertebrate complement factors [53]. Sequence analyses revealed the presence of three TEP genes (TEP1: Cflo_N_g4492t1, TEP2: Cflo_N_g7345t1, and TEP3: Cflo_N_g9745t1) in C. floridanus, with TEP1 and TEP2 containing the CGEQ motif in the deduced amino acid sequences. For the gene encoding TEP3, two alternative transcripts were found (Cflo_N_g9745t1 and Cflo_N_g9745t2). Interestingly, only the CGEQ motif containing TEP1 of C. floridanus is up-regulated upon immune challenge (see below).
Fig. 7.
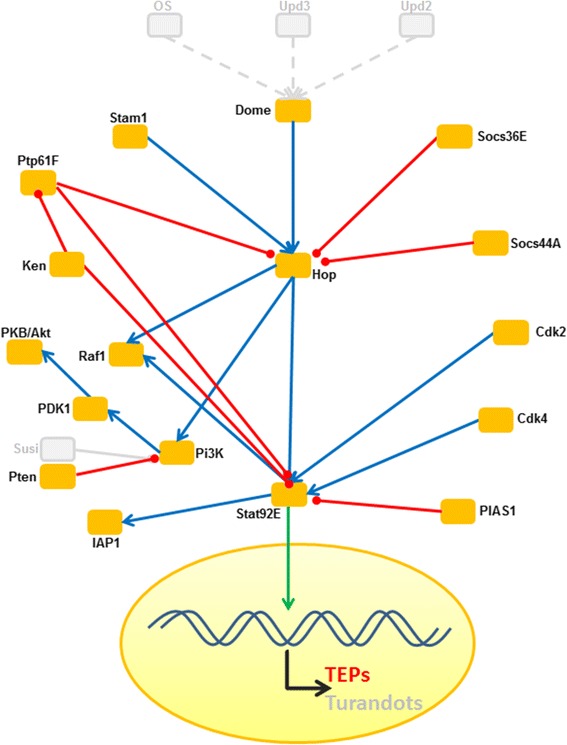
The Jak-Stat pathway of C. floridanus. All identified signalling components are mapped on the comprehensive immune network of D. melanogaster. The names of the factors correspond to the Drosophila designations. Connectivity among nodes is based either on positive attribute (blue arrow) or negative attribute (red arrow). Missing components are shown in grey colour. Nuclear translocation is shown by a green arrow. Factors significantly upregulated on the transcriptional level upon immune-challenge are shown by green boxes
Antimicrobial peptides of C. floridanus and other hymenoptera
The previous analysis of the C. floridanus genome revealed a relatively low number of known AMPs including two defensins, a hymenoptaecin, a tachystatin-like and a crustin-like peptide [56–58]. These peptides are also encoded by most of the other ants [58], however, the distribution pattern of AMPs is in general quite complex (Table 2). For example, several of the ants including C. floridanus lack a gene encoding abaecin, while other ants encode this AMP. Thus, similar to the previously described gain, loss and duplication of defensin genes [56], there is quite an extensive variability in the presence and number of antimicrobial peptide genes in the ant genomes (Table 2). Much alike in the honeybee, the number of predicted or confirmed AMPs appears to be relatively low in ants as compared to the solitary wasp N. vitripennis for which 44 AMPs were described [57]. The apparently low number of AMPs may be compensated by the astonishing gene structure of hymenoptaecin which in the ants is encoded as a huge precursor protein with several repeated hymenoptaecin domains. Proteolytic maturation of this precursor protein leads to a massive amplification of the immune response [56]. In addition, the hymenoptaecin gene is among the most strongly induced genes after immune challenge (see below). The apparently quite low number of AMPs and of PGN recognising PRRs as described above may relate to the social lifestyle of ants and bees as compared to the solitary and parasitic lifestyle of the wasp N. vitripennis, since social life might allow hygienic measures on the colony level [9]. In addition, ants produce a range of antimicrobial secretions that may be used to reduce pathogen pressure externally before an infection of the body occurs. As a consequence these external immune defence strategies may trade off against internal immune defences and may result in a reduction in the number of effector molecules [2, 10].
Table 2.
Antimicrobial peptides of C. floridanus and other hymenoptera
| AMPs | C. floridanus | H. saltator | L. humile | P. barbatus | A. cephalotes | S. invicta | A. echinatior | C. biroi | A. mellifera | N. vitripennis |
|---|---|---|---|---|---|---|---|---|---|---|
| Hymenoptaecin1) | 1 | 2 | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 2 |
| Defensin | 2 | 2 | 1 | 5 | 1 | 2 | 1 | 1 | 3 | 5 |
| Tachystatin-like | 2 | 2 | 3 | 2 | 1 | 3 | 2 | 3 | 1 | 3 |
| Crustin-like2) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | - | - | 1 |
| Abaecin | - | 1 | - | 1 | 1 | - | 1 | 1 | 1 | - |
| Melittin | - | - | - | - | - | - | - | - | 1 | - |
| Apisimin | - | - | - | - | - | - | - | - | 1 | - |
| Apidaecin | - | - | - | - | - | - | - | - | 5 | - |
| Navitripenicin | - | - | - | - | - | - | - | - | - | 4 |
| Nasonin3) | - | - | - | - | - | - | - | - | - | 14 |
| Nabaecin4) | - | - | - | - | - | - | - | - | 4 | |
| Glynavicin3) | - | - | - | - | - | - | - | - | - | 7 |
| Hisnavicin | - | - | - | - | - | - | - | - | - | 5 |
| Nahelixin | - | - | - | - | - | - | - | - | - | 1 |
1)Please note that here the number of genes present in the various species is indicated. In the ants the hymenoptaecin genes encode huge multipeptide precursor proteins which may give rise to several mature peptides (7 in the case of C. floridanus)
2)Adopted from Zhang and Zhu [58]
3)Adopted from Sackton et al. [33]
4)Nabaecin of N. vitripennis is considered as a member of the abaecin family. However, nabaecins belong to different orthologous cluster; therefore we separated nabaecins and abaecins
Prophenoloxidase, serine proteases and serpins
Several immune defence reactions in insects such as phagocytosis, melanisation and nodulation depend on phenoloxidase (PO) activity [59–61]. During the melanisation process toxic intermediates such as reactive oxygen species (ROS) may kill microbial invaders directly. Since phenoloxidase activity can also harm insect cells the enzyme is synthesised as an inactive precursor (Pro-PO). Pro-PO activation involves microbial recognition by PRRs and proteolytic cascades involving terminal serine proteases that finally cleave Pro-PO to its active form [60, 62]. Serpins negatively control the activity of PO and help to avoid overshooting melanisation and dangerous ROS production [63]. Phenoloxidases are related to arylphorins, hemocyanins and hexamerins [64]. Using query sequences from four insect species, BlastP searches resulted in eight significant hits. The first hit corresponds to the C. floridanus prophenoloxidase (Cflo_N_g1918t1), while the other hits are distributed among hemocyanin, arylphorin and hexamerin sequences. Thus, a single prophenoloxidase gene appears to be present in the C. floridanus genome.
In addition, we annotated 34 serine proteases and ten serine protease inhibitors in C. floridanus (Additional file 8: Table S6) using the AutoFACT v3.4 tool for functional annotation of gene models [65]. Among these are five putative immune related serine proteases and four serine protease inhibitors including one serpin that showed differential expression profiles after immune challenge (see below).
Chitinases, glutathione-S-transferases and nitric oxide synthase (NOS)
Enzymes with chitinase activity play an important role in the immune defence of insects by catalysing the breakdown of chitin, a linear polymer found in fungal pathogen cell walls consisting of β-1-4 linked N-acetylglucosamine [66]. Four conserved motifs (KXXXXXGGW, FDGXDLDWEYP, MXYDXXG and GXXXWXXDXD, where X is a non-specified amino acid) have been reported in catalytic domains of insect chitinases [67, 68]. Five prototypic chitinases from different insect species served as a standard for detection [68–71]. In C. floridanus we found putative 13 chitinases containing a variable number of the four conserved sequence motifs (Additional file 9: Table S7). Only two predicted proteins are endowed with all four chitinase signature motifs, while in five sequences no such motif was detected. Overall, there is not much variation in the number of chitinase genes encoded by the different insect species compared here (Additional file 10: Table S8).
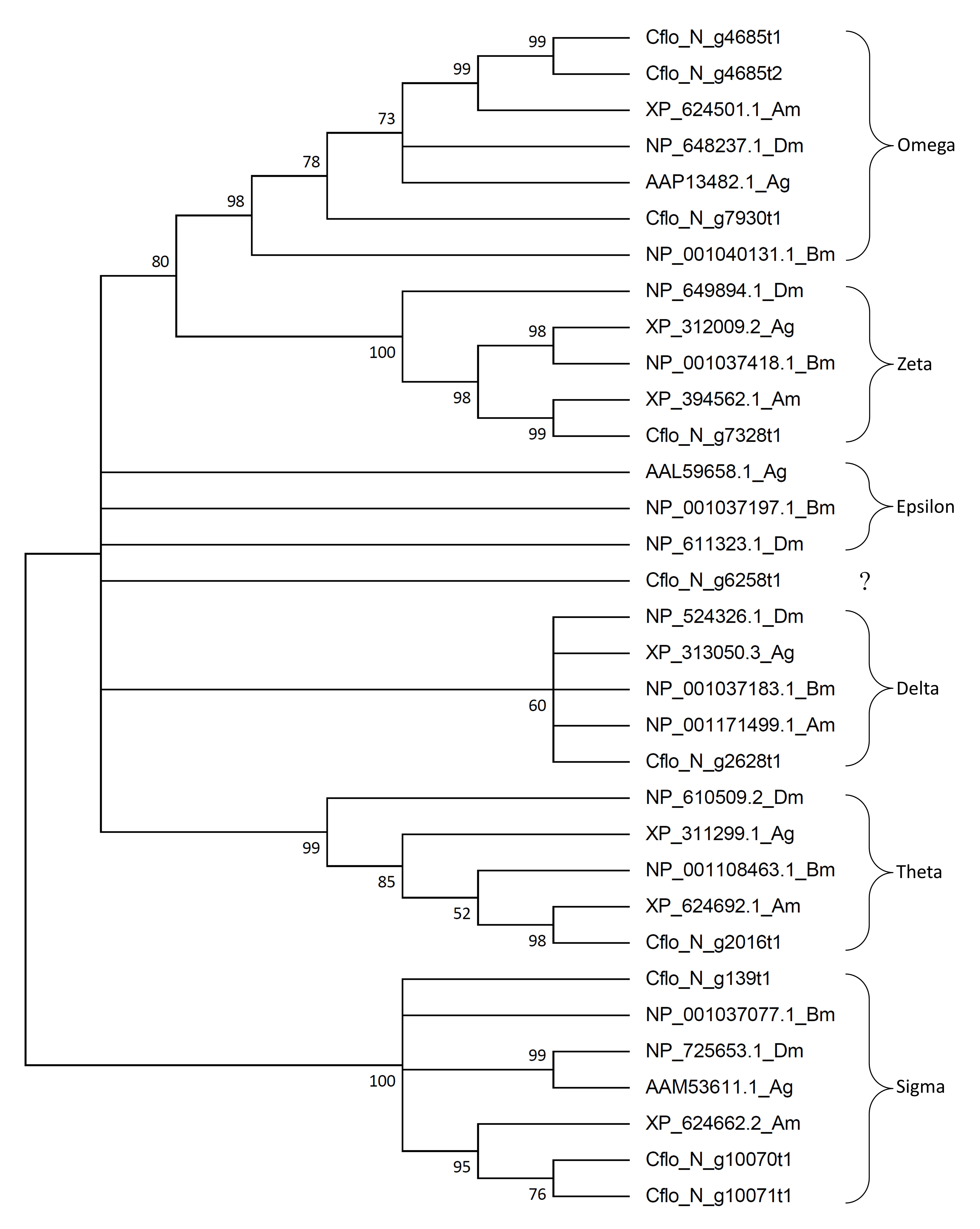
Glutathione S-transferases (GSTs) comprise a diverse family of dimeric enzymes that have attracted attention in insects because of their involvement in the defence towards insecticides [72]. Cytosolic GSTs in insects have been assigned to six classes including delta, epsilon, omega, sigma, theta and zeta [73, 74], and among them the delta and epsilon classes represent over 65 % of the total GST expansions. A recent phylogenetic study of insect GSTs suggested the evolution of the epsilon class from the delta class [66]. We predicted several C. floridanus GSTs that were classified into different classes on the basis of their sequence similarities and phylogenetic relationships with other insect species. Based on these approaches, 9 out of 10 identified C. floridanus GSTs were assigned to five different classes, including three in omega, three in sigma, one in each of delta, theta and zeta and one unclassified GST (Additional file 11: Figure S3). The absence of epsilon class GSTs in C. floridanus is in line with their absence in other Hymenoptera [75]. With the exception of D. melanogaster encoding 20 GSTs, there are only minor differences in the number of GSTs encoded by the other insects (between eight and eleven GSTs) (Additional file 10: Table S8).
NOS belongs to the family of enzymes which form nitric oxide (NO) from L-arginine and makes important contributions to the IMD pathway in activation of Relish, since NOS activity is required for a robust innate immune response to Gram-negative bacteria in Drosophila [76, 77]. In C. floridanus only a single gene (Cflo_N_g5430t1) codes for a protein that matched all criteria of a NOS. With the exception of A. cephalotes and N. vitripennis each encoding two NOS, the other ants and A. mellifera code for a single NOS (Additional file 10: Table S8).
Identification of genes differentially expressed (DEGs) after immune challenge
In the previous sections immune genes were identified mainly based on their similarity to already described immune genes of other insects. To further strengthen this analysis and to identify additional factors possibly involved in immune reactions, the transcriptome analysis presented here was performed with immune challenged and untreated animals (pooling larvae and adult workers). Genes found to be regulated by immune challenge are likely to be involved in immune functions, although it is known that there are many regulatory overlaps with stress responses other than immune challenge [78]. We used two different programmes to evaluate differentially expressed genes between these samples. The combination of the results from Cuffdiff (q-value < 0.05) and DESeq (p-value adjusted < 0.05) allowed the identification of 257 transcripts, which were significantly differentially expressed in response to bacterial challenge. To show at the same time amount of change and statistical significance, Volcano plots summarise the results (Fig. 8). Table S9 (Additional file 12) lists all genes that were up- or down-regulated by at least a factor of 2 including their annotation and log fold change expression value. Among the differentially expressed transcripts, ~20 % of transcripts were identified to code for known immune related proteins or proteins described to be differentially regulated after immune challenge in other insects. The percentage of immune-related genes in the genome is quite low. According to the functional categories shown in Fig. 2 only 0.53 % of genes are related to immune processes. Even when taking into account the limits of the accuracy of functional categories, all other signalling processes together encompass only 5.12 % of all genes. Hence, there is enrichment for immune related genes regarding the much higher percentage within all differentially regulated genes. Genes up-regulated after immune challenge encode well known immune related genes including those that encode PRRs and serine proteases (e.g. Snake and Stubble-like), proteins involved in signalling and transcription (e.g. nuclear factor NF-kappa-B p110 subunit (Relish), NF-kappa-B inhibitor (Cactus), as well as stress-related proteins such as cytochromes P450.
Fig. 8.
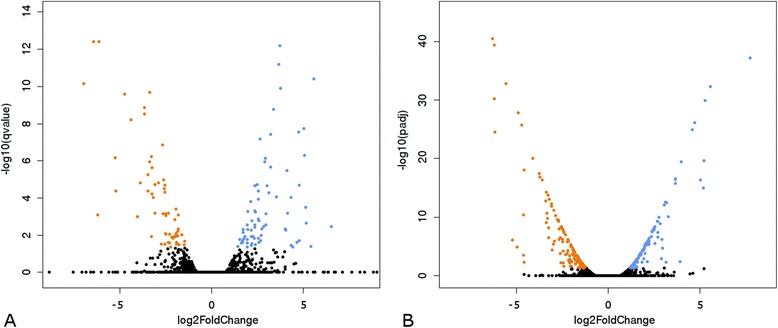
Gene expression changes after immune challenge. Volcano plots show the statistical significance of the difference in expression observed (p-value from a t-test, q-value in case of Cuffdiff and adjusted p-value in case of DEseq; log10 scale). The x-axis indicates the differential expression profiles, plotting the fold-induction ratios in a log-2 scale during immune challenge. The list of significantly differentially expressed protein coding genes can be found in Table S9 (Additional file 12). a Volcano plot from Cuffdiff data. Up-regulated genes (q-value < 0.05 and log2FoldChange ≥1) are shown as blue dots and the down-regulated genes (q-value < 0.05 and log2FoldChange ≤ −1) are shown as orange dots. The top three protein coding genes most up-regulated are Cflo_N_g6748 (hypothetical), Cflo_N_g2827 (voltage-dependent calcium channel type A subunit alpha-1) and Cflo_N_g12631 (serine proteinase Stubble). The three most down-regulated genes include Cflo_N_g2215 (putative chitin binding peritrophin-a domain containing), Cflo_N_g1319 (serine protease inhibitor 3) and Cflo_N_g4308 (lipase member H). b Volcano plot from DEseq data. Up-regulated genes (adjusted p-value < 0.05 and log2FoldChange ≥1) are shown by blue dots and the down-regulated genes (adjusted p-value < 0.05 and log2FoldChange ≤ −1) are shown by orange dots. The top three protein coding genes most up-regulated are Cflo_N_g6748 (hypothetical), Cflo_N_g5222 (hypothetical) and Cflo_N_g531 (aminopeptidase N). The three top down-regulated genes include Cflo_N_g2215 (putative chitin binding peritrophin-a domain containing), Cflo_N_g1319 (serine protease inhibitor 3) and Cflo_N_g907 (chymotrypsin-1)
Further validation of differential expression of 37 of the genes mentioned above was performed using qRT-PCR analysis. Of these 37 genes 15 were up-regulated at least two-fold, and 12 genes down-regulated in the Illumina data. For this analysis we differentiated between immune challenged larvae and adult animals instead of using pooled developmental stages which had been used for generation of the Illumina data. In general, the qRT-PCR analysis confirmed the Illumina data showing that the selected genes are regulated in the same direction in both larvae and workers, or at least in one of the two developmental stages. The corresponding heat map (Fig. 9) visualises the comparison of immune gene expression results obtained by qRT-PCR or Illumina sequencing. In Table S10 (Additional file 13) the respective x-fold changes as well as p- or q-values of these data are shown in detail. The differences between the expression data of immune-related genes gained by Illumina sequencing or qRT-PCR may largely be explained by the fact that the change in expression of several genes after immune-challenge differs substantially between larvae and workers. For example, expression of the genes MPI (metalloproteinase inhibitor), SOCS2 (suppressor of cytokine signaling 2), cact1 (cactus), transf (transferrin), PHR (parathyroid peptide receptor), ester (esterase FE4), PGRP-LB, PGRP-SA, hp67112 (hypothetical protein, Cflo_N_g6748t1), hymenoptaecin, and thioester-containing protein 1 (TEP1) was strongly induced in immune-challenged larvae, but only weakly or not at all in workers (Fig. 9; Additional file 13: Table S10). This is in agreement with previous reports on other insects which indicated that the immune response of larvae might differ from that of adults in holometabolous insects including workers in social insects [79–81]. Thus, in C. floridanus immune induction of gene expression appeared to be much stronger in larvae than in workers. At first glance this is somewhat surprising, since ant larvae are constantly cared for and groomed by nurse workers within the protected nest environment, while in particular foraging adult workers should be exposed more frequently and intensely to a feculent environment. However, larvae may be more vulnerable to pathogen infections due to their relatively thin and soft cuticle and the inability to groom themselves. Therefore, a highly responsive immune gene regulation in particular in larvae may contribute to a long term colony success by ensuring a continuous supply of a large number of healthy offspring. In fact, recent infection experiments with C. pennsylvanicus larvae indicated that their individual immune response is important and brood care by nurses does not alleviate the individual immune competence of immature stages [82]. On the other hand, brood care is also of prime importance since it was shown recently by cross-fostering experiments that in the ant Formica selysi the colony origin of care taker animals contributed to resistance of freshly eclosed animals against an entomopathogenic fungus [83]. In A. mellifera it was also shown that developmental stages differ in immunocompetence – larvae and pupae had the highest haemocyte counts while adult workers had the strongest phenoloxidase activity [84].
Fig. 9.
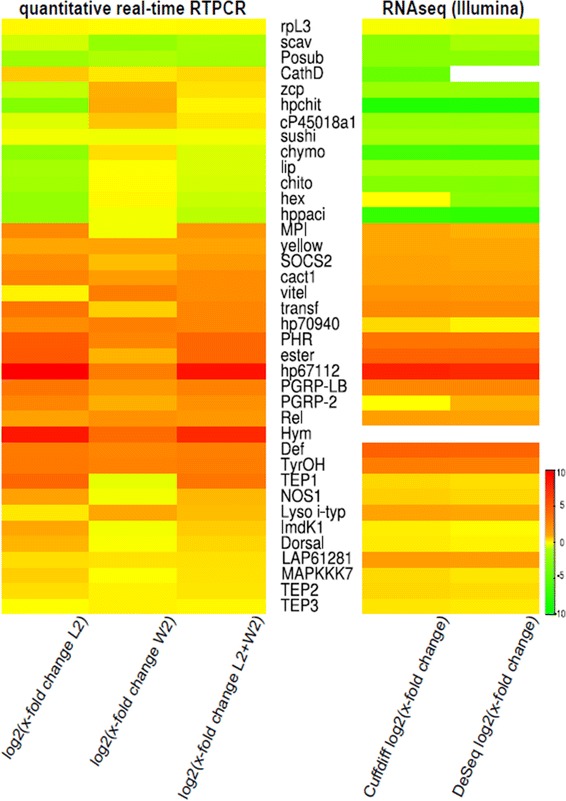
Comparison of expression of 37 selected genes based on the analysis of the Illumina sequencing data using Cuffdiff and DESeq and the corresponding qRT-PCR data. The heat map visualises the expression of 37 regulated immune genes 12 h after pricking of larvae and workers with a 1:1 mix of Gram-negative and Gram-positive bacteria in case of Illumina sequencing (Cuffdifflog2FC / DeSeq log2FC). The corresponding qRT-PCR analysis was performed separately in larvae and workers revealing a stage-specific gene regulation after immune challenge (log2(x-fold change L2 + W2)). Up and down-regulation are colour coded as given in the key highlighting common directions of gene regulation in the different data sets
The gene most strongly induced after immune challenge should also be mentioned here. This gene (Cflo_N_g6748t1) encodes a protein of unknown function. qRT-PCR reveals a more than 700 fold induction after immune challenge in larvae, but only a very moderate (3-fold) induction in workers (Fig. 9; Additional file 13: Table S10). Due to its extreme expression pattern this protein might play an important role in the immune defence of larvae and might merit future attention. Orthologs of this gene are also found in the other ants, in wasps (N. vitripennis and Micropolitis demolitor) and in the termite Zootermopsis nevadensis.
Based on an orthology analysis six common differentially expressed ‘immune genes’ were found when comparing the C. floridanus data with a recently published dataset of immune-stimulated honey bees [85]. These genes encode known immune related proteins such as the serine protease Stubble, NF-kappa-B inhibitor Cactus, Nuclear factor NF-kappa-B p110 subunit Relish, leukocyte elastase inhibitor (Serpin), Tyrosine 3-monooxygenase (involved in melanisation and proPO pathway in Manduca sexta [86]), and a protein NPC2 homolog (involved in microbial recognition in Drosophila melanogaster [87]).
Several genes down-regulated after immune challenge were identified (Fig. 9, Additional file 13: Table S10). Among these genes several encode proteins involved in digestion (e.g. Chymotrypsin, Lipase) and storage (e.g. Hexamerin), which were already described to be down-regulated upon immune-challenge in other insects. This indicates that during infection insects seem to temporarily shut down digestion and synthesis of non-essential proteins in order to use resources for costly defence reactions [88–92].
Comparison of C. floridanus immune proteins with other genome sequenced ants, A. mellifera, N. vitripennis and D. melanogaster
Orthologous clusters were identified using OrthoMCL analysis on the proteomes of 11 insect species (Additional file 14: Table S11). OrthoMCL clustering included the eight ant species as well as A. mellifera, N. vitripennis and D. melanogaster. These proteomes clustered into 18,763 groups from 188,092 protein sequences. All eight ant species cluster into 6620 ortholog groups (Fig. 10; details in Additional file 15: Table S12). Looking at the species distribution we found that 4818 groups were shared by all the species analysed, while 5797 groups were shared by all hymenopterans. 79 groups represent genes that are conserved exclusively among the eight ant species (Additional file 16: Table S13). We further parsed the 18,763 groups with perl scripts to reveal the presence of orthologs of C. floridanus immune proteins in selected species. Table S14 (Additional file 17) lists the identified immune proteins of C. floridanus and their orthologs, if present in the other sequenced ants, A. mellifera and N. vitripennis. Nine differentially regulated genes encoding putative immune-related proteins of C. floridanus do not have any homologs in other insects. KOG annotations of these proteins, listed in Table S15 (Additional file 18), reveal features of some of these proteins including the presence of signal peptides, a chemosensory domain and a DNA-binding domain. Additionally, in a recent study Hamilton and co-workers reported Cathepsin D as a protein that contributes to social immunity in Camponotus pennsylvanicus [12]. C. floridanus also encodes an ortholog of Cathepsin D (Cflo_N_g9172t1) and it will be interesting to investigate a general role of this protein in social immunity in the future.
Fig. 10.
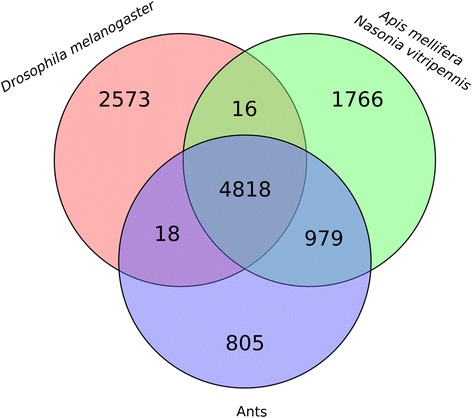
Comparison of insect proteomes. Specific numbers of shared orthologous clusters are indicated (black numbers) comparing the proteomes of 11 insect species (eight ant species (blue) as well as A. mellifera, N. vitripennis (green) and D. melanogaster (red)
Conclusions
We achieved an improved annotation and detailed analysis of the immune gene repertoire of C. floridanus based on the previously published genome sequence of C. floridanus and on an Illumina based transcriptome analysis of immune challenged larvae and worker animals. This analysis allowed us to extend the previously annotated protein repertoire not only by about 20 % (including splicing variants), but was instrumental in analysing the immune response of C. floridanus and to newly identify nine putative Camponotus-specific proteins possibly involved in immune functions or stress response. Furthermore, a comparative overview of immune proteins and pathways is presented which distinguishes between generally conserved parts, ant-specific and Camponotus-specific additions. This analysis shows that the immune gene repertoire of C. floridanus is comparable to that of the other insects. Especially signalling pathways are highly conserved. However, genes encoding PGN recognition proteins and AMPs appear to be present in a reduced number in comparison to solitary insects such as D. melanogaster and N. vitripennis. Whether this apparent reduction in the number of certain immune genes is related to an increased use of social or external immune measures is not clear, since no attenuated selective pressure on immune genes could be observed in ant genomes, at least on several selected genes [93]. Moreover, the recent establishment of the complete genomes of two bumblebee species and their comparison with other bee genomes including a solitary bee did not reveal any correlation between the number of immune genes and the degree of sociality evolved [94]. Interestingly, the gene expression analysis suggests significant stage specific differences in the immune responsiveness of C. floridanus which may be an important feature possibly contributing to colony success, requiring, however, further investigation in the future. The combination of an apparently quite complex innate immune system and social immunity may explain the resilience of these animals against pathogen infestation despite high population densities in their nests and genetic uniformity.
Methods
Sample preparation and RNA extraction
For immune-challenge, late larvae (stage L2) and adult minor workers (stage W2) of colony C90 (for stage definitions see [95]) were pricked with a minutiae needle (Minutiennadel Sphinx V2A 0.1 x 12 mm, bioform), which was previously dipped into a pellet of heat-killed bacteria (1:1 mix of Escherichia coli D31 and Micrococcus luteus) which are commonly used for infection experiments in insects. Afterwards immune-challenged animals as well as non-treated control animals were kept in artificial nests for 12 h. Then total RNA was extracted from each five immune-challenged and five untreated L2 and W2 using TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA) and purified through RNeasy mini kit columns (Qiagen, Hilden, Germany) with on-column DNase digestion (RNase-Free DNase Set, Qiagen) as described in the manufacturer’s procedures. RNA concentration and quality were determined on an Agilent 2100 Bioanalyzer using the Agilent RNA 6000 Nano Chip kit (Agilent Technologies, Böblingen, Germany) according to the manufacturer´s instructions (Additional file 19: Figure S4). Equal amounts of total RNA from immune-challenged L2 and W2 as well as from untreated L2 and W2 were mixed and the obtained two RNA samples (immune-challenged and naïve) were further processed and sequenced with an Illumina HiSeq2000 as 2× 50 bp reads by Eurofins MWG Operon (Ebersberg, Germany). The total number of resulting reads was 125,873,897 and 118,142,837, respectively.
Assembly, detection of new transcripts, and differential gene expression analysis
For the discovery of new transcripts and differential gene expression analysis, the protocol described by Trapnell and co-workers was utilised [96]. All analyses were performed on a HP ProLiant DL580 G5 offering four Intel(R) Xeon(R) CPUs E7440 and 40 Gb of RAM. At the first step the read data of both conditions were mapped onto the reference genome (AEAB01000000 [18]) using Tophat (v2.0.4) [97]. Following this, the programme cufflinks (v2.0.2) was used for the assembly of the expressed transcripts [98]. A merged transcriptome annotation was generated using cuffmerge and the differential gene expression analysis was done by cuffdiff (v2.0.2) and, independently, by DESeq [99]. Cuffdiff algorithm estimates the DEGs at transcript-level resolution by comparing the concentration of transcripts in control and treated samples while DESeq is suitable for identifying significantly differentially expressed genes between two samples based on read counts. The R package cummeRbund (v0.1.3, http://compbio.mit.edu/cummeRbund/) was used for the exploration of differentially expressed genes and for the visualisation of differently spliced genes.
Validation of differential gene expression results by qRT-PCR
For validation of differential gene expression results obtained by transcriptome analysis we performed qRT-PCR. The samples were prepared as described above RNA samples from untreated and immune-challenged animals were prepared in each case from five late larvae (L2) and five adult minor workers (W2) of six different ant colonies (C90, C96, C152, C79, C264, C132) 12 h post infection. RNA was isolated from treated as well as from untreated animals. For each sample cDNA was produced by reverse transcription from 1 μg of total RNA using the RevertAid First Strand cDNA Synthesis Kit (Fermentas). Resulting cDNA was directly diluted to a final concentration of 10 ng/μl.
Expression of candidate genes was analysed by qRT-PCR separately regarding larvae and workers. Oligonucleotide pairs were designed on the chosen genes with Primer3 v. 0.4.0 [100] to yield products of 120–140 bp with Tm values around 56 °C (Additional file 20: Table S16). The qRT-PCR experiments were performed on a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Life Technologies GmbH, Darmstadt, Germany). Therefore, samples contained 1× Absolute™ PerfeCTa™ SYBR® Green FastMix™ (Rox) (Quanta Biosciences, Gaithersburg, MD, USA), gene-specific oligonucleotides (250 nM each), 1 μl of the cDNA and water to a final volume of 20 μl. After 5 min of enzyme activation at 95 °C, 45 cycles of 5 s denaturation at 95 °C, 10 s of annealing at 56 °C and 20 s of extension at 60 °C were run. Fragment specificity was checked in melting curves and each biological sample was run in duplicate in the qRT-PCR. Results were averaged and relative transcription levels were calculated by the ddCt method [101] using ribosomal protein L32 (rpL32) as reference gene.
Statistical analysis was performed using Statistica v10 enterprise ×64. A t-test was used to test whether the relative gene expression of six biological replicates of infected animals (dCT Immune) differs significantly from relative gene expression of control animals (dCT Control). A p-value <0.05 distinguishes a gene as being significantly down- or up-regulated when down-regulation means a ratio <0.5 and up-regulation a ratio >2. Results of the statistical analyses are given in Additional file 21: Table S17.
Identifying repetitive elements
To delineate various repetitive elements present in the C. floridanus genome, RepeatMasker v 4.0 [102] was applied using a custom library comprising a combination of Repbase library [32] and the de novo repeat library customised for the C. floridanus genome constructed with RepeatModeler [103] with default parameters.
Gene annotation
Gene models were predicted with the extended version of Generalized Hidden Markov Model (GHMM) based ab initio predictor Augustus v2.7 [104–106]. We constructed a high-quality training set consisting of 330 genes for the generation of C. floridanus specific parameters for the splice site signals, length distributions, nucleotide composition of exons, introns and intergenic regions. High-confidence gene-models were extracted by combination of a training gene set derived from (a) PASA2 release 2013-06-05 [107] by alignment of cufflinks assembled transcripts on C. floridanus genome using GMAP [108] aligner, (b) Cegma v2.4 [109] which uses ortholog identification of a set of accurately annotated 458 eukaryotic core proteins in the test genome followed by determination of exon-intron structure using a combination of GeneWise [110], HMMER [111] and GeneID [112], and (c) Scipio v1.4 [113] by aligning randomly selected C. floridanus proteins from different GO categories on the C. floridanus genome with BLAT [114] and determining correct exon-intron junctions by hit refinements and filtering with Scipio. We further generated extrinsic evidence (hints) for Augustus predictions from different sources, in particular (a) the raw Illumina reads and (b) the assembled transcripts as well as (c) all EST evidence. Finally, to predict gene structures and estimate alternative transcripts, Augustus predictions with hints were performed on the repeat-masked C. floridanus genome using the tuned optimised parameters of C. floridanus. Detailed sequence analysis of all genes and proteins included different software, databanks and protocols as described previously [115].
Reconstruction of immune signalling pathways
The unidirectional and bidirectional interactions among the components of Toll, Jak-Stat, IMD and JNK pathway of D. melanogaster were mined individually from three pathway databases KEGG [116], FlyReactome (http://fly.reactome.org/) and INOH [117] followed by extensive manual curations from scientific literatures. The proteins and connectivity information were translated into comprehensive immune signalling networks of D. melanogaster. Proteins involved in networks were retrieved from the Entrez protein database available at the National Center for Biotechnology Information (NCBI) (http://ncbi.nlm.nih.gov/) and chosen as queries to perform BlastP search [118] against C. floridanus protein sequences available in Entrez and Uniprot databases [119]. Subsequently, we performed domain analysis with PFAM search [120] for the immune related protein sequences of both of the organisms and accessed the relative domain conservation by global pairwise alignments. The identified homologous immune proteins were mapped onto reconstructed immune signalling networks of D. melanogaster and pathway annotations were transferred if any of the two immune proteins in C. floridanus had corresponding interacting homologs.
Identification of AMPs
The creation of used HMMs of AMPs has been described elsewhere [121]. Briefly, in the AMPer database [121] 1045 mature peptides associated with known antimicrobial activities were considered to create 146 HMMs of mature peptides. All the 146 HMMs of mature peptides were retrieved from the AMPer resource. We further employed HMMsearch module of HMMER3 [121] with an E-value threshold of 1e-03 to scan all the protein isoforms obtained from re-annotation of C. floridanus against HMMs of AMPs. In order to remove the homologous peptides in resulting datasets, a cut-off threshold of 90 % was imposed by using the CD-HIT programme [122]. Furthermore, using known APMs of other insects as a query sequence BlastP and tBlastn, searches were conducted to identify additional AMPs.
Identification of immune effectors
To detect the presence of potential immune effectors including chitinase, lysozymes, prophenoloxidase, nitric oxide synthase, glutathione S-transferase, TEPs and turandots in C. floridanus we implemented the BlastP search using the effector sequences from several insects (D. melanogaster, A. mellifera, Anopheles gambiae, Bombyx mori, Manduca sexta, Aedes aegypti and Phaedon cochleariae) as a query. The GenBank accession numbers of used query sequences are given in Additional file 22: Table S18.
Orthology analysis
We employed OrthoMCLv2.0.9 [123] to examine the presence of orthologs of C. floridanus immune proteins in the proteomes from seven other sequenced ant genomes, including A. cephalotes (leafcutter ant), A. echinatior (Panamanian leafcutter ant), P. barbatus (red harvester ant), H. saltator (Jerdon’s jumping ant), L. humile (Argentine ant), S. invicta (red fire ant), C. biroi (clonal raider ant), A. mellifera (honeybee), N. vitripennis (a parasitic solitary wasp) and D. melanogaster (model insect). OrthoMCL pipeline integrates all-versus-all Blast similarity results and Markov clustering algorithm (MCL) to construct putative orthologous groups, including recent paralogs, across multiple taxa. Blast e-value and MCL inflation index was set to 1e-05 and 1.5 respectively for OrthoMCL clustering.
Availability of supporting data
Additional data for download can be retrieved at www.bioinfo.biozentrum.uni-wuerzburg.de/computing/Camponotus. These files are: Camponotus floridanus augustus gene annotations (.gff format), Camponotus floridanus transcriptome gene coordinates (.gff format), Camponotus floridanus transcripts for the translated regions (fasta format) and Camponotus floridanus predicted proteins (fasta format) for the gene set. All files are zipped to save space. Furthermore, the raw data of the transcriptome sequences (just the Illumina reads, no annotation) are deposited in the NCBI bioproject ID263478 Accession: PRJNA263478 to be released upon acceptance of the manuscript. These data have the NCBI accession numbers [GenBank:SRR1609918] for the immune challenged and [GenBank:SRR1609919] for the unchallenged animals.
Acknowledgements
We gratefully acknowledge funding of this work by the German Research Foundation (DFG-GR1243/8-1). We also would like to thank Annette Laudahn, Flavio Roces and Wolfgang Rössler of the Department of Zoology II of the Biocenter for help with animal breeding and husbandry. We thank Suvagata Roy Chowdhury and Jennifer Heilig for critically reading the manuscript. The publication of this manuscript was funded by the German Research Foundation (DFG) and the University of Würzburg within the funding programme Open Access Publishing.
Additional files
Summary of genes predicted with Augustus run on the repeat masked C. floridanus genome.
Accuracy of trained Augustus on C. floridanus test set sequences.
{kind=link}
Differences in pfam protein domains as deduced from the previous and the new C. floridanus genome annotation.
{kind=link}
Categorisation of 7143 proteins of Camponotus floridanus v3.3set in GO terms (level 2) for (A) biological process, (B) molecular function, and (C) cellular component using a filter score e-value cutoff of 1e-5.
Global changes in level 2 GO class assignment of new and previously annotated Cflov3.3 proteins for the following GO categories: biological process, molecular function and cellular component.
Functional annotation of highly conserved immune proteins shared by D. melanogaster, A. mellifera, N. vitripennis and C. floridanus.
Structural and functional annotation of C. floridanus immune-related proteins.
List of serine proteases and serine protease inhibitors of C. floridanus.
List of putative chitinases of C. floridanus and distribution of conserved motifs in their deduced amino acid sequences.
Number of chitinases, glutathione S-transferases, nitric oxide synthases, thioester-containing proteins, lysozymes and prophenoloxidases encoded by C. floridanus, other ants, A. mellifera, N. vitripennis and D. melanogaster.
{kind=link}
Phylogenetic relationship between GSTs of C. floridanus and of other insects as inferred using the Neighbor Joining algorithm [124]. The statistical reliability of the phylogenetic tree was tested by bootstrap analyses with 10,000 replications. The topology is based on a 50 % condensed tree obtained by bootstrap analysis. The percentage of replicate trees in which the associated nodes clustered together in the bootstrap test is shown next to the branches. Species abbreviations occur after the GenBank accession numbers are as follows: Dm = Drosophila melanogaster, Ag = Anopheles gambiae, Am = Apis mellifera, Bm = Bombyx mori, Aa = Aedes aegypti, Cf = Camponotus floridanus. Evolutionary analyses were conducted in MEGA v5.1 [125].
Complete list of genes of C. floridanus which are differentially expressed after immune challenge. The genes listed were retrieved based on the analysis of the Illumina sequencing data using the programs Cufflinks and DESeq. The table shows significantly up-regulated and down-regulated genes 12 h after picking of larvae and workers with a 1:1 mix of Gram-negative and Gram-positive bacteria. Cufflinks analysis resulted in new gene identifiers (XLOCs), which correspond to one or more of the originally annotated genes (EAGs).
Genes differentially expressed (DEGs) after immune challenge. The table presents a comparison of expression data of 37 selected immune genes based on the analysis of the Illumina sequencing data (Cuffdiff and DESeq) and the corresponding qRT-PCR data. Significance of resulting log2(x-fold change) values is given by asterisks (* p-value/q-value ≤0.05; ** ≤0.01; *** ≤0.001; n.s. for non-significant results)
The source of proteomes of the eleven insect species included in our OrthoMCL analysis.
Comparison of orthologous groups identified by OrthoMCL using an inflation index of 1.5.
Conserved orthologous groups shared among all eight sequenced ant species identified by OrthoMCL analysis.
List of the immune-related proteins of C. floridanus and their orthologs, if also present in the other sequenced ants, and in A. mellifera and N. vitripennis.
List of C. floridanus specific hypothetical genes expressed differentially after immune challenge including KOG annotations.
{kind=link}
Quality control of total RNA samples. (A) The image shows a total RNA gel like-image produced by the Agilent 2100 Bioanalyzer. Lane 1: RNA from C. floridanus workers and larvae (1:1 mix) at 12 h after bacterial challenge, Lane 2: RNA from untreated workers (W2) and larvae (L2) (1:1 mix). (B) Electrophoretic profiles of RNA from immune-challenged (1) and untreated (2) W2 and L2 (1:1 mix). [FU]: Fluorescence units, [s]: seconds.
List of oligonucleotides used for validation of changes of gene expression after immune challenge by qRT-PCR.
Statistical analysis of qRT-PCR results: The tables A (larvae) and B (adult workers) show normalised values (dCT) of immunised and control animals of six different colonies when rpL32 was used as the housekeeping gene. These values were used in a t-test to determine significant differential gene expression (mean ratio as x-fold expression also given here). A p-value <0.05 distinguishes a gene as being significantly down- or up-regulated (highlighted in red).
Accession numbers of the sequentially used query sequences from different insects.
Footnotes
Shishir K. Gupta, Maria Kupper and Carolin Ratzka contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SKG and FF performed the bioinformatics analyses, MK and CR carried out the experiments, HF and MK performed the statistical analysis, SKG, MK and HF were involved in drafting the manuscript, AV, RG and TD conceived the project, supervised the work and wrote the paper. All authors read and agreed to the final version of the manuscript.
Contributor Information
Shishir K. Gupta, Email: shishir.bioinfo@gmail.com
Maria Kupper, Email: maria.kupper@uni-wuerzburg.de.
Carolin Ratzka, carolin.ratzka@googlemail.com.
Heike Feldhaar, Email: feldhaar@uni-bayreuth.de.
Andreas Vilcinskas, Email: Andreas.Vilcinskas@agrar.uni-giessen.de.
Roy Gross, Phone: +49 931 318 4403, Email: roy@biozentrum.uni-wuerzburg.de.
Thomas Dandekar, Phone: +49 931 318 4551, Email: dandekar@biozentrum.uni-wuerzburg.de.
Frank Förster, Email: frank.foerster@biozentrum.uni-wuerzburg.de.
References
- 1.Vallet-Gely I, Lemaitre B, Boccard F. Bacterial strategies to overcome insect defences. Nat Rev Microbiol. 2008;6:302–13. doi: 10.1038/nrmicro1870. [DOI] [PubMed] [Google Scholar]
- 2.Otti O, Tragust S, Feldhaar H. Unifying external and internal immune defences. Trends Ecol Evol. 2014;29:625–34. doi: 10.1016/j.tree.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Bulet P, Stocklin R, Menin L. Anti-microbial peptides: from invertebrates to vertebrates. Immunol Rev. 2004;198:169–84. doi: 10.1111/j.0105-2896.2004.0124.x. [DOI] [PubMed] [Google Scholar]
- 4.Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- 5.Wiesner J, Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence. 2010;1:440–64. doi: 10.4161/viru.1.5.12983. [DOI] [PubMed] [Google Scholar]
- 6.Royet J, Dziarski R. Peptidoglycan recognition proteins: pleiotropic sensors and effectors of antimicrobial defences. Nat Rev Microbiol. 2007;5:264–77. doi: 10.1038/nrmicro1620. [DOI] [PubMed] [Google Scholar]
- 7.Ferrandon D, Imler JL, Hetru C, Hoffmann JA. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat Rev Immunol. 2007;7:862–74. doi: 10.1038/nri2194. [DOI] [PubMed] [Google Scholar]
- 8.Leulier F, Lemaitre B. Toll-like receptors: taking an evolutionary approach. Nat Rev Genet. 2008;9:165–78. doi: 10.1038/nrg2303. [DOI] [PubMed] [Google Scholar]
- 9.Cremer S, Armitage SA, Schmid-Hempel P. Social immunity. Curr Biol. 2007;17:R693–702. doi: 10.1016/j.cub.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Vilcinskas A. Evolutionary plasticity of insect immunity. J Insect Physiol. 2013;59:123–9. doi: 10.1016/j.jinsphys.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 11.Le Conte Y, Alaux C, Martin JF, Harbo JR, Harris JW, Dantec C, et al. Social immunity in honeybees (Apis mellifera): transcriptome analysis of varroa-hygienic behaviour. Insect Mol Biol. 2011;20:399–408. doi: 10.1111/j.1365-2583.2011.01074.x. [DOI] [PubMed] [Google Scholar]
- 12.Hamilton C, Lejeune BT, Rosengaus RB. Trophallaxis and prophylaxis: social immunity in the carpenter ant Camponotus pennsylvanicus. Biol Lett. 2011;7:89–92. doi: 10.1098/rsbl.2010.0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans JD, Aronstein K, Chen YP, Hetru C, Imler JL, Jiang H, et al. Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol Biol. 2006;15:645–56. doi: 10.1111/j.1365-2583.2006.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elsik CG, Worley KC, Bennett AK, Beye M, Camara F, Childers CP, et al. Finding the missing honey bee genes: lessons learned from a genome upgrade. BMC Genomics. 2014;15:86. doi: 10.1186/1471-2164-15-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischman BJ, Woodward SH, Robinson GE. Molecular evolutionary analyses of insect societies. Proc Natl Acad Sci USA. 2011;108:10847–54. doi: 10.1073/pnas.1100301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldhaar H, Straka J, Krischke M, Berthold K, Stoll S, Mueller MJ, et al. Nutritional upgrading for omnivorous carpenter ants by the endosymbiont Blochmannia. BMC Biol. 2007;5:48. doi: 10.1186/1741-7007-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ratzka C, Liang C, Dandekar T, Gross R, Feldhaar H. Immune response of the ant Camponotus floridanus against pathogens and its obligate mutualistic endosymbiont. Insect Biochem Mol Biol. 2011;41:529–36. doi: 10.1016/j.ibmb.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Ratzka C, Gross R, Feldhaar H. Gene expression analysis of the endosymbiont-bearing midgut tissue during ontogeny of the carpenter ant Camponotus floridanus. J Insect Physiol. 2013;59:611–23. doi: 10.1016/j.jinsphys.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 19.Bonasio R, Zhang G, Ye C, Mutti NS, Fang X, Qin N, et al. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science. 2010;329:1068–71. doi: 10.1126/science.1192428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korcsmáros T, Szalay MS, Rovó P, Palotai R, Fazekas D, Lenti K, et al. Signalogs: orthology-based identification of novel signaling pathway components in three metazoans. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reid AJ, Yeats C, Orengo CA. Methods of remote homology detection can be combined to increase coverage by 10 % in the midnight zone. Bioinformatics. 2007;23:2353–60. doi: 10.1093/bioinformatics/btm355. [DOI] [PubMed] [Google Scholar]
- 22.Hegyi H, Gerstein M. Annotation transfer for genomics: measuring functional divergence in multi-domain proteins. Genome Res. 2001;11:1632–40. doi: 10.1101/gr.183801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Apic G, Gough J, Teichmann SA. Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J Mol Biol. 2001;310:311–25. doi: 10.1006/jmbi.2001.4776. [DOI] [PubMed] [Google Scholar]
- 24.Nygaard S, Zhang G, Schiøtt M, Li C, Wurm Y, Hu H, et al. The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming. Genome Res. 2011;21:1339–48. doi: 10.1101/gr.121392.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suen G, Teiling C, Li L, Holt C, Abouheif E, Bornberg-Bauer E, et al. The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oxley PR, Ji L, Fetter-Pruneda I, McKenzie SK, Li C, Hu H, et al. The genome of the clonal raider ant Cerapachys biroi. Curr Biol. 2014;24:451–8. doi: 10.1016/j.cub.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith CD, Zimin A, Holt C, Abouheif E, Benton R, Cash E, et al. Draft genome of the globally widespread and invasive Argentine ant (Linepithema humile) Proc Natl Acad Sci USA. 2011;108:5673–8. doi: 10.1073/pnas.1008617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith CR, Smith CD, Robertson HM, Helmkampf M, Zimin A, Yandell M, et al. Draft genome of the red harvester ant Pogonomyrmex barbatus. Proc Natl Acad Sci USA. 2011;108:5667–72. doi: 10.1073/pnas.1007901108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wurm Y, Wang J, Riba-Grognuz O, Corona M, Nygaard S, Hunt BG, et al. The genome of the fire ant Solenopsis invicta. Proc Natl Acad Sci USA. 2011;108:5679–84. doi: 10.1073/pnas.1009690108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Werren JH, Richards S, Desjardins CA, Niehuis O, Gadau J, Colbourne JK, et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science. 2010;327:343–8. doi: 10.1126/science.1178028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoskins RA, Carlson JW, Wan KH, Park S, Mendez I, Galle SE, et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 2015; gr.185579.114. [DOI] [PMC free article] [PubMed]
- 32.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 33.Sackton TB, Werren JH, Clark AG. Characterizing the infection-induced transcriptome of Nasonia vitripennis reveals a preponderance of taxonomically-restricted immune genes. PLoS One. 2013;8 doi: 10.1371/journal.pone.0083984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffmann JA. The immune response of Drosophila. Nature. 2003;426:33–8. doi: 10.1038/nature02021. [DOI] [PubMed] [Google Scholar]
- 35.Kounatidis I, Ligoxygakis P. Drosophila as a model system to unravel the layers of innate immunity to infection. Open Biol. 2012;2:120075. doi: 10.1098/rsob.120075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aggarwal K, Silverman N. Positive and negative regulation of the Drosophila immune response. BMB Rep. 2008;41:267–77. doi: 10.5483/bmbrep.2008.41.4.267. [DOI] [PubMed] [Google Scholar]
- 37.Qiu P, Pan PC, Govind S. A role for the Drosophila Toll/Cactus pathway in larval hematopoiesis. Development. 1998;125:909–1920. doi: 10.1242/dev.125.10.1909. [DOI] [PubMed] [Google Scholar]
- 38.Valanne S, Wang JH, Rämet M. The Drosophila Toll signaling pathway. J Immunol. 2011;186:649–56. doi: 10.4049/jimmunol.1002302. [DOI] [PubMed] [Google Scholar]
- 39.Gottar M, Gobert V, Matskevich AA, Reichhart JM, Wang C, Butt TM, et al. Dual detection of fungal infections in Drosophila via recognition of glucans and sensing of virulence factors. Cell. 2006;127:1425–37. doi: 10.1016/j.cell.2006.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ashok Y. Drosophila Toll pathway: the new model. Sci Signal. 2009;2:jc1. doi: 10.1126/scisignal.252jc1. [DOI] [PubMed] [Google Scholar]
- 41.Myllymäki H, Valanne S, Rämet M. The Drosophila Imd signaling pathway. J Immunol. 2014;192:3455–62. doi: 10.4049/jimmunol.1303309. [DOI] [PubMed] [Google Scholar]
- 42.Bischoff V, Vignal C, Duvic B, Boneca IG, Hoffmann JA, Royet J. Downregulation of the Drosophila immune response by peptidoglycan-recognition proteins SC1 and SC2. PLoS Pathog. 2006;2 doi: 10.1371/journal.ppat.0020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ertürk-Hasdemir D, Broemer M, Leulier F, Lane WS, Paquette N, Hwang D, et al. Two roles for the Drosophila IKK complex in the activation of Relish and the induction of antimicrobial peptide genes. Proc Natl Acad Sci USA. 2009;106:9779–84. doi: 10.1073/pnas.0812022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rutschmann S, Jung AC, Zhou R, Silverman N, Hoffmann JA, Ferrandon D. Role of Drosophila IKK gamma in a toll-independent antibacterial immune response. Nat Immunol. 2000;1:342–7. doi: 10.1038/79801. [DOI] [PubMed] [Google Scholar]
- 45.Kleino A, Silverman N. The Drosophila IMD pathway in the activation of the humoral immune response. Dev Comp Immunol. 2014;42:25–35. doi: 10.1016/j.dci.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noselli S, Agnès F. Roles of the JNK signaling pathway in Drosophila morphogenesis. Curr Opin Genet Dev. 1999;9:466–72. doi: 10.1016/S0959-437X(99)80071-9. [DOI] [PubMed] [Google Scholar]
- 47.Rämet M, Lanot R, Zachary D, Manfruelli P. JNK signaling pathway is required for efficient wound healing in Drosophila. Dev Biol. 2002;241:145–56. doi: 10.1006/dbio.2001.0502. [DOI] [PubMed] [Google Scholar]
- 48.Jasper H, Benes V, Schwager C, Sauer S, Clauder-Münster S, Ansorge W, et al. The genomic response of the Drosophila embryo to JNK signaling. Dev Cell. 2001;1:579–86. doi: 10.1016/s1534-5807(01)00045-4. [DOI] [PubMed] [Google Scholar]
- 49.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK) - from inflammation to development. Curr Opin Cell Biol. 1998;10:205–19. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 50.Sluss HK, Han Z, Barrett T, Goberdhan DC, Wilson C, Davis RJ, et al. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev. 1996;10:2745–58. doi: 10.1101/gad.10.21.2745. [DOI] [PubMed] [Google Scholar]
- 51.Osman D, Buchon N, Chakrabarti S, Huang YT, Su WC, Poidevin M, et al. Autocrine and paracrine unpaired signaling regulate intestinal stem cell maintenance and division. J Cell Sci. 2012;125:5944–9. doi: 10.1242/jcs.113100. [DOI] [PubMed] [Google Scholar]
- 52.Valanne S. Functional genomics analysis of the Drosophila immune response. Dev Comp Immunol. 2014;42:93–101. doi: 10.1016/j.dci.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 53.Blandin S, Levashina EA. Thioester-containing proteins and insect immunity. Mol Immunol. 2004;40:903–8. doi: 10.1016/j.molimm.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 54.Ekengren S, Tryselius Y, Dushay MS, Liu G, Steiner H, Hultmark D. A humoral stress response in Drosophila. Curr Biol. 2001;11:714–8. doi: 10.1016/s0960-9822(01)00203-2. [DOI] [PubMed] [Google Scholar]
- 55.Bou Aoun R, Hetru C, Troxler L, Doucet D, Ferrandon D, Matt N. Analysis of thioester-containing proteins during the innate immune response of Drosophila melanogaster. J Innate Immun. 2011;3:52–64. doi: 10.1159/000321554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ratzka C, Förster F, Liang C, Kupper M, Dandekar T, Feldhaar H, et al. Molecular characterization of antimicrobial peptide genes of the carpenter ant Camponotus floridanus. PLoS One. 2012;7 doi: 10.1371/journal.pone.0043036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tian C, Gao B, Fang Q, Ye G, Zhu S. Antimicrobial peptide-like genes in Nasonia vitripennis: a genomic perspective. BMC Genomics. 2010;11:187. doi: 10.1186/1471-2164-11-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Z, Zhu S. Comparative genomics analysis of five families of antimicrobial peptide-like genes in seven ant species. Dev Comp Immunol. 2012;38:262–74. doi: 10.1016/j.dci.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 59.Eleftherianos I, Revenis C. Role and importance of phenoloxidase in insect hemostasis. J Innate Immun. 2011;3:28–33. doi: 10.1159/000321931. [DOI] [PubMed] [Google Scholar]
- 60.Lu A, Zhang Q, Zhang J, Yang B, Wu K, Xie W, et al. Insect prophenoloxidase: the view beyond immunity. Front Physiol. 2014;5:252. doi: 10.3389/fphys.2014.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sideri M, Tsakas S, Markoutsa E, Lampropoulou M, Marmaras VJ. Innate immunity in insects: surface-associated dopa decarboxylase-dependent pathways regulate phagocytosis, nodulation and melanization in medfly haemocytes. Immunology. 2008;123:528–37. doi: 10.1111/j.1365-2567.2007.02722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cerenius L, Söderhäll K. The prophenoloxidase-activating system in invertebrates. Immunol Rev. 2004;198:116–26. doi: 10.1111/j.0105-2896.2004.00116.x. [DOI] [PubMed] [Google Scholar]
- 63.Tang H. Regulation and function of the melanization reaction in Drosophila. Fly. 2009;3:105–11. doi: 10.4161/fly.3.1.7747. [DOI] [PubMed] [Google Scholar]
- 64.Hughes AL. The evolution of the arthropod prophenoloxidase/hexamerin protein family. Immunogenetics. 1999;49:106–14. doi: 10.1007/s002510050469. [DOI] [PubMed] [Google Scholar]
- 65.Koski LB, Gray MW, Lang BF, Burger G. AutoFACT: an automatic functional annotation and classification tool. BMC Bioinformatics. 2005;6:151. doi: 10.1186/1471-2105-6-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aronstein KA, Murray KD, Saldivar E. Transcriptional responses in honey bee larvae infected with chalkbrood fungus. BMC Genomics. 2010;11:391. doi: 10.1186/1471-2164-11-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kramer KJ, Muthukrishnan S. Insect chitinases: molecular biology and potential use as biopesticides. Insect Biochem Mol Biol. 1997;27:887–900. doi: 10.1016/s0965-1748(97)00078-7. [DOI] [PubMed] [Google Scholar]
- 68.de la Vega H, Specht CA, Liu Y, Robbins PW. Chitinases are a multi-gene family in Aedes, Anopheles and Drosophila. Insect Mol Biol. 1998;7:233–9. doi: 10.1111/j.1365-2583.1998.00065.x. [DOI] [PubMed] [Google Scholar]
- 69.Choi HK, Choi KH, Kramer KJ, Muthukrishnan S. Isolation and characterization of a genomic clone for the gene of an insect molting enzyme, chitinase. Insect Biochem Mol Biol. 1997;27:37–47. doi: 10.1016/s0965-1748(96)00066-5. [DOI] [PubMed] [Google Scholar]
- 70.Shen Z, Jacobs-Lorena M. Characterization of a novel gut-specific chitinase gene from the human malaria vector Anopheles gambiae. J Biol Chem. 1997;272:28895–900. doi: 10.1074/jbc.272.46.28895. [DOI] [PubMed] [Google Scholar]
- 71.Zhu Q, Deng Y, Vanka P, Brown SJ, Muthukrishnan S, Kramer KJ. Computational identification of novel chitinase-like proteins in the Drosophila melanogaster genome. Bioinformatics. 2004;20:161–9. doi: 10.1093/bioinformatics/bth020. [DOI] [PubMed] [Google Scholar]
- 72.Li X, Schuler MA, Berenbaum MR. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev Entomol. 2007;52:231–53. doi: 10.1146/annurev.ento.51.110104.151104. [DOI] [PubMed] [Google Scholar]
- 73.Chelvanayagam G, Parker MW, Board PG. Fly fishing for GSTs: a unified nomenclature for mammalian and insect glutathione transferases. Chem Biol Interact. 2001;133:256–60. [Google Scholar]
- 74.Ranson H, Rossiter L, Ortelli F, Jensen B, Wang X, Roth CW, et al. Identification of a novel class of insect glutathione S-transferases involved in resistance to DDT in the malaria vector Anopheles gambiae. Biochem J. 2001;359:295–304. doi: 10.1042/0264-6021:3590295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Friedman R. Genomic organization of the glutathione S-transferase family in insects. Mol Phylogenet Evol. 2011;61:924–32. doi: 10.1016/j.ympev.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 76.Foley E, O’Farrell PH. Nitric oxide contributes to induction of innate immune responses to gram-negative bacteria in Drosophila. Genes Dev. 2003;17:115–25. doi: 10.1101/gad.1018503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eleftherianos I, More K, Spivack S, Paulin E, Khojandi A, Shukla S. Nitric Oxide Levels regulate the immune response of Drosophila melanogaster reference laboratory strains to bacterial infections. Infect Immun. 2014;82:4169–81. doi: 10.1128/IAI.02318-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Davies SA, Overend G, Sebastian S, Cundall M, Cabrero P, Dow JA, et al. Immune and stress response ‘cross-talk’ in the Drosophila Malpighian tubule. J Insect Physiol. 2012;58:488–97. doi: 10.1016/j.jinsphys.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 79.Colgan TJ, Carolan JC, Bridgett SJ, Sumner S, Blaxter ML, Brown MJ. Polyphenism in social insects: insights from a transcriptome-wide analysis of gene expression in the life stages of the key pollinator, Bombus terrestris. BMC Genomics. 2011;12:623. doi: 10.1186/1471-2164-12-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fellous S, Lazzaro BP. Potential for evolutionary coupling and decoupling of larval and adultimmune gene expression. Mol Ecol. 2011;20:1558–67. doi: 10.1111/j.1365-294X.2011.05006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Randolt K1, Gimple O, Geissendörfer J, Reinders J, Prusko C, Mueller MJ, et al. Immune-related proteins induced in the hemolymph after aseptic and septic injury differ in honey bee worker larvae and adults. Arch Insect Biochem Physiol. 2008;69:155–67. doi: 10.1002/arch.20269. [DOI] [PubMed] [Google Scholar]
- 82.Rosengaus RB, Malak T, Mackintosh C. Immune-priming in ant larvae: social immunity does not undermine individual immunity. Biol Lett. 2013;9:20130563. doi: 10.1098/rsbl.2013.0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Purcell J, Chapuisat M. Foster carers influence brood pathogen resistance in ants. Proc Royal Soc Lond B. 2014;281:20141338. doi: 10.1098/rspb.2014.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wilson-Rich N, Dres ST, Starks PT. The ontogeny of immunity: Development of innate immune strength in the honey bee (Apis mellifera) J Insect Physiol. 2008;54:1392–9. doi: 10.1016/j.jinsphys.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 85.Richard FJ, Holt HL, Grozinger CM. Effects of immunostimulation on social behavior, chemical communication and genome-wide gene expression in honey bee workers (Apis mellifera) BMC Genomics. 2012;13:558. doi: 10.1186/1471-2164-13-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gorman MA, An C, Kanost MR. Characterization of tyrosine hydroxylase from Manduca sexta. Insect Biochem Mol Biol. 2007;37:1327–37. doi: 10.1016/j.ibmb.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shi XZ, Zhong X, Yu XQ. Drosophila melanogaster NPC2 proteins bind bacterial cell wall components and may function in immune signal pathways. Insect Biochem Mol Biol. 2012;42:545–56. doi: 10.1016/j.ibmb.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Adamo SA. The effects of stress hormones on immune function may be vital for the adaptive reconfiguration of the immune system during fight-or-flight behavior. Integr Comp Biol. 2014;54:419–26. doi: 10.1093/icb/icu005. [DOI] [PubMed] [Google Scholar]
- 89.Aguilar R, Jedlicka AE, Mintz M, Mahairaki V, Scott AL, Dimopoulos G. Global gene expression analysis of Anopheles gambiae responses to microbial challenge. Insect Biochem Mol Biol. 2005;35:709–19. doi: 10.1016/j.ibmb.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 90.Ayres JS, Schneider DS. The role of anorexia in resistance and tolerance to infections in Drosophila. PLoS Biol. 2009;7 doi: 10.1371/journal.pbio.1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lourenco AP, Martins JR, Bitondi MM, Simoes ZL. Trade-off between immune stimulation and expression of storage protein genes. Arch Insect Biochem Physiol. 2009;71:70–87. doi: 10.1002/arch.20301. [DOI] [PubMed] [Google Scholar]
- 92.Meng Y, Omuro N, Funaguma S, Daimon T, Kawaoka S, Katsuma S, et al. Prominent down-regulation of storage protein genes after bacterial challenge in eri-silkworm, Samia cynthia ricini. Arch Insect Biochem Physiol. 2008;67:9–19. doi: 10.1002/arch.20214. [DOI] [PubMed] [Google Scholar]
- 93.Roux J, Privman E, Moretti S, Daub JT, Robinson-Rechavi M, Keller L. Patterns of positive selection in seven ant genomes. Mol Biol Evol. 2014;31:1661–85. doi: 10.1093/molbev/msu141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barribeau SM, Sadd BM, du Plessis L, Brown MJ, Buechel SD, Cappelle K, et al. A depauperate immune repertoire precedes evolution of sociality in bees. Genome Biol. 2015;16:83. doi: 10.1186/s13059-015-0628-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stoll S, Feldhaar H, Fraunholz MJ, Gross R. Bacteriocyte dynamics during development of a holometabolous insect, the carpenter ant Camponotus floridanus. BMC Microbiol. 2010;10:308. doi: 10.1186/1471-2180-10-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 101.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 102.Smit AF, Hubley R, Green P. RepeatMasker 3.0 repeatmasker.org, http://www.repeatmasker.org/webrepeatmaskerhelp.html (1996–2013).
- 103.Smit AF, Hubley R. RepeatModeler 1.05.repeatmasker.org, http://www.repeatmasker.org/RepeatModeler.html (2011).
- 104.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;Suppl 2:ii215–25. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 105.Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;3(4):W435–9. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Jr, Hannick LI, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–66. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–75. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 109.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–7. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 110.Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–95. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7 doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Parra G, Blanco E, Guigó R. GeneID in Drosophila. Genome Res. 2000;10:511–5. doi: 10.1101/gr.10.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Keller O, Odronitz F, Stanke M, Kollmar M, Waack S. Scipio: using protein sequences to determine the precise exon/intron structures of genes and their orthologs in closely related species. BMC Bioinformatics. 2008;9:278. doi: 10.1186/1471-2105-9-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kent WJ. BLAT-the BLAST-like alignment tool. Genome Res. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gaudermann P, Vogl I, Zientz E, Silva FJ, Moya A, Gross R, et al. Analysis of and function predictions for previously conserved hypothetical or putative proteins in Blochmannia floridanus. BMC Microbiol. 2006;6:1. doi: 10.1186/1471-2180-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007;36:D480–4. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yamamoto S, Sakai N, Nakamura H, Fukagawa H, Fukuda K, Takagi T. INOH: ontology-based highly structured database of signal transduction pathways. Database (Oxford). 2011:bar052. [DOI] [PMC free article] [PubMed]
- 118.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, et al. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–9. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, et al. The PFAM protein families database. Nucleic Acids Res. 2010;38:D211–22. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fjell CG, Hancock REW, Cherkasov A. AMPer: a database and an automated discovery tool for antimicrobial peptides. Bioinformatics. 2007;23:1148–55. doi: 10.1093/bioinformatics/btm068. [DOI] [PubMed] [Google Scholar]
- 122.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 123.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 125.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]