

Abstract
Herpes simplex virus 1 (HSV-1) microRNAs (miRNAs) mostly located in transcription-associated transcript (LAT) region have been identified that play critical roles in the intricate host-pathogen interaction networks. Increasing evidences throw new insight into the role of miRNA-mediated miRNA-mRNA cross-talk in HSV-1 latent or acute infection. In the present study, we found that hsv-1 miR-H4-5p (here termed as miR-H4b) can down-regulate the expression of cyclin-dependent kinase inhibitor 2A (CDKN2A, p16) in neuroblastoma (SHSY5Y) cell lines. Decreased expression of miR-H4b was directly related to attenuated cell proliferation and invasion as well as malfunction of cell cycle in recombinant SHSY5Y cells that stably expressing miR-H4b. Bioinformatics analysis and luciferase assays demonstrated miR-H4b can directly target p16 mRNA. MiR-H4b exerts its pro-proliferation function through inhibition of the p16-related PI3K-Akt pathways. Our findings provide, for the first time, significant clues regarding the role of herpesvirus-encoded miRNAs as a viral modulator to host cells.
Keywords: Viral microRNA, p16, host-pathogen interaction
Introduction
Viral miRNAs are small RNA molecules which have recently gained widespread attention as critical regulators in host-pathogen interaction networks [1]. These miRNAs regulate translation of messenger RNA (mRNA) also by binding to it depending on the extent of sequence complementarity with the target [2]. So far, more than 140 herpesvirus-encoded miRNAs have been identified. Modest initial reports have experimentally validated these miRNAs targets. Moreover, most of these herpesvirus-encoded miRNAs have been shown to target either viral gene mRNA or cellular gene binding sites, except for miR-UL112-1 encoded by HCMV which can target both immediate-early transactivator of viral genes and cellular NK cell ligand gene [3,4].
A hallmark of herpesvirus biology is their ability to establish lifelong latency in the host nervous system and reactivate in the area served by these sensory neurons [5]. The noncoding latency-associated transcript (LAT) plays a crucial role partly via LAT-encoded miRNAs in maintaining latent infection and reactivation [6]. However, the specific regulatory mechanisms of most HSV-encoded miRNAs are still unknown [7]. Deciphering the role of these viral miRNAs require the identification of corresponding targets and experimentally verified [8].
HSV-1 miR-H4-5p briefly termed as miR-H4b was sliced from 5’ arm of microRNA precursor. We confirmed miR-H4 can efficiently target the expression of infected cell protein 34.5 (ICP34.5). But whether this miRNA can regulate cellular gene expression is currently not known. Here, we determined that a cyclin-dependent kinase inhibitor 2A named as p16 is downregulated in SHSY5Y cells by miR-H4b, and we found an inverse correlation between the levels of miR-H4b and p16 protein. MiR-H4b directly targets the 3’-UTR of the p16 transcript that related to cell proliferation, invasion and cell cycle. MiR-H4b mediates its proliferation activator function, at least in part, by regulating PI3K-AKT pathway.
Materials and methods
Construction of recombinant plasmid
To construct a plasmid expressing miRNA-H4b, a fragment containing miRNA-H4 precursors was amplified from HSV-1 strain 17syn+ genomic DNA. For miR-H4b overexpression, the amplified fragments were then cloned into a pcDNA3.0 vector, which is here termed pcDNA/H4b. The empty vector was used as the blank control. Scrambled miRNAs were inserted into pcDNA3.0 in the same way and termed as pcDNA/NC. Scramble sequences: GGTACTGCAAAGTTCTCAA TGC. To construct wild 3’-UTR reporter plasmids (luc-wild), the 3’-UTR fragments of human p16 mRNA containing the putative miR-H4b binding sites were amplified from 5Y cDNA, and cloned into the Kpn I and Hind III sites of the firefly luciferase vector (Promega, U.S.). Similarly, for luc-mut vectors, in which six-point-mutations were introduced into the seed regions of the miRNA-H4b binding sites as a mutant control, primers of the seed sequences were mutated and amplified using a PCR approach. Empty luciferase vectors were inserted into luciferase vector to construct negative control (psi-CHECK, luc-NC).
Cell lines, cell transfection and infection
This study employed human neuroblastoma cell line SHSY5Y which obtained from medical laboratory department of our hospital. SHSY5Y4b cells stably expressing miR-H4b were established after recombinant plasmids pcDNA/H4b were transfected into cultured SHSY5Y cells and screened by G418 as references in our laboratory. Control vector expressing a scrambled miRNA were tansfected into SHSY5Y cells and selected with 400 mg/l G418 to generate a negative control (5YNC) cell line. SHSY5Y was nominated as 5Y cell here and SHSY5Y4b cell line containing miR-H4b precursor sequences was briefly named as 5Y4b for conveniences. All cell lines, including 5Y, 5Y4b, HEK 293, were grown in Dulbecco’s Modified Eagle’s medium (DMEM, GIBCO) supplemented with 10% fetal calf serum (FCS) and 100 U/ml penicillin-streptomycin. All cells were maintained at 37°C under an atmosphere of 5% CO2. The inhibitors antimiR-H4b (anti-4b) and its negative control of scrambled nucleotides (anti-NC) chemically synthesized were also transfected into 5Y4b cells using Lipofectamine 2000 (Invitrogen) to establish stable miR-H4b knockdown model.
Quantitative RT-PCR analysis (qPCR)
Total RNAs were extracted from cells with TRIzol reagent (Invitrogen). For the detection of relative expression of miR-H4b and p16 mRNA, cDNA was synthesized according to the manufacturer’s instructions of reverse reaction kit (Promega). QPCR reactions were performed by means of a qSYBR-green-containing PCR kit (Invitrogen), and β-actin was used as an endogenous control for miRNA detection. Expression of each gene was quantified by measuring cycle threshold (Ct) values using the 2-ΔΔCt method.
MTT assay for assessment of cell proliferation
Approximately 0.5 g/L tetrazolium MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) was added into the cells cultured in 96-well microtitre plates at a concentration of 5×104 cells per well. Following 0 h, 24 h, 48 h, 72 h of culture after anti-4b and anti-NC transfection, cells were incubated with 150 ml of MTT for 4 h in a 37°C incubator with 5% CO2. The absorbance at 490 nm was measured using automatic thermo Scientific Multiskan FC multifunctional enzyme-labeled instrument (thermo fisher).
Cell proliferation assay
After transfection of indicated inhibitors, cells in different groups were plated into six-well plates at the desired cell concentrations. Following 0 h, 24 h, 48 h and 72 h of culture, cell counts were estimated by trypsinizing the cells and performing analysis in triplicate with a Coulter counter (Beckman Coulter, Fullerton, CA).
Cell invasion assay
At 48 hours post transfection, cells were seeded onto the basement membrane matrix (EC matrix, Chemicon, Temecula, CA) present in the insert of a 24-well culture plate. FBS was added to the lower chamber as a chemoattractant. After a further 48 hours, the non-invading cells and EC matrix were gently removed with a cotton swab. Invasive cells located on the lower side of the chamber were stained with Crystal Violet, air-dried and photographed.
Luciferase assay
Luciferase reporter assays were performed using the dual luciferase reporter assay system (Promega) according to the manufacturer’s instructions. Luc-wt, Luc-mut and Luc-NC were constructed as indicated methods above. Luc-wt, Luc-mut and Luc-NC were co-transfected with pcDNA/H4b or pcDNA/NC into Heck cells. In addition, Luc-wt was cotransfected with miR-H4b inhibitor into Heck cells. Luciferase activity was measured in cell lysates 48 hours after transfection. Results were normalized against Renilla luciferase activity, which were performed at least three times.
Flow cytometry analysis
Cells (5Y4b or 5Y) transfected for 48 h with inhibitor were harvested and then stained with 200 μl for 20 min as Guava Cell Cycle Reagent Kit (Millipore) instructions. Analysis of cell cycle analysis was performed according to the manufacturer’s protocol. Flow cytometry analysis was performed using the Beckman Coulter flow cytometer and EXPO32 software (Beckman).
Western blotting
Western blotting was carried out as described previously [9]. Anti-p16 antibody was obtained from Sigma-Aldrich. Antibodies against β-actin, CDK4, CDK6, AKT, GSK3β, phosphorylated CDK4 and CDK6, phosphorylated AKT, phosphorylated GSK3 band antibodies were obtained from Cell Signaling Technology. β-actin was used as internal control.
Statistical analysis
Student’s unpaired t-tests were used to assess statistical significance. Data are expressed as means ± S. E. M. P<0.05 was considered statistically significant.
Results
miR-H4b promotes cell proliferation, invasion and boost cell cycle in S phase in 5Y4b cell lines
We establish a new cell line 5Y4b stably expressed high level of miR-H4b contrast to 5YNC cells and 5Y cells after HSV-1 infection using qPCR detection (Figure 1A). To investigate whether anti-4b can suppress the expression of miR-H4b, after transfecting of anti-4b into 5Y4b cells, the level of miR-4b expression in 5Y4b was reduced dramatically compared with the level of expression without inhibitors (Figure 1A).
Figure 1.
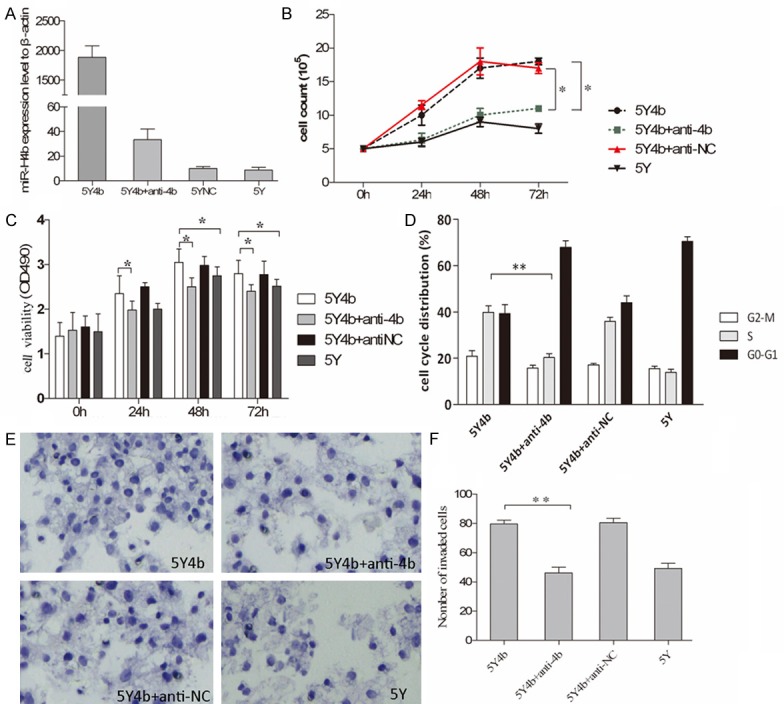
miR-4b promotes cell proliferation, invasion and cell cycle progression. A. qPCR analysis of the expression levels of miR-H4b in 5Y or 5Y4b cells. Anti-4b nucleotides were transfected into 5Y4b cells to detect its downregulation role on miR-4b. The expression values of β-actin are set at 1 and the relative expression levels of miR-H4b were determined by using the 2-ΔΔCt method. B. Proliferation of 5Y4b cells was measured by cell counting assay after transfecting either anti-4b or scramble anti-NC. The cell proliferation was determined by cell counting 0, 24, 48 and 72 h following transfection. The results are presented as the mean number of cell lines at different time points. The experiment was performed in triplicate. Anti-4b interfering RNA decreased the proliferation capacity of 5Y4b cells. C. Cell viability was measured by the MTT assay post 0 h, 24 h, 48 h, 72 h of transfection. The histogram shows mean values of A490 from three independent experiments. Anti-4b or anti-NC were transfected into 5Y4b cell separately. Compared to 5Y4b cells, cell viability of 5Y cells and 5Y4b cells with anti-4b was dramatically decreased. *P<0.05. D. The proportion of 5Y4b cells stayed in S-phase was lager than 5Y cells. 5Y4b cells transfected with anti-4b or anti-NC were subjected to cell cycle analysis. *P<0.05. **P<0.01. E. miR-4b increased the invasive capacity of 5Y cells. 5Y cells, 5Y4b cells transfected with anti-4b or anti-NC were loaded into the Matrigel-coated upper chambers of Transwell plates. Cells were counted under a microscope in five random fields at ×200 magnification. F. The compelling number of invaded cell was illustrated in histogram. *P<0.05, 5Y4b cells vs. 5Y4b cells with anti-4b.
The effects of miR-H4b on cell growth, invasion and cell cycle were determined via several assays. Transfection of anti-4b significantly reduced cell proliferation in 5Y4b cell lines, compared to control transfections of anti-NC (Figure 1B). There was no significant difference in levels of proliferation ability to cells transfected with anti-H4b and 5Y cells without treatment. MTT assay confirmed that transfection of anti-4b can effectively decreased cell activity than other control groups (Figure 1C). The effects of miR-H4b on cell invasion were determined with the Matrigel-based transwell invasion assay. 5Y4b cells treated with anti-4b inhibitors were distinctively less migratory than cells transfected with anti-sn and similar to 5Y cells with no treatment (Figure 1E, 1F). Additionally, a lager proportion of 5Y4b cells stayed in S phase than 5Y cells, and anti-4b can effectively reduced this ration (Figure 1D).
miR-H4b directly targets and inhibits p16 expression
To explore the function of miR-H4b, three computational algorithms, Target Scan, RNAhybrid and miRanda, were used to search for potential miR-H4b cellular target genes and a large number of different target genes were predicted [10]. Among these candidate target genes, P16, which was predicted by all three algorithms, attracted our attention immediately. P16 is a master regulator and is known to be essential in regulating cell growth, proliferation, carcinogenesis and cell cycle.
The ability of miR-H4b to regulate the 3’UTR region of p16 mRNA was evaluated via luciferase reporter assay. HEK 293 cells, which have been identified did not express miR-H4b (data not shown), were used to verify this effect. There was perfect base pairing between the seed sequence of mature miR-H4b and the 3’-UTR of p16 mRNA (Figure 2A). The 3’UTR targets sites of p16 were cloned into psiCHECK-2-control vector, downstream of a luciferase minigene. We observed that addition of pcDNA/H4b plasmids, but not pcDNA/NC including scramble control, dramatically suppressed the luciferase activity upon co-transfection of the luciferase vector (wild-type, mutant or negative control), demonstrating specificity of the target sequence for p16 (Figure 2B). The ability of miR-H4b to influence the endogenous p16 mRNA and protein were also tested in 5Y cells. Western blotting and qPCR assay showed that p16 mRNA and protein were greatly decreased in 5Y4b cells than those transfected with anti-4b and cells without any treatment (Figure 2C and 2D). These findings are consistent with D Niederacher et.al report that inactivation of p16 could lead to uncontrolled cell growth [9].
Figure 2.
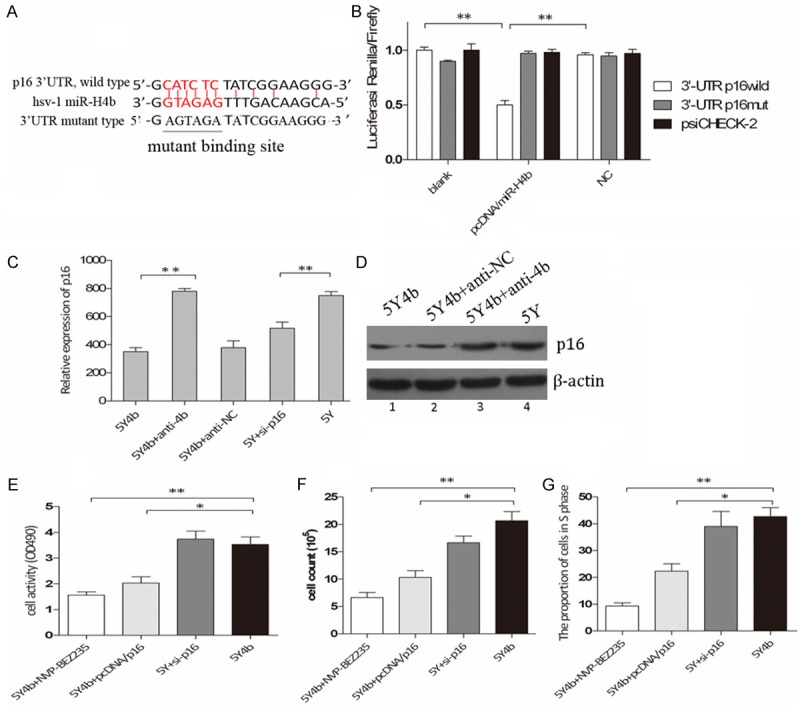
CDKN2A is target of miR-H4b. A. MiR-H4b target site resides at nucleotides of the p16 3’-UTR. Binding sites are indicated in red letters and the corresponding mutants are labeled in a solid line. All the complementary pairing bases are indicated in the vertical curve. B. Luciferase activity assay for direct targeting of the 3’-UTR of p16 by miR-H4b. Two copies of wild-type and mutant miR-H4b target sequences of p16 were fused with luciferase reporter and respectively co-transfected into HEK 293 cells with recombinant plasmid containing miR-4b precursor, and luciferase activity measured 48 hours post transfection. **P<0.01. C. P16 mRNA levels analyzed in 5Y4b cells tranfected with anti-4b by qRT–PCR; D. P16 protein levels analyzed by western blot. P16 expression was lower in 5Y4b with or without anti-NC cells compared with 5Y or 5Y4b cells with anti-4b. Similar results were observed in cells cotransfected with siRNA for p16 into 5Y cells. E. Promoting proliferation ability of miR-H4b mediated by inhibiting target genes p16. The cell viability of 5Y4b cells added with either NVP-BEZ235 at 1 nM for 60 h or pcDNA/p16 plasmids (without 3’-UTR binding sites) was much lower than that of 5Y4b cells or 5Y cells with siRNA for p16. F. Proliferation of cells in different groups was measured by cell counting assay. NVP-BEZ235 and pcDNA/p16 in 5Y4b cells can effectively decrease the proliferation capacity compared to 5Y cells with si-p16. G. PcDNA/p16 and NVP-BEZ235 rescued the uncontrolled cell cycle, and 5Y4b cells or 5Y cells with si-p16 showed a higher proportion of cells in s phase than other groups. **P<0.01.
Transfection of pcDNA/P16 contributed to attenuate uncontrolled cellular behavior
Transfection of p16-encoding vector in 5Y4b cells rescued the abnormal level of p16 to investigate the whether introduction of p16 can suppress uncontrolled biological function. Cell proliferation assay, cell invasion assay and flow cytometry analysis were also performed at 60 h after transfection. In 5Y4b cells, in vitro overexpression of p16 we found that the effect of miR-H4b was partially attenuated and cell invasion, proliferation and cell cycle were inhibited (Figure 2E-G). These data confirmed that the pro-proliferation effect of miR-H4b was mediated by inhibiting target genes p16.
To investigate the biological functions of p16 in 5Y cells, we determined whether inhibition of p16, similar to miR-H4b overexpression, promoted 5Y4b cell invasion, proliferation. In 5Y cells, knockdown of p16 promoted cell invasion, cell growth and cell cycle progression. To further test this, we transfected 5Y cells with siRNA for p16 mRNA and found that the capacity of cell invasion, proliferation and cell cycle progression was advanced. Additionally, NVP-BEZ235 was added into 5Y4b cells to explore the potential signaling pathway of this effect. Similar to those in 5Y4b cells with anti-4b, the effect of miR-H4b promoting effect was partially attenuated by NVP-BEZ235 (Figure 2E-G). NVP-BEZ235 is well-known as an effective activity inhibitor of PI3K and Mtor [11]. These data confirmed that the promoting effect of miR-H4b was mediated by inhibiting target genesp16 and may involve in PI3K signaling pathway.
miR-H4b inhibits the PI3K-AKT pathways
P16 protein acts as a negative cell cycle regulator which can bind to CDK4 and prevents the association of CDK4 with cyclin D1 [12]. Phosphorylation of substrates essential for G1 transition of the cell cycle and cell growth is subsequently inhibited [13]. Inactivation of the CDKN2A gene may therefore lead to uncontrolled cell growth. P16 activation can trigger several important signaling pathways, such as the PI3K-AKT, p14ARF-mdm2-p53 and p16INK4a-CDK4 (CDK6)-pRb pathways, most of which regulate cell proliferation, invasion and apoptosis [14]. Therefore, we investigated the possibility that miR-H4b regulates those pathways by targeting p16. Downregulation of miR-H4b, through transfection of anti-4b, in 5Y4b cells decreased the phosphorylation levels of PI3K and its downstream target Akt (Figure 3A, 3B). We also observed that overexpression of miR-H4b, through transfection of miR-H4b mimics; in 5Y cells increased the levels of phosphorylated AKT and GSK-3β (Figure 3A). These western blotting results demonstrated that miR-H4b is negative regulator of PI3K pathways. Subsequently, rescue experiments were performed by overexpressing the p16 vector (without an endogenous 3’-UTR) in 5Y4b cells: 5Y4b cells were first transfected with p16-encoding vector. The miR-H4b-induced downregulation of P16 was rescued upon the introduction of P16, and the phosphorylation levels of AKT and mTOR were altered in a similar manner. The downregulation of phosphorylated AKT and phosphorylated mTOR by miR-H4b could be rescued by re-expression of P16. These observations suggest that miR-H4b inhibits the PI3K-AKT and mTOR pathways by targeting P16.
Figure 3.
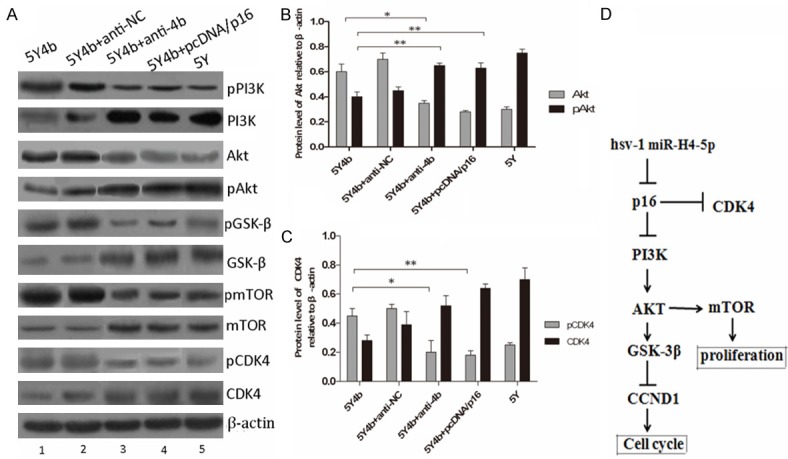
miR-H4b inhibits PI3K-AKT and mTOR pathways by targeting p16. A. miR-H4b overexpression reduced the activity of PI3K and mTOR pathways in 5Y4b cells. Knockdown of miR-H4b by anti-4b increased the activity of PI3K and mTOR signaling in 5Y4b cells. PcDNA/p16 plasmids were transfected into 5Y4b cells to investigate whether re-expression of p16 can rescue these effects. PI3K-AKT-(GSK-3β)-mediated pathway activity was measured by examining expression of phosphorylated AKT (pAKT), phosphorylated PI3K and phosphorylated GSK3β (pGSK3β), whereas PI3K-AKT-mTOR pathway activity was measured by examining expression of mTOR and phosphorylated mTOR. Protein level of phosphorylated CDK4 decreased in 5Y4b cells after transfecting with anti-4b. B-C. The bar chart showed the ratio of Akt or pAkt and CDK4 or pCDK4 to β-actin at each group. These data are means ± SEM. (n=3, *P<0.05, **P<0.01). D. Schematic regulation of hsv-1 miR-H4-5p on PI3K-Akt pathway and Akt-mediated mTOR pathway. Arrows means promopting effect, and T type arrow means inhibiting effect on downstream.
Discussion
The major causes of refractoriness from herpes simplex are complications arising from latency [15]. During recent years, HSV-encoded miRNAs have been found to involve in a series of biological processes of herpesvirus-host interaction, providing a new perspective on the mechanism of latent infection. Both HSV-1 and HSV-2 are neurotropic virus and only non-coding latency-associated transcript are easily detected during latent infection [16]. Most miRNAs are located within this region and have been demonstrated closely related to the establishment of latency and recurrence. Herpesviruses establish infections that persist for the life of the host via by regulating itself or host genes expression, an intricate balance therefore exists between host immune surveillance and virus immune evasion [17]. Recently, four miRNAs were identified in HSV-1 LAT and three in HSV-2 LAT. In HSV-1, miR-H2 is antisense relative to ICP0, whereas both miR-H3 and miR-H4 are antisense relative to ICP34.5 [18]. Based on this antisense positioning it was predicted that LAT-encoded miRNAs might target these genes, but only miR-H2 has been shown experimentally to repress ICP0 protein production [15]. Targeting of ICP0 in HSV-2 was also confirmed experimentally for miR-H2 that is antisense relative to ICP0. Whether these miRNAs also target host genes in a seed-sequence specific fashion is currently not known [19,20]. Therefore, we believe more effort should be made, not only towards the identification of relevant miRNAs but also into the specific mechanisms by which they accomplish their specific functions.
Considering barely detectable expression level of miR-H4b in guinea pig dorsal root ganglia during latent infection, pcDNA/H4b plasmids were transfected into neuroblastoma cells 5Y and recombinant cells were screened by G418 to establish overexpression model [21]. We used invasion assay, cell proliferation assay and flow cytometry analysis to observe the differences of biological behavior. In 5Y4b cells, the increase in ability of cell proliferation and invasion as well as the proportion of cells stayed in S phase was several times greater than that in 5Y cells or 5Y4b cells with miR-H4b inhibitor. The levels of miR-H4b were found to be positively correlated to proliferation and invasion, implying that potential cellular genes may be regulated by miR-H4b.
To explore the possible targets of miR-H4b in nerve cells, different computational algorithms were used to predict the potential target. CDKN2A as a candidate target of miR-H4b attracted our attention immediately for its regulatory role in cell growth and apoptosis, which was further confirmed in luciferase activity assays and miR-H4b-mediated p16 expression analysis. The luciferase activity assays demonstrated that miR-H4b could bind to the 3’-UTR of p16 mRNA. When we transfected miR-H4b inhibitors into 5Y4b cell an inverse biological behavior of invasion, cell growth and cell cycle were observed. Introduction of overexpression can rescue aberrant cell properties. CDKN2A gene inactivation is involved in the tumorigenesis of ovarian cancer. Importantly, we know CDKN2A gene can encode two regulatory protein p16INK4a and p14ARF which have been known involved in cellular bioprocess via cyclinD-CDK4/6-pRb-E2F and p14ARF-mdm2-p53 pathways [22,23]. Binding of p16 to CDK4 prevents the association of CDK4 with cyclin D1, and p16 acts as a negative regulator in cyclin D-dependent CDK4/6-pRb-E2F feedback regulation [24]. On the other hand, downregulation of p14ARF will directly cause the degradation of p53 protein well-known as tumor suppressor. Phosphorylation level of Rb and the level of p53 expression were verified and consisted with previously report.
Activation of the p16 signal has been well documented in various cancer cells. Besides CDK4/6-pRb-E2F and p14ARF-mdm2-p53 pathway, PI3K-AKT is the best typical pathway downstream of p16 [25]. The ability to target CDKN2A transcripts signifies that miR-H4b might be a potential regulator of PI3K-AKT pathways in 5Y cells. Our results demonstrated that miR-4b can inhibit the phosphorylation of PI3K, Akt, mTOR and GSK-3β, whereas expression of exogenous p16 (without an endogenous 3’-UTR) can rescue this inhibition. CCND1 protein is a key regulator to control the entering of cell cycle into S phase. mTOR protein was also included in this study as a downstream regulator of Akt, which it controls cell proliferation and invasion. These results were consistent with the effect of NVP-BEZ235 on 5Y4b cells and further demonstrating that the promoting effects of miR-H4b affect cellular behavior via PI3K-Akt pathway [26].
It is increasingly clear that viral miRNAs can positively or negatively influence viral replication. Viral miRNAs can directly affect host physiology by restricting cellular gene expression to reshape cellular environment for virus replication or latency [8,27]. Here, our findings suggest that miR-H4b functions as cell cycle promoter, affecting 5Y cell proliferation and invasion by targeting p16 and subsequently suppressing downstream AKT and mTOR signaling pathways. Although low expression level of miR-H4b can not obviously affect pathological processes in nerve cells during HSV-1 infection, some functions of miR-H4b in infected cells will undoubtedly turn out to be important for virus replication or the establishment of latency in vivo and may therefore become targets for therapeutic intervention.
Acknowledgements
This work was supported by a grant from the reviewers for their helpful comments on this paper.
Disclosure of conflict of interest
None.
References
- 1.Boss IW, Plaisance KB, Renne R. Role of virus-encoded microRNAs in herpesvirus biology. Trends Microbiol. 2009;17:544–553. doi: 10.1016/j.tim.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. microRNA expressed by herpes simplex virus1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–3. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, Horwitz E, Prokocimer Z, Prichard M, Hahn G, Goldman-Wohl D, Greenfield C, Yagel S, Hengel H, Altuvia Y, Margalit H, Mandelboim O. Host immune system gene targeting by a viral miRNA. Science. 2007;317:376–81. doi: 10.1126/science.1140956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cosman D, Müllberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14:123–33. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- 5.Zheng SQ, Li YX, Zhang Y, Li X, Tang H. MiR-101 regulates HSV-1 replication by targeting ATP5B. Antiviral Res. 2011;89:219–226. doi: 10.1016/j.antiviral.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, Coen DM. Numerous conserved and divergent micrornas expressed by herpes simplex viruses 1 and 2. J Virol. 2010;84:4659–4672. doi: 10.1128/JVI.02725-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cullen BR. Viral and cellular messenger RNA targets of viral microRNAs. Nature. 2009;457:421–425. doi: 10.1038/nature07757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottwein E, Cullen BR. Viral and cellular micrornas as determinants of viral pathogenesis and immunity. Cell Host Microbe. 2008;3:375–387. doi: 10.1016/j.chom.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niederacher D, Yan HY, An HX, Bender HG, Beckmann MW. CDKN2Agene inactivation in epithelial sporadic ovarian cancer. Br J Cancer. 1999;80:1920–1926. doi: 10.1038/sj.bjc.6690621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X, Zhang Z, Sun L, Chai N, Tang S, Jin J, Hu H, Nie Y, Wang X, Wu K, Jin H, Fan D. microRNA-499-5p promotes cellular invasion and tumor metastasis in colorectal cancer by targeting FOXO4 and PDCD4. Carcinogenesis. 2011;32:1798–1805. doi: 10.1093/carcin/bgr213. [DOI] [PubMed] [Google Scholar]
- 11.Moon du G, Lee SE, Oh MM, Lee SC, Jeong SJ, Hong SK, Yoon CY, Byun SS, Park HS, Cheon J. NVP-BEZ235, a dual PI3K/mTOR inhibitor synergistically potentiates the antitumor effects of cisplatin in bladder cancer cells. Int J Oncol. 2014;45:1027–35. doi: 10.3892/ijo.2014.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Giorgi V, Savarese I, D’Errico A, Gori A, Papi F, Colombino M, Cristina Sini M, Stanganelli I, Palmieri G, Massi D. CDKN2A mutations could influence the dermoscopic pattern of presentation of multiple primary melanoma: a clinical dermoscopic genetic study. J Eur Acad Dermatol Venereol. 2015;29:574–80. doi: 10.1111/jdv.12643. [DOI] [PubMed] [Google Scholar]
- 13.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 14.Danielsen SA, Eide PW, Nesbakken A, Guren T, Leithe E, Lothe RA. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim Biophys Acta. 2014;1855:104–121. doi: 10.1016/j.bbcan.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Tang S, Patel A, Krause PR. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J Virol. 2008;83:1433–1442. doi: 10.1128/JVI.01723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent. Nature. 2008;454:780–3. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang S, Patel A, Krause PR. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J Virol. 2009;83:1433–42. doi: 10.1128/JVI.01723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–3. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui C, Griffiths A, Li G, Silva LM, Kramer MF, Gaasterland T, Wang XJ, Coen DM. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J Virol. 2006;80:5499–508. doi: 10.1128/JVI.00200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertke AS, Patel A, Imai Y, Apakupakul K, Margolis TP, Krause PR. Latency-associated transcript (LAT) exon 1 controls herpes simplex virus species-specific phenotypes: reactivation in the guinea pig genital model and neuron subtype-specific latent expression of LAT. J Virol. 2009;83:10007–15. doi: 10.1128/JVI.00559-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abraham J. PI3K/AKT/mTOR pathway inhibitors: the ideal combination partners for breast cancer therapies? Expert Rev Anticancer Ther. 2015;15:51–68. doi: 10.1586/14737140.2015.961429. [DOI] [PubMed] [Google Scholar]
- 23.Yin S, Deng W, Zheng H, Zhang Z, Hu L, Kong X. Evidence that the nonsense-mediated mRNA decay pathway participates in X chromosome dosage compensation in mammals. Biochem Biophys Res Commun. 2009;383:378–82. doi: 10.1016/j.bbrc.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 24.DiRocco DP, Bisi J, Roberts P, Strum J, Wong KK, Sharpless N, Humphreys BD. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am J Physiol Renal Physiol. 2014;306:F379–88. doi: 10.1152/ajprenal.00475.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng T, Peng L, Chao C, Fu B, Wang G, Wang Y, Zhu X. IRE1alpha-TRAF2-ASK1 complex-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to CXC195-induced apoptosis in human bladder carcinoma T24 cells. Biochem Biophys Res Commun. 2015 doi: 10.1016/j.bbrc.2014.12.112. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 26.Oishi T, Itamochi H, Kudoh A, Nonaka M, Kato M, Nishimura M, Oumi N, Sato S, Naniwa J, Sato S, Shimada M, Kigawa J, Harada T. The PI3K/mTOR dual inhibitor NVP-BEZ235 reduces the growth of ovarian clear cell carcinoma. Oncol Rep. 2014;32:553–8. doi: 10.3892/or.2014.3268. [DOI] [PubMed] [Google Scholar]
- 27.Scaria V, Hariharan M, Maiti S, Pillai B, Brahmachari SK. Host-virus interaction: a new role for microRNAs. Retrovirology. 2006;3:68. doi: 10.1186/1742-4690-3-68. [DOI] [PMC free article] [PubMed] [Google Scholar]