

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired hematopoietic stem cell (HSC) disorder arising from a somatic mutation of the X-linked phosphatidylinositol glycan complementation class A gene (PIG-A) which leads to partial or complete deficiency of glycosyl-phosphatidylinositol (GPI)-linked membrane proteins and causes intravascular hemolysis. Its pathophysiological links with aplastic anemia (AA) and myelodysplastic syndrome (MDS) have been described frequently, and few acute leukemia are proved to be derived from PNH. However, PNH with transformation to chronic myeloid leukemia (CML) has never been reported. Here, we report a patient initially diagnosed with PNH while 11 years later, Ph chromosome and BCR/ABL fusion gene were detected and the patient was eventually confirmed the diagnosis of CML. Here, the diagnosis and management of the interesting case, as well as questions regarding pathogenesis, are discussed.
Keywords: Paroxysmal nocturnal hemoglobinuria (PNH), chronic myeloid leukemia (CML), clonal evolution
Introduction
PNH is a clonal hematopoietic stem cell disorder that harbors a PIG-A mutation which leads to partial or complete deficiency of GPI-linked membrane proteins (CD55 and CD59), resulting in the high sensitivity of red blood cells to the activated complement. Previous studies have always focused on its pathophysiological links with some specific hematopoietic stem cell disorders, such as AA, MDS, and very few acute leukemia. In this paper, we report a case of a patient initially presented with a clonal disorder of PNH and finally transformed to another clonal disorder of CML 11 years later. From PNH to CML transformation has, to our knowledge, never been reported previously.
Case report
A 52-year-old man was admitted to our hospital for fatigue and dark-colored urine in 2003. Physical examination revealed moderate anemia with no lymph node enlargement, hepatomegaly or splenomegaly. Complete blood examination revealed white blood cell (WBC) 3.8×109/L; hemoglobin (Hb) 72 g/L; platelet (Plt) 21×109/L and reticulocyte ratio 2.5%. Bone marrow aspiration showed hypercellular with relative erythroid hyperplasia (10%). Coombs test was negative while Ham’s test showed positive. Flow cytometry analysis of peripheral blood cells revealed that 13.1% of leucocytes and 12.4% of erythrocytes were CD55 deficiency and 23.7% of leucocytes and 11% of erythrocytes were CD59 deficiency. Thus, the patient was diagnosed with PNH. Administration of cyclosporin A, prednisolone, erythropoietin and andriol proved effective. One month later, the blood examination revealed that WBC, Hb, Plt increased to 7.01×109/L, 113 g/L, 76×109/L, respectively, and reticulocyte ratio decreased to 0.4%. Bone marrow aspiration showed hypercellular with normal erythrocyte. Regular follow-up demonstrated slight pancytopenia recurred from time to time. With the treatments of cyclosporine A and prednisone, the peripheral blood cells could recovered to near-normal.
In year 2014, the patient was found to have an elevated WBC (30.0×109/L) but normal Hb and Plt. Physical examination revealed slight splenomegaly. Flow cytometry analysis of peripheral blood cells revealed a normal expression of CD55 and CD59 in leucocytes and erythrocytes. Bone marrow aspiration showed chronic myeloproliferative neoplasm and cytogenetic analysis revealed the presence of a t (9;22) (q34;q11) in all examined metaphases. RT-PCR for BCR/ABL transcript was positive, and fluorescence in situ hybridization (FISH) also confirmed the BCR/ABL fusion gene (Figure 1). These findings were consistent with the chronic phase of CML. After imatinib 400 mg daily for a month, the complete hematological remission was achieved, 6 months later, a partial cytogenetic remission was reached. He sticks to the therapy at present.
Figure 1.
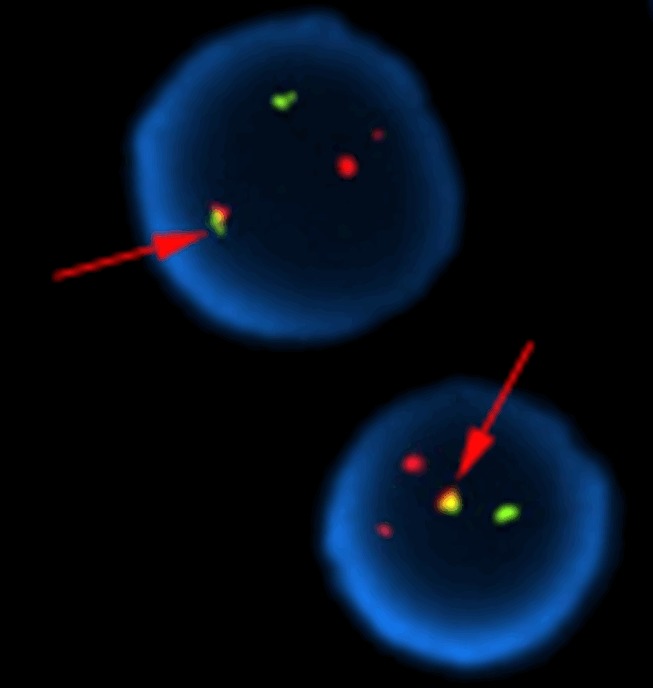
Fluorescence in situ hybridization (FISH) pattern of BCR/ABL fusion gene: FISH using orange-labeled ABL (9q34) and green-labeled BCR (22q11.2) probes. The cell shows 1 yellow fusion signal (arrow pointed), which indicates rearrangement between the ABL and BCR genes.
Discussion
We describe here a patient first diagnosed with a clonal disease PNH and finally transformed to another malignant clonal disorder CML 11 years later. To our knowledge, this is the first report that one patient with PNH transformed to CML ultimately.
PNH is a disease originating from a multipotent hematopoietic stem cell, caused by a somatic mutation in the X-linked PIG-A gene, which abrogates synthesis of the GPI-anchored proteins (GPI-AP), resulting in the deficiency of cell surface CD55 and CD59. Absence of CD55 and CD59 in particular explains the intravascular hemolysis, due to failure to inactivate the late components of complement [1,2]. It was originally recognized as a hemolytic anemia, but bone marrow failure and developed to MDS and acute leukemia were also related to PNH [1,3], demonstrating that PNH is a clonal disorder.
The coexistence of PNH and AA was first reported in 1944 by Dacie JV [4]. PNH may arise de novo or in the setting of AA [5]. In long-term survived severe AA patients, GPI anchor-deficient hematopoietic cell populations emerge at a high frequency (29% to 52%), suggesting a pathological link between these two disorders [6]. In contrast to classic PNH patients, AA patients typically have a lower percentage of PNH clones, fewer than 10% GPI-AP-deficient leucocytes are detected at diagnosis, but occasionally they may have bigger clones [7]. Some studies show that the presence of a minor population of PNH clones represents a reliable marker of a positive response to immunosuppressive therapy and a favorable prognosis factor for AA patients [8]. DNA sequencing of the GPI-AP-deficient cells from AA patients reveals clonal PIG-A gene mutations [9]. Although many AA patients exhibit no signs of PNH when the PNH clone size is small, most, but not all, will experience further expansion of the PIG-A mutant clone and progress to classic PNH [10].
PNH clone has also been detected in MDS patients. By high-resolution flow cytometry, approximately 15%-20% of low-risk MDS patients have been found to have a detectable GPI-AP-deficient erythrocytes and leucocytes, but sequencing of the PIG-A gene to establish clonality has not been performed in many of these studies [3,8,11-14]. MDS patients with PNH clone tend to be classified as refractory anemia and often have the following characteristics: moderate to severe thrombocytopenia, a hypocellular bone marrow, HLA-DR15 positive, normal cytogenetics, and a high likelihood of response to immunosuppressive therapy [3,8,11]. The association between PNH and MDS is specific for lower risk disease categories. PNH cells are not observed in higher risk categories [12].
A minority of PNH patients have been described to progress to acute leukemia. Most of the cases are acute myeloid leukemia (AML) and only 4 known cases of acute lymphoblastic leukemia (ALL) have been previously described [15-18]. So far no cases of PNH with transformation to CML have been reported yet. In the present case, the patient experienced 11 years from PNH to CML transformation. When he finally developed CML, Ph chromosome and BCR/ABL fusion gene emerged while PNH clones disappeared.
In most PNH transformed AML, the leukemic clone was derived from the PNH clone, genetic instability of PNH might be the underlying mechanism [19]. Ishihara S et al. reported two PNH patients who were transformed from AA, and sequentially progressed to MDS and leukemia. As the disease progressing, first the PNH clones appeared and expanded, gradually, these clones were replaced by MDS clones (monosomy 7), then MDS clones and their subclones expanded with additional chromosomal abnormalities. By the time when leukemia developed, WT1 (Wilms’ tumor gene) expression increased, showing the clonal evolution process [20]. Tanaka et al. reported an AML patient who developed from AA-PNH syndrome. Flow cytometry analysis showed that the leukemic cells were CD59 deficiency and P-glycoprotein was positive. Mutation analysis disclosed a frame shift mutation in exon 2 of the PIG-A gene, which generated truncated PIG-A protein. Based on these findings, the leukemic cells were confirmed to evolve from the affected PNH clone. Normal chromosome, -7, and -7 plus -20 were detected in the stage of AA-PNH, AML, and relapsed stage of AML, respectively, showing the strong evidence of a clonal evolution [21].
CML is one of the most in-depth explored malignant diseases that originates from an abnormal hematopoietic stem cell harboring the Philadelphia (Ph) chromosome. This chromosomal abnormality is generated by a reciprocal translocation involving the long arm of a chromosome 9 and the short arm of a chromosome 22 [t (9;22) (q34;q11)], which results in the formation of the BCR-ABL fusion gene and the chimeric BCR-ABL protein that functions as a constitutively active tyrosine kinase. This aberrant BCR-ABL oncoprotein constitutes the molecular basis of CML and makes cell cancerous [22].
To our knowledge, no cases of PNH with transformation to CML have been reported yet. Although both PNH and CML are clonal hematopoietic stem cell disorders, it is not clear yet whether there exists any association between them. One possible explanation for the transformation of our presenting case is that CML blasts were derived from affected PNH clones or the abnormal bone marrow micro-enviroment of PNH. In the clinical management of PNH patients, we should always be aware of the possible malignant transformation of PNH clones, and thus strengthen the follow-up in order to recognize abnormal clones in time for the diagnosis and treatment switch.
Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant 81170489.
Disclosure of conflict of interest
None.
References
- 1.Hernandez-Campo PM, Martin-Ayuso M, Almeida J, Lopez A, Orfao A. Comparative analysis of different flow cytometry-based immunophenotypic methods for the analysis of CD59 and CD55 expression on major peripheral blood cell subsets. Cytometry. 2002;50:191–201. doi: 10.1002/cyto.10072. [DOI] [PubMed] [Google Scholar]
- 2.Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 3.Wang H, Chuhjo T, Yasue S, Omine M, Nakao S. Clinical significance of a minor population of paroxysmal nocturnal hemoglobinuria-type cells in bone marrow failure syndrome. Blood. 2002;100:3897–902. doi: 10.1182/blood-2002-03-0799. [DOI] [PubMed] [Google Scholar]
- 4.Dacie JV, Gilpin A. Refractory anaemia (Fanconi type): Its Incidence in Three Members of One Family, with in One Case a Relationship to Chronic Haemolytic Anaemia with Nocturnal Haemo-Globinuria (Marchiafava-Micheli Disease or Nocturnal Haemoglobinuria) Arch Dis Child. 1944;19:155–162. doi: 10.1136/adc.19.100.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, Hillmen P, Luzzatto L, Young N, Kinoshita T, Rosse W, Socié G International PNH Interest Group. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699–3709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azenishi Y, Ueda E, Machii T, Nishimura J, Hirota T, Shibano M, Nakao S, Kinoshita T, Mizoguchi H, Kitani T. CD59-deficient blood cells and PIG-A gene abnormalities in Japanese patients with aplastic anaemia. Br J Haematol. 1999;104:523–529. doi: 10.1046/j.1365-2141.1999.01214.x. [DOI] [PubMed] [Google Scholar]
- 7.Mukhina GL, Buckley JT, Barber JP, Jones RJ, Brodsky RA. Multilineage glycosylphosphatidylinositol anchor-deficient haematopoiesis in untreated aplastic anaemia. Br J Haematol. 2001;115:476–482. doi: 10.1046/j.1365-2141.2001.03127.x. [DOI] [PubMed] [Google Scholar]
- 8.Sugimori C, Chuhjo T, Feng X. Minor population of CD55-CD59- blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107:1308–1314. doi: 10.1182/blood-2005-06-2485. [DOI] [PubMed] [Google Scholar]
- 9.Nagarajan S, Brodsky RA, Young NS, Medof ME. Genetic defects underlying paroxysmal nocturnal hemoglobinuria that arises out of aplastic anemia. Blood. 1995;86:4656–4661. [PubMed] [Google Scholar]
- 10.Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood. 2009;113:6522–6527. doi: 10.1182/blood-2009-03-195966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishiyama K, Chuhjo T, Wang H, Yachie A, Omine M, Nakao S. Polyclonal hematopoiesis maintained in patients with bone marrow failure harboring a minor population of paroxysmal nocturnal hemoglobinuria-type cells. Blood. 2003;102:1211–1216. doi: 10.1182/blood-2002-12-3706. [DOI] [PubMed] [Google Scholar]
- 12.Sugimori C, Mochizuki K, Qi Z, Sugimori N, Ishiyama K, Kondo Y, Yamazaki H, Takami A, Okumura H, Nakao S. Origin and fate of blood cells deficient in glycosylphosphatidylinositol-anchored protein among patients with bone marrow failure. Br J Haematol. 2009;147:102–112. doi: 10.1111/j.1365-2141.2009.07822.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang SA, Pozdnyakova O, Jorgensen JL, Medeiros LJ, Stachurski D, Anderson M, Raza A, Woda BA. Detection of paroxysmal nocturnal hemoglobinuria clones in patients with myelodysplastic syndromes and related bone marrow diseases, with emphasis on diagnostic pitfalls and caveats. Haematologica. 2009;94:29–37. doi: 10.3324/haematol.13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn DE, Tanawattanacharoen P, Boccuni P, Nagakura S, Green SW, Kirby MR, Kumar MS, Rosenfeld S, Young NS. Paroxysmal nocturnal hemoglobinuria cells in patients with bone marrow failure syndromes. Ann Intern Med. 1999;131:401–408. doi: 10.7326/0003-4819-131-6-199909210-00002. [DOI] [PubMed] [Google Scholar]
- 15.Ariel I, Weiler-Ravell D, Stalnikowicz R. Preleukemia in acute lymphoblastic leukemia. Acta Haematol. 1981;66:50–52. doi: 10.1159/000207092. [DOI] [PubMed] [Google Scholar]
- 16.Katahira J, Aoyama M, Oshimi K, Mizoguchi H, Okada M. Paroxysmal nocturnal hemoglobinuria terminating in TdT-positive acute leukemia. Am J Hematol. 1983;14:79–87. doi: 10.1002/ajh.2830140110. [DOI] [PubMed] [Google Scholar]
- 17.Shaheen SP 2nd, Talwalkar SS, Simons R, Yam L. Acute lymphoblastic leukemic transformation in a patient with chronic idiopathic myelofibrosis and paroxysmal nocturnal hemoglobinuria: a case report and review of the literature. Arch Pathol Lab Med. 2005;129:96–99. doi: 10.5858/2005-129-96-ALLTIA. [DOI] [PubMed] [Google Scholar]
- 18.Isoda A, Ogawa Y, Matsumoto M, Sawamura M. Coexistence of paroxysmal nocturnal hemoglobinuria (PNH) and acute lymphoblastic leukemia (ALL): Is PNH a prodrome of ALL. Leuk Res. 2009;33:e3–e5. doi: 10.1016/j.leukres.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 19.Mortazavi Y, Tooze JA, Gordon-Smith EC, Rutherford TR. N-RAS gene mutation in patients with aplastic anemia and aplastic anemia/paroxysmal nocturnal hemoglobinuria during evolution to clonal disease. Blood. 2003;95:646–650. [PubMed] [Google Scholar]
- 20.Ishihara S, Nakakuma H, Kawaguchi T, Nagakura S, Horikawa K, Hidaka M, Asou N, Mitsuya H. Two cases showing clonal progression with full evolution from aplastic anemia-paroxysmal nocturnal hemoglobinuria syndrome to myelodysplastic syndromes and leukemia. Int J Hematol. 2003;72:206–209. [PubMed] [Google Scholar]
- 21.Tanaka H, Imamura N, Oguma N, Shintani T, Tanaka K, Hyodo H, Oda K, Kimura A. Acute myelogenous leukemia with PIG-A gene mutation evolved from aplastic anemia-paroxysmal nocturnal hemoglobinuria syndrome. Int J Hematol. 2001;73:206–212. doi: 10.1007/BF02981939. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Li S. Molecular mechanisms for survival regulation of chronic myeloid leukemia stem cells. Protein Cell. 2013;4:186–196. doi: 10.1007/s13238-013-2115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]