

Abstract
Streptococcus gordonii, a primary colonizer of the tooth surface, interacts with salivary α-amylase via amylase-binding protein A (AbpA). This enzyme hydrolyzes starch to glucose, maltose, and maltodextrins that can be utilized by various oral bacteria for nutrition. Microarray studies demonstrated that AbpA modulates gene expression in response to amylase, suggesting that the amylase-streptococcal interaction may function in ways other than nutrition. The goal of this study was to explore the role of AbpA in gene regulation through comparative transcriptional profiling of wild-type KS1 and AbpA− mutant KS1ΩabpA under various environmental conditions. A portion of the total RNA isolated from mid-log-phase cells grown in 5% CO2 in (i) complex medium with or without amylase, (ii) defined medium (DM) containing 0.8% glucose with/without amylase, and (iii) DM containing 0.2% glucose and amylase with or without starch was reverse transcribed to cDNA and the rest used for RNA sequencing. Changes in the expression of selected genes were validated by quantitative reverse transcription-PCR. Maltodextrin-associated genes, fatty acid synthesis genes and competence genes were differentially expressed in a medium-dependent manner. Genes in another cluster containing a putative histidine kinase/response regulator, peptide methionine sulfoxide reductase, thioredoxin protein, lipoprotein, and cytochrome c-type protein were downregulated in KS1ΩabpA under all of the environmental conditions tested. Thus, AbpA appears to modulate genes associated with maltodextrin utilization/transport and fatty acid synthesis. Importantly, in all growth conditions AbpA was associated with increased expression of a potential two-component signaling system associated with genes involved in reducing oxidative stress, suggesting a role in signal transduction and stress tolerance.
INTRODUCTION
It is well known that dental plaque is involved in the etiology of the two most common oral diseases, caries, and periodontal disease. Streptococcus gordonii is one of the pioneer bacteria that initiate the formation of dental plaque on tooth surfaces. Dental plaque formation is a complex process that involves the participation of a variety of salivary components (1). Salivary α-amylase is the most abundant enzyme in saliva and is best known for its ability to degrade starch by hydrolyzing 1,4-glycosidic linkages with subsequent formation of maltose, maltotriose, and limit dextrins as the main products (2). Amylase binds to a number of oral streptococcal species, collectively referred to as the amylase-binding streptococci (ABS) (3–6). Once bound to streptococcal cells, amylase retains enzymatic activity to mediate the hydrolysis of starch to fermentable oligosaccharides (7–9). Thus, streptococcus-bound salivary amylase hydrolyzes dietary starch that can be further metabolized for streptococcal nutrition. It is also possible that S. gordonii and other ABS contribute to oral microbial colonization by metabolizing dietary starch and providing nutrition for non-ABS species within the dental plaque.
S. gordonii binds salivary amylase to its surface with high efficiency and specificity via the surface-expressed 20-kDa amylase-binding protein A (AbpA), which is maximally expressed during the mid-log phase of bacterial growth (4, 10, 11). Previous in vitro studies demonstrated that amylase promotes the adhesion of ABS to surfaces and plays a role in biofilm formation (8). Studies using a rat model, however, showed that the ability to bind amylase did not correlate with colonization of the oral cavity (12). Thus, the amylase-streptococcus interaction may function in ways other than promoting nutrition, adhesion, or biofilm formation.
Recent microarray analysis showed that 33 genes of S. gordonii grown in chemically defined medium containing 0.8% glucose were differentially expressed after exposure to purified salivary amylase and that mutation of abpA eliminated the amylase-dependent gene response (13). In another study, the expression of both the abpA gene and its cognate protein were significantly increased after incubation in defined medium containing 0.2% glucose supplemented with starch and amylase (14). Based on these experiments we now postulate that AbpA may directly or indirectly participate in a signaling pathway that enables AbpA-modulated gene expression in response to amylase.
The RNA-sequencing (RNA-Seq) method permits examination of differential gene expression with greater sensitivity and less technical variability than microarrays and results in a deeper, more accurate assessment of transcriptomes (15, 16). It has now been widely used for the global transcriptome analysis of many microorganisms (17–19). Thus, the goals of the present study were to use RNA-Seq to (i) determine the effect of abpA deletion on gene expression of S. gordonii when grown in different media, (ii) determine the transcriptional changes of S. gordonii after binding to human salivary amylase in different growth media, and (iii) determine the effect of amylase and starch on the transcriptional changes in S. gordonii. The results of this study validate the concept that S. gordonii “senses” the oral environment, in this case amylase and starch, resulting in specific changes in bacterial gene expression that may affect the fitness of the bacteria to survive in the oral cavity.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
A kanamycin-resistant derivative of S. gordonii strain CH1 (20), designated KS1, that carries a chromosomal aphIII gene replacing a phage integrase gene (SGO_2076) was constructed previously for use in animal studies (21). An AbpA-deficient derivative of KS1 (KS1ΩabpA) was constructed by inserting the tet(M) gene, encoding tetracycline resistance (EMBL accession no. X56353) into the snaB1 site of the chromosomal abpA gene (21).
All bacterial strains were cultured from frozen stock to plates containing tryptic soy broth supplemented with 0.5% yeast extract and 1.5% Bacto agar (TSBY; Becton Dickinson, Sparks, MD) and grown for 48 h at 37°C in a candle jar. Strains were initially isolated on plates containing kanamycin at 750 μg/ml. For routine experiments, bacteria were cultured in TSBY medium or chemically defined medium (DM) with glucose, as indicated, without antibiotic supplementation (22). All bacterial strains used in the present study are listed in Table S1 in the supplemental material.
Construction of complemented strain.
To complement KS1ΩabpA, abpA and its promoter were first amplified by PCR using the primers Hind3C′abpAF and Hind3C′abpAR containing engineered HindIII sites (see Table S2 in the supplemental material). The PCR product was cloned into HindIII-digested plasmid pVA749 (23) and transformed into wild-type S. gordonii CH1 cells made competent, as previously described (24, 25). Transformants were selected by plating the competent cells on Todd-Hewitt agar supplemented with 5 μg of erythromycin/ml and confirmed by extracting the plasmid (26), digesting it with HindIII, and then analyzing it on an ethidium bromide-stained agarose gel. After confirming the fidelity of the insertion in pFS001 by nucleotide sequencing using plasmid-specific pVA749-F and pVA749-R primers (see Table S2 in the supplemental material), plasmid pFS001 was transformed into competent AbpA-deficient S. gordonii KS1ΩabpA cells. Transformants carrying plasmid pFS001 were selected by extraction of the plasmid and confirmed by running the HindIII-digested plasmids on an ethidium bromide-stained agarose gel.
Purification of human salivary α-amylase.
Human parotid saliva was collected from several healthy donors, as previously described, with slight modifications (27). The University at Buffalo Human Subjects Institutional Review Board approved the saliva collection protocol. Saliva was clarified by centrifugation at 12,800 × g for 10 min. The supernatant was extensively dialyzed against distilled water, lyophilized, resuspended in chromatography buffer, and subjected to BioGel P60 gel filtration chromatography, as previously described (6). The peaks corresponding to nonglycosylated amylase were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and an amylase ligand-binding assay using anti-amylase antibody, as described previously (4). Selected peaks corresponding to nonglycosylated amylase were pooled, dialyzed, lyophilized, and stored at −20°C. For RNA sequencing (RNA-Seq) experiments requiring amylase-treated samples, nonglycosylated amylase was resuspended in either simulated salivary buffer (21 mM sodium phosphate buffer, 36 mM NaCl, 0.96 mM CaCl2) (27) or DM, as described below, and was used at 0.4 mg/ml, corresponding to its salivary concentration (28).
RNA-Seq experiments. (i) RNA-Seq1: transcriptional profiling of TSBY-grown S. gordonii exposed to salivary amylase.
S. gordonii KS1 and KS1ΩabpA were cultured statically in 30 ml of TSBY broth at 37°C in a candle jar to mid-log phase corresponding to an optical density at 600 nm (OD600) of 0.5 to 0.7. Each mid-log-phase bacterial culture was then divided into three aliquots of equal volume. Experiments were conducted in parallel with cells from KS1 and KS1ΩabpA. Bacterial cells from all aliquots were pelleted by centrifugation at 6,000 × g in a Sorvall RC6 centrifuge at 18°C for 5 min. One aliquot of each culture (KS1 and KS1ΩabpA) was directly used for RNA isolation. The other aliquots were treated with either amylase or denatured amylase. The pellets were washed once with simulated salivary buffer prewarmed to 37°C. Simulated salivary buffer (2 ml) containing 0.4 mg of purified, nonglycosylated salivary amylase/ml and prewarmed to 37°C was added to the cells of one aliquot. As a negative control, simulated salivary buffer containing 0.4 mg of denatured salivary amylase/ml, following heating to 100°C for 15 min and cooling to 37°C just prior to use, was added to the cells of another aliquot. Each aliquot was incubated statically for 15 min at 37°C in a candle jar. Three independent experiments were conducted on different days.
Total RNA was immediately isolated using the FastRNA Blue Kit (MP Biomedicals LLC, Solon, OH) according to the manufacturer's instructions except that the cells were disrupted using a Mini-Bead Beater 8 (BioSpec Products, Bartlesville, OK) for three 1-min cycles. Total RNA was then DNase-treated with Turbo DNA-free (Ambion, Austin, TX) to remove any contaminating DNA. The remaining contaminants were removed by using a Qiagen RNeasy MiniElute cleanup kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Total RNA was quantified using the NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE), and RNA integrity was determined by 1% agarose gel electrophoresis stained with ethidium bromide. The total RNA was then treated with a Ribo-Zero rRNA removal kit (gram-positive bacteria; Epicentre, Madison, WI) to remove rRNA. The quantity and quality of the RNA was analyzed using the NanoDrop 2000 spectrophotometer and an Agilent 2100 electrophoresis bioanalyzer (Agilent Technologies, Santa Clara, CA). All purified RNA samples had a RNA integrity number of ≥8. An Illumina TruSeq RNA sample preparation kit was used to prepare cDNA libraries from rRNA-depleted RNA samples. Briefly, the mRNA was cleaved into fragments, the first strand was reverse transcribed to cDNA using SuperScript II reverse transcriptase and random primers (Invitrogen, Carlsbad, CA), followed by second-strand cDNA synthesis using the Second Strand master mix supplied with the kit. After end repair, the addition of a single ‘A’ base, and ligation with adapters, the products were enriched and purified with PCR to create the final cDNA library according to per manufacturer's protocol. The cDNA products were then sequenced using the Illumina HiSeq 2000 or HiSeq 2500 at the New York State Center for Excellence in Bioinformatics and Life Sciences, UB Genomics and Bioinformatics Core, Buffalo, NY.
(ii) RNA-Seq2: transcriptional profiling of DM–0.8% glucose-grown S. gordonii exposed to salivary amylase.
S. gordonii KS1 and KS1ΩabpA were cultured statically in 30 ml of DM containing 0.8% (wt/vol) glucose (DM–0.8% glucose) at 37°C in a candle jar to mid-log phase corresponding to an OD600 of 0.6 to 0.8. All of the other preparation procedures were the same as for RNA-Seq1.
(iii) RNA-Seq3: transcriptional response of DM–0.2% glucose-grown S. gordonii after exposure to amylase with or without starch.
S. gordonii KS1 and KS1ΩabpA were cultured statically in 40 ml of DM containing 0.2% (wt/vol) glucose (DM–0.2% glucose) at 37°C in a candle jar to mid-log phase corresponding to an OD600 of 0.5 to 0.6. Each mid-log-phase bacterial culture was then divided into four aliquots of equal volume. Bacterial cells from all aliquots were pelleted by centrifugation at 6,000 × g in a Sorvall RC6 centrifuge at 18°C for 5 min, and the supernatants were removed. Experiments were conducted in parallel with cells from KS1 and KS1ΩabpA. Pellets from one aliquot of each strain were resuspended in fresh DM–0.2% glucose (2 ml) alone (no amylase control), and pellets from the other three aliquots were resuspended in fresh DM–0.2% glucose (2 ml) supplemented with 0.4 mg of amylase/ml to allow amylase to bind to the cell surface. Each aliquot was incubated statically for 15 min at 37°C in a candle jar. All cell suspensions were then pelleted by centrifugation at 6,000 × g at 18°C for 5 min; the aliquots of bacteria treated with DM–0.2% glucose alone and one aliquot treated with DM–0.2% glucose plus amylase were used directly for RNA isolation. The two remaining aliquots treated with amylase were washed once with simulated salivary buffer. Cells from one aliquot were resuspended in fresh DM–0.2% glucose supplemented with 1% starch (0.05 g/ml) and, as a control, cells from the other aliquot were resuspended in fresh DM–0.2% glucose alone. These two aliquots were incubated statically for 15 min at 37°C in a candle jar. RNA was then isolated from cells of these two aliquots. The experiment was repeated on three different days. The other preparation procedures were the same as RNA-Seq1.
RNA-Seq data analysis.
Raw RNA-Seq reads for each sample were mapped to the reference genome from the National Center of Biotechnology Information (Streptococcus gordonii Challis CH1, RefSeq ID NC_009785, http://www.ncbi.nlm.nih.gov/genome) using TopHat v2.0.7 software (http://ccb.jhu.edu/software/tophat). The resulting alignment files were supplied to Cuffdiff v2.1.1 software (http://cole-trapnell-lab.github.io/cufflinks/), which calculates expression levels based on the input gene annotation file and tests the statistical significance of observed changes. Annotations were considered significant if the adjusted P value was <0.05 after Benjamini-Hochberg correction. Replicate samples were processed independently and then pooled for analysis of gene expression between tests and control samples for each growth condition tested.
cDNA synthesis.
DNase-treated total RNA obtained from each sample was also used immediately for cDNA synthesis. Before cDNA synthesis, PCR was performed using 5 μl of each total RNA sample (328 to 654 ng/μl) as the template and a primer pair (SGO_1174/1175-F and SGO_1174/1175-R; see Table S6 in the supplemental material) spanning the intergenic region between SGO_1174 and SGO_1175 to confirm the absence of contaminating genomic DNA. The total RNA was then reverse transcribed to cDNA using a modified protocol from the Pathogen Functional Genomics Resource Center (PFGRC) at the J. Craig Venter Institute (ftp://ftp.jcvi.org/pub/data/PFGRC/MAIN/pdf_files/protocols/M007.pdf). Briefly, 4 μg of total RNA was reverse transcribed using Superscript III (Invitrogen) reverse transcriptase in a deoxynucleotide mixture. After alkaline hydrolysis to remove the RNA template, cDNA was purified with QIAquick PCR columns (Qiagen). cDNA samples were stored at −80°C prior to use.
Quantitative real-time PCR (qRT-PCR).
qRT-PCR was performed to validate the transcriptional expression of several highly differentially expressed genes identified by RNA-Seq. The concentration of cDNA was determined using the NanoDrop 2000 spectrophotometer, and for qRT-PCR each sample template was standardized to 1 to 2 ng/μl dependent upon primer efficiency. Gene-specific primers were designed using primer BLAST from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov) and synthesized by Invitrogen. To ensure that the efficiency of the assay was between 90 and 110%, qRT-PCR was performed using serial dilutions of the cDNA template with each primer set. Each 25-μl reaction mixture contained 5 μl of template, 12.5 μl of Power SYBR green PCR master mix (Applied Biosystems, Foster City, CA), 2 μl each of the forward and reverse primers (160 nM), and 3.5 μl of ultrapure water. The qRT-PCR assay was performed on the ABI 7500 thermal cycler (Applied Biosystems), using uniform cycling conditions (95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C) for Power SYBR green (Applied Biosystems). Dissociation curve analysis was performed at the conclusion of the each run to verify the amplification of a single product. Assay controls for each run included (i) amplification with a primer set for gyrase A (gyrA) as the endogenous control and (ii) amplification with a reaction mixture without a template. All reactions were run in triplicate and were repeated with three independent biological replicates.
qRT-PCR statistical analysis.
The 2−ΔΔCT method was used to calculate the fold difference in gene expression between the AbpA mutant relative to wild-type KS1. The threshold cycle (CT) value was averaged over the technical replicates to compute the CT for each biological sample. For the RNA-Seq1 (TSBY) and RNA-Seq2 (DM–0.8% glucose) experiments, two sample t tests (P < 0.05) were used to compare the 2−ΔΔCT values for the target genes SGO_1174-1179 and srtB between the AbpA mutant using wild-type KS1 as the calibrator. The RNA-Seq3 (DM–0.2% glucose and amylase with or without salivary amylase) experiment used paired t tests to compare 2−ΔΔCT values for target genes SGO_0100 to SGO_0104 between the growth conditions as that experiment began with each biological sample divided in half for each growth condition. The R programming language was used to perform all calculations.
Reverse transcription-PCR (RT-PCR).
To determine whether genes SGO_1173 to SGO_1180 and genes SGO_2103 to SGO_2106 were cotranscribed as polycistronic messages, RT-PCR was performed with cDNA obtained from KS1 (RNA-Seq2 in DM–0.8% glucose) and primers designed to amplify regions spanning the adjacent gene junctions. The primers were designed using primer BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and synthesized by Invitrogen. In a 25-μl PCR, 6 μl of cDNA standardized to 2 ng/μl was used as the template along with 250 nM each primer. The following PCR program was used: preincubation at 95°C for 3 min; followed by denaturing at 95°C for 30 s, annealing at 55°C for 1 min, and extension at 72°C for 1 min for 30 cycles; followed in turn by a 5-min incubation at 72°C. PCR negative (no template) and positive controls containing 200 ng of DNA from S. gordonii KS1, prepared as previously described (29), were included in each experiment.
Effect of hydrogen peroxide on growth. (i) Determination of MIC.
Bacterial strains were cultured from freezer stocks onto TSBY agar plates. Broth cultures of KS1 and KS1ΩabpA were prepared in TSBY supplemented as appropriate with 750 μg of kanamycin/ml and 10 μg of tetracycline/ml and grown statically overnight at 37°C in a candle jar. Overnight cultures of S. gordonii KS1 and KS1ΩabpA were diluted in TSBY medium without antibiotics to a starting OD600 of 0.01. Experiments to determine strain sensitivity to H2O2 were performed as previously described (30). In a 96-well microplate, 48 wells were filled with 100 μl of standardized culture of KS1 or KS1ΩabpA. For each strain, six replicate wells were treated with 100 μl of different concentrations of H2O2 in TSBY (0, 1.25, 2.5, 5, 10, 20, 40, or 80 mM). Hence, the final H2O2 concentrations were 0, 0.625, 1.25, 2.5, 5, 10, 20, or 40 mM. Bacterium-free medium controls (200 μl) for each H2O2 concentration were included. The microplates were covered by sterile Nunc sealing tape (Thermo Scientific) and incubated at 37°C for 17 h in a candle jar. The OD600 of each well was determined on a microplate reader (Beckman Coulter AD340). To determine the growth in each well, the mean OD600 of the bacterium-free medium control was subtracted from the OD600 of each well at the same concentration of H2O2. The MIC was determined as the lowest concentration of H2O2 that inhibited visible growth.
(ii) Effect on cell viability.
Broth cultures of KS1 and KS1ΩabpA were prepared in TSBY supplemented with appropriate antibiotics and grown statically overnight at 37°C in a candle jar. Overnight cultures were diluted in DM–0.8% glucose without antibiotics to a starting OD600 of 0.1. When the cells reached mid-log phase (OD600 of 0.5 to 0.6), 1 ml of each culture was added to six microcentrifuge tubes and centrifuged at 8,000 × g for 5 min. After the supernatant was discarded, the pellets were resuspended in DM–0.8% glucose with different concentrations of H2O2 (0, 1.25, and 2.5 mM). Two tubes with a given H2O2 concentration were prepared for each strain. One set of tubes was incubated statically at room temperature for exactly 5 min, and the other set of tubes was incubated for exactly 30 min. At the end of the incubation, treatment was terminated by centrifugation for 5 min at 8,000 × g and resuspension of the cell pellets in phosphate-buffered saline (pH 7.5). The drop plate method (31) was used to determine cell viability. The CFU were counted after overnight growth on TSBY agar at 37°C in a candle jar. The relative viability at each H2O2 concentration at a given incubation time was calculated as (CFU with peroxide/CFU without peroxide) × 100. The statistical significance was determined by using a Student t test (P < 0.05).
RNA-Seq data accession number.
RNA-Seq data were deposited in the Sequence Read Archive database at NCBI and are accessible through BioProject number PRJNA199999.
RESULTS AND DISCUSSION
S. gordonii, like all bacteria, adapts to different environmental conditions by altering gene expression to optimize its survival. The ability of S. gordonii to bind salivary α-amylase through AbpA has been shown to play a role in this gene response. Recent mass spectrometric analysis of the whole secretome and amylase-precipitated secretome proteins from S. gordonii grown in TSBY showed the secretion of AbpA, sortase B, and other potential signal transduction proteins (32). To further define the role of AbpA in gene regulation, RNA-Seq was used to compare the global transcriptional responses of S. gordonii KS1 and the AbpA− mutant KS1ΩabpA. In light of the fact that AbpA is subject to catabolite repression (33), bacterial cells were grown not only in DM–0.8% glucose and TSBY to further extend previous findings but also in DM–0.2% glucose to better simulate the nutrient-limited environment of the oral cavity and to minimize the effect of glucose on abpA expression. Gene expression was also compared to the addition of amylase, the predominant protein in saliva. Starch, the primary substrate of amylase and a potential nutrient source, was also added to the DM–0.2% glucose experiments in an attempt to mimic the exposure of bacteria to a dietary starch challenge. As described below, the choice of in vitro growth medium had a pronounced effect on global gene expression in S. gordonii.
Transcriptional response in complex versus minimal culture media. (i) TSBY.
TSBY is a complex growth medium containing 0.25% glucose that provides bacteria with a rich mix of nutrients. When gene transcription of KS1ΩabpA was compared to KS1 as the control following growth in this medium, relatively few genes were differentially expressed (Tables 1 and 2). These data suggest that when the nutritional needs of the bacteria are met, in the absence of extracellular amylase, AbpA affects transcription of only a small number of genes.
TABLE 1.
RNA-Seq: effect of growth medium on the number of differentially expressed genes
| Growth medium and gene status | Condition and no. of genesa |
|||
|---|---|---|---|---|
| WT vs AbpA− mutant |
Amylase vs control |
|||
| KS1ΩabpA vs KS1 | KS1ΩabpA vs KS1 | KS1 | KS1ΩabpA | |
| TSBY | No amylase | Amylase | Denatured amylase | Denatured amylase |
| Genes upregulated | 0 | 1 | NS | NS |
| Genes downregulated | 12 | 9 | NS | NS |
| DM–0.8% glucose | No amylase | Amylase | Denatured amylase | Denatured amylase |
| Genes upregulated | 97 | 34 | 54 | NS |
| Genes downregulated | 69 | 39 | 51 | NS |
| DM–0.2% glucose | No amylase | Amylase | Buffer | Buffer |
| Genes upregulated | ND | 3 | 13 | 2 |
| Genes downregulated | ND | 11 | 4 | 3 |
For KS1ΩabpA-versus-KS1 values, the control was the KS1 parental strain. For the amylase-versus-control values, the control was heat-denatured amylase (100°C for 15 min) diluted in simulated salivary buffer when using denatured amylase or simulated salivary buffer (21 mM sodium phosphate buffer, 36 mM NaCl, 0.96 mM CaCl2) when using buffer. NS, not significant after Benjamini-Hochberg correction (P > 0.05); ND, analysis not done.
TABLE 2.
RNA-Seq1: genes downregulated in KS1ΩabpA mutant compared to KS1 grown in TSBY medium
| Category and NCBI locus | Protein encoded | Fold change |
|---|---|---|
| Signal transduction: two-component systems | ||
| SGO_1174 | Two-component system histidine kinase | −6.02 |
| SGO_1175 | Response regulator | −6.5 |
| Protein modification and repair | ||
| SGO_1176 | Peptide methionine sulfoxide reductase-like protein | −9.51 |
| Energy metabolism: electron transport | ||
| SGO_1177 | Thioredoxin family protein | −9.38 |
| SGO_1179 | Cytochrome c-type biogenesis protein (CcdA1) | −11.79 |
| Cell envelope | ||
| SGO_1178 | Putative lipoprotein | −10.93 |
| SGO_0208 | Glycosyl hydrolase family LPXTG cell wall surface protein | −3.84 |
| SGO_0316 | Serine protease subtilase family LPXTG cell wall surface protein | −3.05 |
| SGO_0317 | Serine protease subtilase family LPXTG cell wall surface protein | −3.05 |
| Transport and binding proteins | ||
| SGO_0878 | ABC transporter ATP-binding protein | −3.56 |
| SGO_2105 | Amylase-binding protein (AbpA) | −12.38 |
| Protein and peptide secretion and trafficking | ||
| SGO_2104 | Sortase B (SrtB) | −24.59 |
(ii) DM–0.8% glucose.
In contrast, when the strains were grown in a minimal medium with higher glucose concentration (0.8%), several additional genes were differentially transcribed (Table 1 and see Table S3 in the supplemental material). Interestingly, abpB, which encodes amylase-binding protein B and functions as a dipeptidase, was upregulated in KS1ΩabpA. Four genes (SGO_0099 to SGO_0102) encoding proteins involved in maltodextrin utilization and transport were upregulated 5- to 7-fold, as well as 11 genes (SGO_0043, SGO_0045 to SGO_0049, and SGO_1890 to SGO_1893) belonging to phosphotransferase systems (PTS), which are major carbohydrates transport system. The gene cluster (SGO_1759 to SGO_1771) was also upregulated, which includes several glycosyl hydrolases and ABC sugar transporters. Overall, the genes predominantly upregulated under this condition are involved in energy metabolism (31.3%) and transport and binding (20.8%), as well as genes predominantly downregulated function in transport and binding (21.8%), and genes involved in the cell envelope (13.0%) and in purine, pyrimidine, nucleoside, and nucleotide biosynthesis (13%). Figure 1 depicts the functional categories of genes upregulated or downregulated in KS1ΩabpA relative to KS1 cultured in DM–0.8% glucose. These data suggest that in minimal medium with high glucose, even without amylase, AbpA may influence the induction of maltose utilization, carbohydrate transport systems, and energy production and may possess a signaling function.
FIG 1.

RNA-Seq2: categories of genes upregulated (A) or downregulated (B) in KS1ΩabpA mutant relative to KS1 grown in DM with 0.8% glucose.
(iii) abpA expression.
As expected, amylase-binding protein A (abpA, SGO_2105) grown in either TSBY or DM–0.8% glucose was downregulated 12.4- and 19.7-fold, respectively, in KS1ΩabpA compared to KS1 (see Table S3 in the supplemental material). However, sortase B (srtB, SGO_2104), which is located immediately downstream of abpA (Fig. 2A), was also significantly downregulated approximately 25- and 47-fold in TSBY and DM–0.8% glucose, respectively, in this strain. It has been suggested that SrtB plays a role in the attachment of AbpA to the cell wall of S. gordonii, although AbpA does not have a classic SrtB recognition domain (34). The present study found that regardless of the growth medium, deletion of abpA resulted in significant downregulation of srtB (see Table S3 in the supplemental material). In a previous Northern blot study, a biotinylated abpA probe bound predominantly to an ∼600-bp (abpA) band of S. gordonii RNA, as well as a lighter band of greater size (33), suggesting the possibility of cotranscription. In the present study, qRT-PCR studies confirmed that srtB expression in KS1ΩabpA was significantly downregulated −4.5892-fold (P < 0.00387) in TSBY relative to KS1, indicating that insertion of tet(M) possessing a weak terminator (35) inhibited the transcription of abpA through to srtB, resulting in an apparent polar effect. RT-PCR analysis of gene junctions using primers listed in Table S6 in the supplemental material supports the cotranscription of abpA and srtB as a polycistronic message (Fig. 2B). These results demonstrate that transcription of abpA and srtB is linked. It has been observed that surface proteins are often located in the same transcriptional unit with a sortase gene and that the two genes presumably comprise a enzyme-substrate pair (36). AbpA is expressed on the cell surface of S. gordonii (10), and SrtB likely functions to anchor it to the cell wall. We postulate that following cleavage of the N-terminal signal peptide AbpA acts as the substrate for sortase B, which binds to the LPXTG-like sorting motif located near the C terminus of AbpA. Sortase acting as a membrane-anchored transpeptidase enables AbpA to be transiently anchored to the cell wall, not unlike SvpA in Listeria monocytogenes (37). Studies are ongoing to determine the relationship of abpA to srtB in S. gordonii and how it affects the localization of AbpA and its subsequent functions.
FIG 2.

RT-PCR of adjacent genes for detecting polycistronic messages. (A) Schematic of gene cluster SGO_2103 to SGO_2106 with gene junction primers indicated by black arrows (not to scale). (B) Amplicons of SGO_2103 to SGO_2106 from RNA-Seq2 samples using gene junction primers: 1-kb DNA ladder, KS1 cDNA, AbpA− cDNA, and genomic (g) CH1 DNA (positive control). A robust amplicon was observed only at the SGO_2104-SGO_2105 gene junction in KS1 cDNA. (C) Schematic of redox gene cluster SGO_1173 to SGO_1180 with gene junction primers indicated by black arrows (not to scale). (D) One-kilobase DNA ladder, no-template control (NTC, negative control), KS1 cDNA, and CH1 genomic DNA (positive control). The cDNAs between the seven gene junctions from SGO_1173 to SGO_1180 were amplified using gene junction primers. Amplicons were found between all genes except for SGO_1175 and SGO_1176, suggesting two polycistronic messages.
(iv) Redox gene cluster.
In both TSBY and DM–0.8% glucose, a cluster of genes potentially involved in signal transduction and oxidative stress was differentially downregulated in the KS1ΩabpA compared to KS1. Two genes encoding a putative histidine kinase (SGO_1174) and response regulator (SGO_1175) comprise a potential two-component signal transduction system. Contiguous genes SGO_1176 (peptide methionine sulfoxide reductase-like protein), SGO_1177 (thioredoxin family protein), SGO_1178 (putative lipoprotein), and SGO_1179 (cytochrome c-type biogenesis protein) were also downregulated. Functional annotation of the genes in this cluster suggest their potential role in electron transport and adjustment to redox conditions and have thus been designated the “redox gene cluster” for descriptive purposes. qRT-PCR using primers listed in Table S3 in the supplemental material validated that SGO_1174 to SGO_1179 were significantly downregulated in both TSBY (Table 3) and DM–0.8% glucose (Table 4). In order to obtain preliminary evidence of a polycistronic message containing genes of this cluster (SGO_1173 to SGO_1180, Fig. 2C), transcript analysis was performed at each individual gene junction of these genes using primers listed in Table S6 in the supplemental material. Robust amplicons obtained from cDNA using the gene junction primers SGO_1173/1174 and SGO_1174/1175 suggest that the cytochrome c-type protein gene ccdA2 and the histidine kinase and response regulator genes are transcribed together (Fig. 2D). The SGO_1175/1176 region failed to produce an amplicon. Additional robust amplicons were obtained from SGO_1176/SGO_1177, SGO_1177/SGO_1178, and SGO_1178/SGO_1179, while a light band was obtained from SGO_1179/SGO_1180 (Fig. 2D). It appears the peptide methionine sulfoxide reductase, the thioredoxin family protein, the putative lipoprotein, and the cytochrome c-type protein ccdA1 genes are cotranscribed. However, it is unclear whether the histidine kinase (SGO_1180) gene is part of this cluster since it was only faintly detectable by PCR, is only differentially expressed under one condition and is upregulated, whereas the contiguous cluster is downregulated. In each assay where a cDNA amplicon was obtained, PCR using the RNA template was negative (data not shown), confirming the lack of genomic DNA contamination of the RNA samples. These results support the polycistronic nature of at least two groups of genes within this redox gene cluster. Further experiments will need to be conducted to verify the role of AbpA gene regulation within this region.
TABLE 3.
qRT-PCR results for the redox gene cluster from the TSBY experiment
| Gene locus | RQa | Log2 (ratio) | P |
|---|---|---|---|
| SGO_1174 | 0.0621 | −4.0119 | 0.006 |
| SGO_1175 | 0.0455 | −4.4591 | 0.0101 |
| SGO_1176 | 0.0257 | −5.2814 | 0.0038 |
| SGO_1177 | 0.0203 | −5.6254 | 0.003 |
| SGO_1178 | 0.0128 | −6.2857 | 0.0006 |
| SGO_1179 | 0.0166 | −5.9097 | 0.0003 |
RQ, relative quantification of the gene in the AbpA-deficient mutant relative to KS1 from three independent assays.
TABLE 4.
qRT-PCR results for the redox gene cluster from the DM–0.8% glucose experiment
| Gene locus | RQa | Log2 (ratio) | P |
|---|---|---|---|
| SGO_1174 | 0.0532 | −4.232 | 0.001 |
| SGO_1175 | 0.0488 | −4.3569 | 0.021 |
| SGO_1176 | 0.0349 | −4.8393 | 0.012 |
| SGO_1177 | 0.0362 | −4.7893 | 0.026 |
| SGO_1178 | 0.0306 | −5.0289 | 0.042 |
| SGO_1179 | 0.0273 | −5.1942 | 0.032 |
RQ, relative quantification of the gene in the AbpA-deficient mutant relative to KS1 from three independent assays.
Transcriptome response to amylase in different culture media. (i) TSBY with amylase.
The potential for host salivary amylase to serve as an environmental signal for signal transduction was examined. When nonglycosylated α-amylase (0.4 mg/ml) was added to the complex culture medium TSBY, there was little difference in gene expression between KS1ΩabpA and KS1, except for abpA, srtB, and the redox gene cluster (Table 1; see also Table S3 in the supplemental material). There was also no significant difference in gene expression in either KS1 or KS1ΩabpA when the addition of amylase was compared to denatured amylase (control) (Table 1; see also Table S4 in the supplemental material), suggesting that amylase activity is not required and that the observed effects are due to the lack of AbpA.
(ii) DM–0.8% glucose with amylase.
In contrast, when mid-log-phase bacteria grown in DM–0.8% glucose were incubated for 15 min with amylase, many more genes were differentially up- and downregulated in KS1ΩabpA compared to KS1 (Table 1; see also Table S2 in the supplemental material). Notably, genes involved in maltodextrin utilization and transport were no longer differentially upregulated since they were in this medium without amylase. As shown in Fig. 3A, genes involved in transport (27.8%) and energy metabolism (13%) were again the primary upregulated functions in the AbpA− mutant. In addition, genes involved in the cell envelope (11.1%) and fatty acid and phospholipid metabolism (7.4%) were also upregulated. Other genes involved in the cell envelope (20%), energy metabolism (18%), transport and binding (16%), and amino acid biosynthesis (10%) were functional categories most downregulated (Fig. 3B).
FIG 3.

RNA-Seq2: categories of genes upregulated (A) or downregulated (B) in KS1 grown in DM–0.8% glucose treated with amylase.
The genes within the redox cluster in KS1ΩabpA were also downregulated 20.5- to 42.2-fold after exposure to amylase. In fact, genes in this cluster were the most differentially expressed under these conditions compared to any of the other conditions tested (see Table S3 in the supplemental material). These conditions also triggered a 4.5-fold downregulation of methionine sulfoxide reductase, msrA (SGO_0278), as well as a ∼2-fold upregulation of oxidoreductases (SGO_0841 and SGO_1007), further emphasizing the potential role of AbpA in adaptation to oxidative stress.
(iii) Fatty acid synthesis genes.
The expression of several genes in KS1 was altered in the presence of amylase versus denatured amylase in DM–0.8% glucose (see Table S4 in the supplemental material). Importantly, genes involved in the initiation of fatty acid synthesis and product elongation, including SGO_1694 (fabD), SGO_1695, SGO_1698 (fabH), SGO_1699 (fabT), and SGO_1700, encoding enoyl-coenzyme A hydratase, which has a role of in the beta-oxidation pathway of fatty acid metabolism, were upregulated in the presence of amylase. These findings are consistent with our previous microarray analysis of KS1 grown under the same conditions (DM–0.8% glucose; see Table S4 in the supplemental material) (13). Here, RNA-Seq analysis revealed the upregulation of additional fatty acid synthesis genes within the locus in the presence of amylase relative to denatured amylase (control). However, in the AbpA− mutant (KS1ΩabpA) the level of expression of these genes was not significantly different with the addition of amylase or denatured amylase. This validates previous qRT-PCR studies of fatty acid synthesis gene expression in KS1ΩabpA (13). Thus, in this growth medium in the presence of amylase, the expression of these genes does not appear to be significantly altered in S. gordonii without AbpA. However, when AbpA is present in KS1, a variety of genes are differentially expressed upon exposure to amylase, particularly those involved in fatty acid synthesis (see Table S4 in the supplemental material).
However, when KS1ΩabpA was compared to KS1 (control), there was no differential expression of these fatty acid genes (see Tables S3 and S7 in the supplemental material) with the addition of amylase. The greatest differential expression was found between KS1 with amylase compared to exposure to denatured amylase. These observations indicate that in the presence of amylase, fatty acid synthesis gene transcription can proceed at nearly the same level with or without AbpA. However, if AbpA is present without amylase (or with denatured amylase), expression of these genes is downregulated (see Table S7 in the supplemental material). Interestingly, fatty acid synthesis genes were not differentially expressed in KS1 with addition of amylase in any of the other growth medium tested here with the exception of upregulation of 3-oxyacyl-ACP synthase (SGO_1698) in the AbpA− mutant in DM–0.2% glucose with amylase and starch. Fatty acids and phospholipids are key components of bacterial membranes. Previously, we found that increased synthesis of fatty acids associated with the binding of amylase to AbpA did not alter the proportion of fatty acids in membranes, but increased cell growth, survival at low pH, and resistance to triclosan (13). Taken together, these data suggest that AbpA can affect the expression of many genes, especially fatty acid synthesis genes, but these changes appear to be growth medium specific.
(iv) DM–0.2% glucose with amylase.
AbpA is subject to glucose catabolite repression. High glucose is known to bind to the catabolite repressible element (cre) located 153 bp downstream of the translational start site of abpA to downregulate transcription (33). In RNA-Seq3, a glucose concentration of 0.2% in DM was used to minimize the inhibitory effect of glucose on abpA expression. Exposure of KS1ΩabpA and KS1 grown in DM–0.2% glucose to amylase alone resulted in minimal differential gene expression apart from abpA, srtB, and the redox gene cluster (Table 1; see also Table S4 in the supplemental material). Thus, in nutritionally limited medium with minimal glucose, the addition of amylase does not affect many changes in gene expression, not unlike the results in nutrient rich TSBY. Overall, host amylase itself does not appear to trigger numerous changes in gene expression in S. gordonii strains, which are predominantly growth medium dependent.
Transcriptome response to starch in minimal medium. (i) DM–0.2% glucose with amylase and starch.
In order to determine the role of host salivary amylase together with starch as a potential nutrient source, transcriptional changes of S. gordonii were monitored after a brief exposure to salivary amylase, followed by washing and then incubation with 1% starch. Relatively few genes were up- or downregulated in either KS1 or KS1ΩabpA incubated with amylase and starch (Table 5; see also Table S4 in the supplemental material) compared to cells treated with amylase and salivary buffer. The greatest differential expression of genes occurred when KS1ΩabpA was compared to KS1 when cells were cultured in DM–0.2% glucose in the presence of both amylase and starch (Table 5; see also Tables S3 and S5 in the supplemental material). The predominant functional categories of the 110 upregulated genes were purine, pyrimidine, nucleoside, and nucleotide synthesis (21.8%), transport and binding (17.3%), hypothetical proteins (15.5%), and energy metabolism (12.7%); the predominant downregulated genes were involved in transport and binding (25.5%), hypothetical proteins (26.5%), and energy metabolism (12.5%). The redox gene cluster (SGO_1173 to SGO_1180), as well as peptide methionine sulfoxide reductase (msrA and msrB, SGO_0278), also associated with redox regulation, was similarly downregulated.
TABLE 5.
RNA-Seq: effect of DM–0.2% glucose with or without starch on the number of differentially expressed genes
| Gene status | No. of genesa |
||
|---|---|---|---|
| Starch |
Starch vs buffer |
||
| KS1ΩabpA vs KS1 | KS1 | KS1ΩabpA | |
| Genes upregulated | 110 | 9 | 19 |
| Genes downregulated | 136 | 2 | 0 |
The growth medium was DM–0.2% glucose for all of these experiments. Amylase was used for all of these experiments.
(ii) abpA expression.
A recent study found that the expression of abpA in KS1 was significantly increased after 40 min of incubation in DM–0.2% glucose supplemented with starch and amylase (14). However, the RNA-Seq study did not find abpA to be upregulated in S. gordonii after 15 min of incubation with amylase, followed by washing the cells, and followed by incubation with starch for 15 min. As concluded in Nikitkova et al. (14), the expression of AbpA in culture is time dependent. Transcription of abpA in vitro may also be dependent on the time of exposure to amylase, as well as to starch.
(iii) Maltose utilization.
With the production of maltose and maltodextrins in this medium from the hydrolysis of starch by amylase, several genes from the maltodextrin utilization pathway were upregulated. Although only pulA2 (SGO_0099), malD (SGO_0102), malQ (SGO_0105), and glgP2 (SGK_0106) were upregulated in KS1 comparing starch with buffer, the entire gene cluster, including malR (SGO_0100) involved in maltose operon regulation, malE (SGO_0101) involved in maltose utilization, and Mal-like ABC transporters malC (SGO_0103), malD (SGO_0102), and malE (SGO_0104) were upregulated and to a greater degree in the AbpA− mutant (see Table S4 in the supplemental material). These findings were confirmed by qRT-PCR (Table 6) using the primers listed in Table S2 in the supplemental material. Thus, in low-glucose minimal medium, starch triggers S. gordonii cells primed by exposure to amylase, with or without AbpA, to upregulate maltose-associated genes. Similarly, the malQ and glgP genes of Streptococcus mutans, which lacks ABPs, participate in the metabolism of starch degradation products induced by salivary amylase (38). However, differential expression of the maltose utilization and transport genes between KS1ΩabpA and KS1 was even more pronounced in DM–0.8% glucose without amylase and starch, suggesting that this alteration in maltose-utilization genes has more to do with the amount of carbohydrate in the environment, regardless of the presence of amylase (see Table S3 in the supplemental material). Overall, AbpA may be involved in modulating maltose uptake and utilization.
TABLE 6.
qRT-PCR analysis of the maltodextrin-associated gene cluster from DM–0.2% with amylase in the buffer-versus-starch experiment
| Gene locus | RQa | Log2 (ratio) | P |
|---|---|---|---|
| SGO_0100 | 2.6634 | 1.4133 | 0.0003 |
| SGO_0101 | 2.6666 | 1.415 | 0.0011 |
| SGO_0102 | 2.9261 | 1.549 | 0.0089 |
| SGO_0103 | 2.8531 | 1.5126 | 0.0407 |
| SGO_0104 | 2.3722 | 1.2462 | 0.0575 |
RQ, relative quantification of the gene in the AbpA-deficient mutant relative to KS1 from three independent assays.
Biological implications. (i) Oxidative stress.
Perhaps the most interesting new finding of the present study is the association of AbpA expression with genes coding for redox proteins, designated here the redox gene cluster. This gene cluster was significantly downregulated in the KS1ΩabpA mutant regardless of growth in complex or minimal medium and in the presence of amylase with or without starch. In each case, genes within this cluster were progressively downregulated with SGO_1179 decreased the greatest and SGO_1174 decreased the least. In the presence of AbpA, the proteins encoded by this gene cluster may provide protection from external reactive oxygen species such as H2O2, which is produced by other streptococci as well as S. gordonii. To test this hypothesis, when exponential-phase cells of KS1 and KS1ΩabpA grown in DM–0.8% glucose were treated with 1.25 or 2.5 mM H2O2 for 5 min, viability decreased for both strains (data not shown). However, by 30 min a clear differential reduction in viability was observed at the sublethal H2O2 (1.25 mM) concentration, where KS1 decreased ∼15% compared to 95% for KS1ΩabpA. Complementation of KS1ΩabpA with a low-copy-number plasmid expressing abpA restored resistance to the sublethal H2O2 concentration to wild-type levels (Fig. 4). Similar results were obtained in TSBY medium (data not shown). Thus, AbpA may provide some protection to oxidative stress produced by sublethal concentrations of H2O2, which is produced by many commensal streptococcal species within dental plaque biofilms. Further studies are required to determine the exact target of this gene cluster and how it relates to the expression of AbpA protein.
FIG 4.
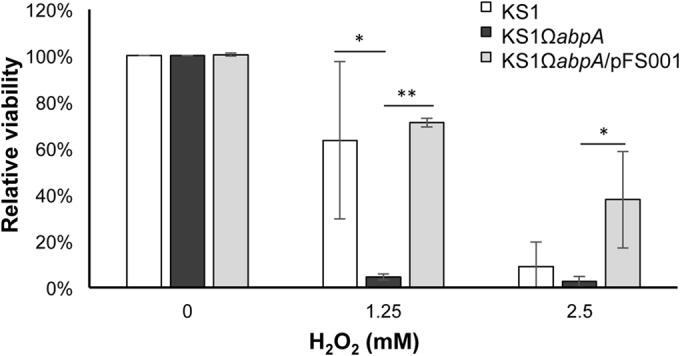
Effect of H2O2 treatment on exponential-phase KS1, KS1ΩabpA, and KS1ΩabpA/pFS001 cells after 30 min of treatment in DM–0.8% glucose.
(ii) PTS.
In the oral cavity, streptococci utilize dietary carbohydrates as an important source of energy. Bacteria have various carbohydrate-specific phosphotransferase systems (PTS) to make efficient use of the type of carbohydrate available in the environmental milieu. Here, we found that various PTS genes were differentially expressed in the various media tested. In DM–0.8% glucose, fructose/mannose-specific PTS genes (SGO_1889 to SGO_1893) and general PTS genes (SGO_0045 to SGO_0049), as well as a gene cluster (SGO_1759 to SGO_1771), purportedly involved in sugar transport and metabolism (glycosyl hydrolases) were upregulated in KS1ΩabpA. When amylase was added, many of the genes in this cluster (SGO_1759 to SGO_1771) were still significantly upregulated in KS1ΩabpA but to a lesser fold change. However, the nine PTS genes were no longer differentially expressed. Therefore, through the binding of host amylase (itself a glycosyl hydrolase) to the S. gordonii cell surface, AbpA may play a role in regulating other sugar transporters depending on specific sugar availability.
Bacterial PTSs are not only responsible for the binding, transmembrane transport, and phosphorylation of numerous sugar substrates but also play a substantial role in the regulation of carbon metabolism (39). Some PTS systems of S. gordonii may be involved in other physiological processes, such as the synthesis of cell surface glycoproteins and/or polysaccharides involved in adhesion and biofilm formation (40), and this may explain why the AbpA-deficient mutant produced greater biofilm than the wild-type strain grown under static conditions in the presence of sucrose (41). Multiple PTS systems have also been shown previously to be involved in signal transduction in Gram-positive bacteria (42). Therefore, it is possible that AbpA could interact with the PTS on the cell surface and initiate signaling through PTS.
(iii) Signal transduction.
Our previous microarray study found that a total of 33 genes of S. gordonii KS1 were differentially expressed after exposure to purified salivary amylase, and mutation of AbpA eliminated the amylase-dependent gene response of some genes (13). How can AbpA induce differential gene expression of S. gordonii? If AbpA is a cell wall-associated protein transiently localized at the cell surface, how does it convey a signal upon binding salivary amylase or other environmental stimuli? The results of the present study showed that abpA deletion downregulates a two-component signal transduction system (SGO_1174 and SGO_1175) within the redox gene cluster. Two-component signal transduction systems enable bacteria to sense, respond, and adapt to changes in their environment. Each two-component system consists of a membrane-bound sensor protein-membrane kinase (HK) and a cytosolic response regulator (RR). In the typical two-component pathway, the sensor HK autophosphorylates its own conserved His residue in response to a signal(s) in the environment. Subsequently, the phosphoryl group of HK is transferred onto a specific Asp residue on the RR. The activated RR can then effect changes in cellular physiology, often by regulating gene expression. Two-component pathways often enable cells to sense and respond to stimuli by inducing changes in transcription (43). Consequently, abpA may influence the expression of a two-component signal transduction system, which may provide a pathway for AbpA-modulated gene expression in response to amylase or other environmental signal. Yet, this signal transduction system is downregulated in the AbpA− mutant in all growth media studied, regardless of the presence of amylase. What then is the environmental signal? This question is open to future studies.
The only other gene downregulated in KS1ΩabpA under nearly all conditions tested, besides abpA and srtB, was SGO_0878 encoding an ATP-binding ABC transporter. Based on its functional domains, this protein may be involved in the export of lipoprotein to the cell membrane. The SGO_1178 in the redox cluster encodes a putative lipoprotein. Whether these genes are functionally related remains to be determined.
(iv) Adaptation to the environment.
Like other bacteria, S. gordonii adapts to different growth media by altering gene expression, which optimizes its survival potential in the ever-changing oral environment. AbpA appears to play a role in this response. The interplay of AbpA, amylase and carbohydrates is complex. The addition of salivary amylase to DM–0.8% glucose increased fatty acid synthesis and utilization genes. When both amylase and starch were added to DM–0.2% glucose, the generation of maltodextrins upregulated the maltose utilization operon. However, under all conditions tested, a histidine/response regulator within a redox gene cluster was upregulated when AbpA was expressed. Is AbpA a major component of this signal transduction system or does it have an ancillary role? What environmental signals, in addition to amylase, are being detected and what is the outcome? These questions are the current focus of investigation.
Summary.
We validated here the notion that oral commensal species such S. gordonii are able to sense host proteins and dietary components. We show that in complex medium AbpA affects the transcription of a small group of genes, whereas in minimal medium the transcription of a greater number of genes, especially those involved in maltose utilization, carbohydrate transport, and energy production, is affected. Host amylase itself does not appear to trigger extensive changes in gene expression in S. gordonii, which are by-and-large growth medium dependent. Amylase-binding protein A (abpA) and sortase B (srtB) genes are cotranscribed. The transcription of abpA in S. gordonii in vitro depends on the time of exposure to amylase, as well as starch. AbpA affects adaptation to oxidative stress in a medium-independent manner. AbpA affects the expression of genes involved in maltose utilization, carbohydrate transport, and fatty acid synthesis in a medium-dependent fashion. Finally, AbpA is a potential regulatory protein. Together, these findings inform our understanding of the complex networks that control the adaptation of the oral microbiome to the host.
Supplementary Material
ACKNOWLEDGMENTS
We thank Meg Vickerman for insightful discussions and Jonathan Bard at the UB Next Generation Sequencing and Expression Analysis core for assistance with the bioinformatic analysis of the RNA-Seq data.
Funding for this study was provided through grant R01DE022673 from the National Institute of Dental and Craniofacial Research.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01221-15.
REFERENCES
- 1.Scannapieco FA, Solomon L, Wadenya RO. 1994. Emergence in human dental plaque and host distribution of amylase-binding streptococci. J Dent Res 73:1627–1635. [DOI] [PubMed] [Google Scholar]
- 2.Robyt JF. 2008. Starch: structure, properties, chemistry, and enzymology, p 1437–1472. In Fraser-Ried BO, Tatsuta K, Thiem J, Cote GL (ed), Glycoscience. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 3.Douglas CW. 1983. The binding of human salivary alpha-amylase by oral strains of streptococcal bacteria. Arch Oral Biol 28:567–573. doi: 10.1016/0003-9969(83)90003-1. [DOI] [PubMed] [Google Scholar]
- 4.Douglas CW. 1990. Characterization of the alpha-amylase receptor of Streptococcus gordonii NCTC 7868. J Dent Res 69:1746–1752. doi: 10.1177/00220345900690110701. [DOI] [PubMed] [Google Scholar]
- 5.Kilian M, Nyvad B. 1990. Ability to bind salivary alpha-amylase discriminates certain viridans group streptococcal species. J Clin Microbiol 28:2576–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scannapieco FA, Bergey EJ, Reddy MS, Levine MJ. 1989. Characterization of salivary alpha-amylase binding to Streptococcus sanguis. Infect Immun 57:2853–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Douglas CW, Heath J, Gwynn JP. 1992. Enzymic activity of salivary amylase when bound to the surface of oral streptococci. FEMS Microbiol Lett 71:193–197. [DOI] [PubMed] [Google Scholar]
- 8.Rogers JD, Palmer RJ Jr, Kolenbrander PE, Scannapieco FA. 2001. Role of Streptococcus gordonii amylase-binding protein A in adhesion to hydroxyapatite, starch metabolism, and biofilm formation. Infect Immun 69:7046–7056. doi: 10.1128/IAI.69.11.7046-7056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scannapieco FA, Bhandary K, Ramasubbu N, Levine MJ. 1990. Structural relationship between the enzymatic and streptococcal binding sites of human salivary alpha-amylase. Biochem Biophys Res Commun 173:1109–1115. doi: 10.1016/S0006-291X(05)80900-3. [DOI] [PubMed] [Google Scholar]
- 10.Scannapieco FA, Haraszthy GG, Cho MI, Levine MJ. 1992. Characterization of an amylase-binding component of Streptococcus gordonii G9B. Infect Immun 60:4726–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Tanzer JM, Scannapieco FA. 2002. Identification and analysis of the amylase-binding protein B (AbpB) and gene (abpB) from Streptococcus gordonii. FEMS Microbiol Lett 212:151–157. doi: 10.1111/j.1574-6968.2002.tb11259.x. [DOI] [PubMed] [Google Scholar]
- 12.Tanzer JM, Baranowski LK, Rogers JD, Haase EM, Scannapieco FA. 2001. Oral colonization and cariogenicity of Streptococcus gordonii in specific pathogen-free TAN:SPFOM(OM)BR rats consuming starch or sucrose diets. Arch Oral Biol 46:323–333. doi: 10.1016/S0003-9969(00)00126-6. [DOI] [PubMed] [Google Scholar]
- 13.Nikitkova AE, Haase EM, Vickerman MM, Gill SR, Scannapieco FA. 2012. Response of fatty acid synthesis genes to the binding of human salivary amylase by Streptococcus gordonii. Appl Environ Microbiol 78:1865–1875. doi: 10.1128/AEM.07071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikitkova AE, Haase EM, Scannapieco FA. 2012. Effect of starch and amylase on the expression of amylase-binding protein A in Streptococcus gordonii. Mol Oral Microbiol 27:284–294. doi: 10.1111/j.2041-1014.2012.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. 2008. RNA-Seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Vliet AH. 2010. Next generation sequencing of microbial transcriptomes: challenges and opportunities. FEMS Microbiol Lett 302:1–7. doi: 10.1111/j.1574-6968.2009.01767.x. [DOI] [PubMed] [Google Scholar]
- 17.Cattoir V, Narasimhan G, Skurnik D, Aschard H, Roux D, Ramphal R, Jyot J, Lory S. 2013. Transcriptional response of mucoid Pseudomonas aeruginosa to human respiratory mucus. mBio 3:e00410-12. doi: 10.1128/mBio.00410-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandlik A, Livny J, Robins WP, Ritchie JM, Mekalanos JJ, Waldor MK. 2011. RNA-Seq-based monitoring of infection-linked changes in Vibrio cholerae gene expression. Cell Host Microbe 10:165–174. doi: 10.1016/j.chom.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scherr TD, Roux CM, Hanke ML, Angle A, Dunman PM, Kielian T. 2013. Global transcriptome analysis of Staphylococcus aureus biofilms in response to innate immune cells. Infect Immun 81:4363–4376. doi: 10.1128/IAI.00819-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tardif G, Sulavik MC, Jones GW, Clewell DB. 1989. Spontaneous switching of the sucrose-promoted colony phenotype in Streptococcus sanguis. Infect Immun 57:3945–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanzer JM, Thompson A, Sharma K, Vickerman MM, Haase EM, Scannapieco FA. 2012. Streptococcus mutans out-competes Streptococcus gordonii in vivo. J Dent Res 91:513–519. doi: 10.1177/0022034512442894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loo CY, Corliss DA, Ganeshkumar N. 2000. Streptococcus gordonii biofilm formation: identification of genes that code for biofilm phenotypes. J Bacteriol 182:1374–1382. doi: 10.1128/JB.182.5.1374-1382.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macrina FL, Tobian JA, Jones KR, Evans RP. 1982. Molecular cloning in the streptococci, p 195–210. In Hollander A, DeMoss R, Kaplan S, Konisky J, Savage D, Wolfe R (ed), Genetic engineering of microorganisms for chemicals. Plenum Publishing Corp, New York, NY. [Google Scholar]
- 24.Lawson JW, Gooder H. 1970. Growth and development of competence in the group H streptococci. J Bacteriol 102:820–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sulavik MC, Tardif G, Clewell DB. 1992. Identification of a gene, rgg, which regulates expression of glucosyltransferase and influences the Spp phenotype of Streptococcus gordonii Challis. J Bacteriol 174:3577–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehrenfeld EE, Clewell DB. 1987. Transfer functions of the Streptococcus faecalis plasmid pAD1: organization of plasmid DNA encoding response to sex pheromone. J Bacteriol 169:3473–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradway SD, Bergey EJ, Jones PC, Levine MJ. 1989. Oral mucosal pellicle: adsorption and transpeptidation of salivary components to buccal epithelial cells. Biochem J 261:887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobsen N, Melvaer KL, Hensten-Pettersen A. 1972. Some properties of salivary amylase: a survey of the literature and some observations. J Dent Res 51:381–388. doi: 10.1177/00220345720510022501. [DOI] [PubMed] [Google Scholar]
- 29.Chaudhuri B, Paju S, Haase EM, Vickerman MM, Tanzer JM, Scannapieco FA. 2008. Amylase-binding protein B of Streptococcus gordonii is an extracellular dipeptidyl-peptidase. Infect Immun 76:4530–4537. doi: 10.1128/IAI.00186-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrews JM. 2001. Determination of minimum inhibitory concentrations. J Antimicrob Chemother 48(Suppl 1):S5–S16. [DOI] [PubMed] [Google Scholar]
- 31.Chen CY, Nace GW, Irwin PL. 2003. A 6×6 drop plate method for simultaneous colony counting and MPN enumeration of Campylobacter jejuni, Listeria monocytogenes, and Escherichia coli. J Microbiol Methods 55:475–479. doi: 10.1016/S0167-7012(03)00194-5. [DOI] [PubMed] [Google Scholar]
- 32.Maddi A, Haase EM, Scannapieco FA. 2014. Mass spectrometic analysis of whole secretome and amylase-precipitated secretome proteins from Streptococcus gordonii. J Proteomics Bioinform 7:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogers JD, Scannapieco FA. 2001. RegG, a CcpA homolog, participates in regulation of amylase-binding protein A gene (abpA) expression in Streptococcus gordonii. J Bacteriol 183:3521–3525. doi: 10.1128/JB.183.11.3521-3525.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang X, Chen Y, Wu H. 2011. Sortase B assemble amylase-binding protein A to streptococcal cell surface, p 72 89th Gen Session Int Assoc Dent Res IADR, San Diego, CA. [Google Scholar]
- 35.Su YA, He P, Clewell DB. 1992. Characterization of the tet(M) determinant of Tn916: evidence for regulation by transcription attenuation. Antimicrob Agents Chemother 36:769–778. doi: 10.1128/AAC.36.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pallen MJ, Lam AC, Antonio M, Dunbar K. 2001. An embarrassment of sortases: a richness of substrates? Trends Microbiol 9:97–102. doi: 10.1016/S0966-842X(01)01956-4. [DOI] [PubMed] [Google Scholar]
- 37.Ton-That H, Marraffini LA, Schneewind O. 2004. Protein sorting to the cell wall envelope of Gram-positive bacteria. Biochim Biophys Acta 1694:269–278. doi: 10.1016/j.bbamcr.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 38.Sato Y, Okamoto-Shibayama K, Azuma T. 2013. The malQ gene is essential for starch metabolism in Streptococcus mutans. J Oral Microbiol doi: 10.3402/jom.v5i0.21285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kotrba P, Inui M, Yukawa H. 2001. Bacterial phosphotransferase system (PTS) in carbohydrate uptake and control of carbon metabolism. J Biosci Bioeng 92:502–517. doi: 10.1016/S1389-1723(01)80308-X. [DOI] [PubMed] [Google Scholar]
- 40.Kilic AO, Tao L, Zhang Y, Lei Y, Khammanivong A, Herzberg MC. 2004. Involvement of Streptococcus gordonii beta-glucoside metabolism systems in adhesion, biofilm formation, and in vivo gene expression. J Bacteriol 186:4246–4253. doi: 10.1128/JB.186.13.4246-4253.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaudhuri B, Rojek J, Vickerman MM, Tanzer JM, Scannapieco FA. 2007. Interaction of salivary alpha-amylase and amylase-binding-protein A (AbpA) of Streptococcus gordonii with glucosyltransferase of S. gordonii and Streptococcus mutans. BMC Microbiol 7:60. doi: 10.1186/1471-2180-7-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lengeler JW, Jahreis K. 2009. Bacterial PEP-dependent carbohydrate: phosphotransferase systems couple sensing and global control mechanisms. Contrib Microbiol 16:65–87. doi: 10.1159/000219373. [DOI] [PubMed] [Google Scholar]
- 43.Stock AM, Robinson VL, Goudreau PN. 2000. Two-component signal transduction. Annu Rev Biochem 69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.