

Abstract
Skeletal muscle harbors several types of cells, among which a population of progenitors committed to the adipogenic lineage has only recently been identified. Potential sources of white and brown adipocytes, the latter representing a potential target to treat obesity, are of considerable interest to the field. Fluorescence-activated cell sorting (FACS) provides an elegant strategy for prospective isolation of closely defined cell populations. Here we describe a flow cytometric method to isolate muscle-resident adipogenic progenitor cells with a default potential to undergo white adipogenesis. We further describe an approach to induce commitment to a lineage of brown-like adipocytes upon exposure to bone morphogenetic protein 7 (BMP7).
Keywords: Skeletal muscle, Adipogenic progenitors, Fluorescence-activated cell sorting, Cell surface marker antibodies, Brown adipogenesis, Bone morphogenetic protein 7
1 Introduction
Skeletal muscle contains various populations of progenitor cells from different developmental ancestries. The most noted precursor cells that possess myogenic potential have been termed satellite cells and were originally described by Mauro [1]. Principally, myogenic progenitors are quiescent in adult muscle, but in response to injury represent the major source of regenerating myofibers (reviewed in [2, 3]). In contrast to satellite cells that reside underneath the myofiber basal lamina, adipogenic progenitor cells are predominantly localized in the interstitial space between mature myofibers [4]. Developmental lineage tracing studies using the Cre/loxP recombination system have revealed that myogenic cells commonly arise from an embryonic lineage of stem/progenitor cells that express myogenic transcription factors such as myogenic factor 5 (Myf5). On the other hand, adipogenic progenitors within skeletal muscle derive from a lineage that never expresses these transcription factors (Fig. 1) [5–7]. These muscle- resident adipogenic progenitor cells (MusAPCs) are therefore functionally and developmentally distinct from satellite cells and were described in two independent studies by their characteristic expression of the surface markers platelet-derived growth factor receptor (PDGFR)-α [4] and stem cell antigen 1 (Sca1) [5]. Both populations display adipogenic capacity in vivo and in vitro. Additional analysis of Sca1-expressing cells isolated from muscle revealed that these populations of adipogenic cells within skeletal muscle are in fact identical and homogenously express a set of surface markers commonly found in progenitors derived from regular adipose tissues, altogether suggesting functional similarities [6]. Specifically, the adipogenic Sca1+ population is negative for the hematopoietic markers Integrin αM (CD11b), Leukocyte common antigen (CD45), and c-Kit (CD117), and the endothelial marker PECAM-1 (CD31) and expresses typical pre-adipocyte markers such as Integrin β1 (CD29), CD34, PDGFRα (CD140a), and PDGFRβ (CD140b) [6]. Taken together, these studies collectively indicate that MusAPCs are non-myogenic and committed towards an adipogenic lineage and do not require further lineage commitment steps besides a regular adipogenic induction that is commonly used for bone marrow- or adipose-derived progenitor cells. Hence, this unique surface molecule signature can be used for identification and prospective isolation of muscle-resident adipose progenitors.
Fig. 1.

MusAPCs are not derived from a Myf5-expressing lineage. Animals expressing Cre recombinase under control of the Myf5-promoter are crossed to ROSA26-YFP (yellow fluorescent protein) reporter mice. Transient Myf5-expression during development (that will therefore also lead to expression of Cre) leads to permanent Cre-mediated DNA recombination that removes the loxP-flanked transcriptional stop cassette in the ROSA26 locus. Subsequently, expression of the reporter YFP is detectable in Myf5-expressing cells and, importantly, all their progeny independent of actual Myf5 expression
In healthy muscle tissue, differentiation of adipogenic progenitors is strongly inhibited by direct cell–cell interaction with myofibers suggesting that the local microenvironment (also known as the stem cell niche) triggers their developmental fate [4]. However, in response to muscle damage [4] and absence of functional Interleukin (IL)-4/IL-13 signaling [8], they contribute to ectopic fat infiltration of skeletal muscle. Muscle-resident pre-adipocytes, belonging to a distinct developmental lineage, do not generate myofibers themselves (Fig. 1) [4, 6, 7], but enhance differentiation of adjacent satellite cells in injured muscle [5, 8, 9]. Besides their regulatory function in myogenesis by direct cell–cell interactions, cell fate decisions might be driven by external factors. Schulz et al. were able to demonstrate the capacity of MusAPCs to differentiate into adipocytes with a brown adipocyte-like phenotype, the so-called beige or brite adipocytes, in response to stimulation with bone morphogenetic protein 7 (BMP7) [6]. The notion of muscle-resident brown fat is entirely consistent with previous observations that skeletal muscle of obesity-resistant mouse strains contains large depots of UCP1-expressing brown adipocytes [10]. Consequently, MusAPCs might be not constitutively committed to a white adipogenic lineage, but could indeed give rise to either white or beige/brite adipocytes after exposure to specific inductive cues.
One of the first methods to isolate muscle-derived stem cells (MDSC) is the pre-plate technique [11–13] by separating cells based on adhesion characteristics. In order to dissociate MDSC from their niche, steps of mincing, enzymatic digestion, and repetitive trituration have to be performed. Various protocols for muscle digestion have been frequently described in the literature [14, 15]. Substantial efforts to further improve the pre-plate technique to purify enriched populations of distinct cell fractions, such as satellite cells, were made [16, 17]. A more specific protocol for isolation of satellite cells according to their anatomical localization and subsequent purification by fluorescence-activated cell sorting (FACS) has previously been described [7, 18, 19].
Here, we describe a modified approach based on this method that enables the separation of two distinct muscle cell fractions: Interstitial cells and myofiber-associated cells. An enriched population of progenitor cells with adipogenic potential (MusAPCs) is purified by flow cytometry using antibodies directed against a specific set of surface antigens. Besides pre-adipocytes, this technique provides a promising strategy to isolate different, highly purified cell populations within the general population of mononucleated cells isolated from skeletal muscle by including additional surface markers. We further describe a method to differentiate this highly adipogenic population of cells into bona fide brown adipocytes of the recruitable type under in vitro conditions. While many studies suggest that the default commitment of these cells is white adipogenic, our studies also indicate that mouse strain-specific genetic differences determine whether these progenitor cells do or do not express a signature of genes that is typical for brown adipocytes, such as UCP1 [6].
2 Materials
2.1 Isolation of Progenitors from Murine Skeletal Muscle
70 % ethanol.
50 mL conical tubes.
Water bath.
Fetal bovine serum (FBS).
Phosphate-buffered saline (PBS).
10 cm petri dish.
37 °C incubator.
10 mL serological pipette.
Digestion solution 1: 0.2 % Collagenase type 2 (Life Technologies; see Note 1) in Dulbecco's modified Eagle's medium (DMEM; high glucose, l -glutamine with sodium pyruvate, Life Technologies). Freshly prepare an appropriate volume; approximately 10 mL per 2 g of muscle wet weight (see Note 2). Filter the solution through a 0.22 μm filter.
Digestion solution 2: 0.1 % Dispase 2, 0.025 % Collagenase 2 in F-10 Nutrient Mixture (all purchased from Life Technologies). Prepare approximately 5–6 mL per mouse. Filter the solution through a 0.22 μm filter. Prepare on day of isolation and keep refrigerated until use.
F-10 Nutrient Mixture supplemented with 20 % FBS.
ACK lysis buffer: 0.15 M ammonium chloride, 0.01 M potassium bicarbonate, 0.01 M ethylenediaminetetraacetic acid (EDTA) in 1 L distilled deionized water. Pass through a sterile 0.22 μm filter. Solution can be stored at room temperature.
Sorting medium: PBS supplemented with 2 % FBS.
Glass Pasteur pipette: Use a diamond pen to prepare pipette with desired opening width for trituration of skeletal muscle pieces. Flame with gas burner to blunt sharp edges and sterilize the pipette.
Fisherbrand cell strainer 40, 70, and 100 μm (Thermo Fisher Scientific).
2.2 FACS Staining
Compensation beads: OneComp eBeads (eBioscience).
Calcein blue stock solution (10 mM): 1 mg calcein blue (Life Technologies) in 215 μL dimethylsulfoxide (DMSO).
Propidium iodide (PI, Sigma-Aldrich): Use a final concentration of 1 μg/mL from a 1,000× stock in water.
Fluorochromes conjugated to anti-mouse antibodies (eBioscience) at final concentrations indicated in Table 1.
Table 1. Mouse monoclonal antibodies used for FACS isolation of MusAPCs.
| Antibody | Fluorochrome | Clone | Isotype | Concentration (μg/mL) |
|---|---|---|---|---|
| Anti-mouse-Sca1 | APC | D7 | Rat IgG2a | 0.5 |
| Anti-mouse-CD11b | FITC | M1/70 | Rat IgG2b | 2.5 |
| Anti-mouse-CD45 | FITC | 30-F11 | Rat IgG2b | 2.5 |
| Anti-mouseCD31 | PE/Cy7 | 390 | Rat IgG2a | 1 |
APC allophycocyanin, FITC fluorescein isothiocyanate, PE-Cy7 phycoerythrin/Cy7
2.3 Cell Culture
24- and 48-well cell culture plates.
Matrigel-coated dishes: Thaw a Matrigel aliquot at 4 °C overnight and resuspend in cold F-10 medium to a final concentration of 2 % under sterile conditions (see Note 3). Pipette an aliquot of the 2 % solution onto culture plate/well and aspirate. Leave coated plates to dry for several hours. To ensure sterility, dried plates can be exposed to UV light in the cell culture hood for 20 min.
Growth medium (modified from [20]; (Table 2)): 60 % DMEM with low glucose, 40 % MCDB201 (Sigma-Aldrich), 100 U/mL penicillin and 1,000 U/mL streptomycin (Life Technologies), 2 % FBS, 1× insulin-transferrin-selenium mix, 1× linoleic acid conjugated to bovine serum albumin (BSA), 1 nM dexamethasone, and 0.1 mM l - Ascorbic acid 2-phosphate (all purchased from Sigma-Aldrich). Pass through a sterile filter; store at 4 °C. Immediately before use, add the following growth factors to the medium: 10 ng/mL epidermal growth factor (EGF; PeproTech), 10 ng/mL leukemia inhibitory factor (LIF; Millipore), 10 ng/mL platelet-derived growth factor BB (PDGFR-BB; PeproTech), and 5 ng/mL basic fibroblast growth factor (bFGF; Sigma- Aldrich) and pass through a sterile filter.
Adipogenic induction medium: Growth medium without growth factors, 5 μg/mL human insulin (Roche Applied Science), 50 μM indomethacin, 1 μM dexamethasone, 0.5 μM isobutylmethylxanthine (IBMX), 1 nM 3,3′,5-triiodo-l-thyronine (T3) (all purchased from Sigma-Aldrich). Prepare stock solutions for all chemicals in the adipogenic induction medium and store at −20 °C (Table 3).
Adipogenic differentiation medium: Growth medium without growth factors, 5 μg/mL human insulin and 1 nM T3.
Gentamycin.
Trizol reagent (or similar).
25 μL Hamilton syringe.
Table 2. Growth medium for culture of MusAPCs.
| Stock concentration | Final concentration | Volume (500 mL) | |
|---|---|---|---|
| DMEM (low glucose) | – | 60 % | 300 mL |
| MCDB201 media | – | 40 % | 200 mL |
| FBS | – | 2 % | 10 mL |
| Penicillin/streptomycin | 100× | 1× | 5 mL |
| Dexamethasone | 10 μM | 1 nM | 50 μL |
| l-Ascorbic acid-2P | 50 mM | 0.1 mM | 1,000 μL |
| ITS MIX | 100× | 1× | 5 mL |
| Linoleic acid-albumin | 100× | 1× | 5 mL |
Table 3. Adipogenic induction medium.
| Stock concentration | Final concentration | |
|---|---|---|
| Insulin (human) | 10 mg/mL (water) | 5 μg/μL |
| Indomethacin | 30 mM (methanol) | 50 μM |
| IBMX | 5 mM (methanol) | 0.5 μM |
| Dexamethasone | 2 mg/mL (5 mM, ethanol) | 1 μM |
| T3 | 10 μM (water) | 1 nM |
3 Methods
3.1 Isolation of Progenitors from Murine Skeletal Muscle
Sacrifice mouse by cervical dislocation.
Spray dead animals extensively with 70 % ethanol. Create a transversal incision through the skin in the abdominal region. Peel the flap of skin to completely expose underlying tissue and the abdominal area of the animal. Harvest skeletal muscles of hind limbs (e.g., Soleus, Gastrocnemius, Tibialis anterior, Extensor digitorum longus, Quadriceps femoris). Ensure that mostly intact muscles are collected; this is critical for the subsequent digestion step.
Remove as much contaminating non-muscle tissue as possible (e.g., connective tissue and the small adipose tissue depots). Place intact muscles into a 50 mL conical tube with digestion solution 1, seal tube with Parafilm, and incubate at 37 °C in a water bath with gentle agitation for 90 min (see Note 4).
Stop enzymatic digestion by adding 10 % FBS, incubate for 10 min at room temperature.
Carefully discard liquid and floating debris without disturbing muscle pieces. Fill the tube with F-10 supplemented with 20 % FBS. Before decanting, let pieces sink to the bottom (see Note 5). Repeat washing twice using PBS to remove residual serum.
Single myofibers are separated mechanically; add muscle pieces with an appropriate volume of PBS to a 10 cm petri dish. The muscle is triturated/minced with a flamed glass Pasteur pipette by gently pipetting the digest up and down. The pipette tip has been cut further up to increase its opening width to approximately 3 mm (see Note 6). When PBS becomes turbid with single fibers and debris, collect supernatant into a clean 50 mL conical tube. Ensure that no intact muscle pieces are collected and add more PBS to the dish (approximately 5–10 mL each time). Repeat this step as many times as necessary to completely mince muscle. In the end, only connective tissue should remain.
Interstitial cells and myofibers are separated by differential centrifugation (no more than 50 × g for 1 min; all subsequent centrifugations are performed in a cooled centrifuge at 4 °C) leaving interstitial cells in the supernatant and fibers as pellet. Collect approximately 25 mL of the supernatant in another 50 mL conical tube. For preparation of interstitial cells proceed with Part B.
Wash single myofibers with PBS and repeat centrifugation (step 7).
After the second centrifugation step, remove most of the supernatant, but ensure that the myofiber-pellet is not disturbed. Wash myofibers again with PBS, invert two times, and place in a 37 °C incubator for 10–15 min to let fibers settle by sedimentation. Repeat this procedure until supernatant remains more or less clear, i.e., free of interstitial cells and debris. Usually, 3–4 repeats are sufficient.
Remove supernatant carefully to less than 5 mL residual volume including the pellet of myofibers (see Note 7). For preparation of myofiber-associated cells proceed with Part A.
Part A: Preparation of Myofiber-Associated Cells
Add 5–6 mL of digestion solution 2, seal cap with Parafilm, and incubate for 30 min in a 37 °C water bath with gentle agitation.
To inactivate enzymatic digestion, add FBS to a final concentration of approximately 10 %. Pipette fibers several times with a 10 mL serological pipette to dissociate myofiber-associated cells from myofibers. Use high speed mode on the pipettor to generate mechanical strain to break pre-digested myofibers. Differential centrifugation (no more than 50 × g for 1 min) sediments most of the larger debris, but leaves myofiber-associated cells in the supernatant.
Collect supernatant and filter through a 100 μm cell strainer. Discard leftover debris and wash strainer with an additional volume of sorting medium. Centrifuge at 300 × g for 5 min. Resuspend the pellet and repeat filtration with a 40 μm cell strainer. Spin down the cells and resuspend in sorting medium to transfer suspension to a 5 mL sorting tube for staining (5 min, 300 × g).
Part B: Preparation of Interstitial Cells
Centrifuge the supernatant containing interstitial cells at 300 × g for 5 min.
To remove red blood cells, resuspend pellet in 2–3 mL ACK lysis buffer and incubate on ice for 3 min. Stop lysis by adding 10 mL of sorting medium.
After centrifugation at 300 × g for 5 min, resuspend cells in sorting medium and pass through a 100 μm cell strainer, and subsequently through a 40 μm cell strainer (see step 3 of Part A).
Spin down as before and resuspend the pellet in sorting medium and transfer the suspension to a 5 mL sorting tube for staining.
3.2 Purification of Adipogenic Progenitors by FACS
For FACS purification of adipogenic progenitors, isolated muscle cells are stained with fluorophore-tagged monoclonal antibodies directed against Sca1, the lineage markers CD11b, CD45, and CD31, and optionally CD29 to enrich myogenic cells (see Note 8).
Sample preparation: Centrifuge myofiber-associated cells and interstitial cells at 300 × g for 5 min. Resuspend pellets in a small defined volume (e.g., 250–150 μL). Take small aliquots of interstitial cells for preparing staining controls as indicated in steps 2–4.
Use staining controls for voltage adjustments according to specific flow cytometer parameters, depending on the instrument used for sorting (see Note 9).
Compensation controls: Prepare an unstained sample as autofluorescence control as well as single color controls for each fluorochrome used. This includes antibody-coupled fluorochromes as well as live/dead-staining fluorochromes (see Note 10). To generate each control, use a 100 μL aliquot of a control cell suspension that contains cells that are comparable to the cell types occurring in the regular samples. Since compensation controls with cells are not always completely effective, compensation beads which contain microparticles that bind to mouse isotype antibodies (positive compensation control) and microparticles with no binding capacity (negative compensation control) can be used alternatively. Add individual fluorochromes at concentrations indicated in Table 1.
Fluorescence-minus-one (FMO) controls: FMOs are used for setting the threshold of negative and positive signals. In each FMO control, all antibodies (not for other non-antibody fluorochromes) used in the multicolor stain are included except the individual antibody for which the threshold is to be determined. Hence, if four antibodies are being used in a four color stain, four FMOs, each lacking one antibody, need to be generated.
Antibody preparation: Add all antibodies at the indicated concentrations (Table 1) to samples and staining controls (see Note 11).
Incubate samples and controls on ice and in the dark for at least 20 min.
Wash cells and centrifuge at 300 × g for 5 min and resuspend in 200 μL sorting medium.
Immediately before sorting, filter cell suspensions through a 70 μm cell strainer to avoid clogging of the tubing of the flow cytometer.
Live cells are isolated by positive selection for calcein blue staining and negative selection for propidium iodide staining. The dyes can be added before or after final filtration (see Note 12).
Analyze samples on a flow cytometer that is equipped with sorting capacity. Start data acquisition on flow cytometer by creating a forward scatter (FSC) and sideward scatter (SSC) plot to adjust the settings until the proceeded events are clearly delineated. Set the photomultiplier tube (PMT) voltage of the FITC, APC, PE/Cy7 detectors using the unstained sample and single color controls (see Note 13).
Run the single color sample to correct the potential spectral overlap between color filters and fluorochromes using software compensation.
After compensation, run FMO controls for defining negative signal threshold.
Gating strategy (Fig. 2): To select and sort defined populations of cells, a gating strategy to remove debris, cell aggregates, and dead cells will be used before selection of certain cell populations according to surface marker expression.
To exclude debris, define a muscle cell population in the FSC versus SSC plot by means of a polygonal region (R1) drawn around the events that represent cells (Fig. 2a).
To exclude duplets, the selected population is gated for single cells (R2) in a forward scatter-height (FSC-H) versus forward scatter-area (FSC-A) plot (Fig. 2b).
The single cells are further analyzed for their uptake of calcein blue or propidium iodide to determine live (Calcein- positive) versus dead (PI-positive) cells and cellular debris (double negative) (Fig. 2c).
Set gates according to Fig. 2d, e to select for MusAPCs. Within the viable population (R3), gate for cells negative for lineage markers CD31, CD11b, and CD45 (R4) in the PE/Cy7 versus FITC plot (see Fig. 2d). Finally, select cells residing in Sca1 positive gate (R6) in the FSC-H versus APC plot for sorting and collection (Fig. 2e). For additional analysis of myogenic progenitors see Note 8.
Fig. 2.
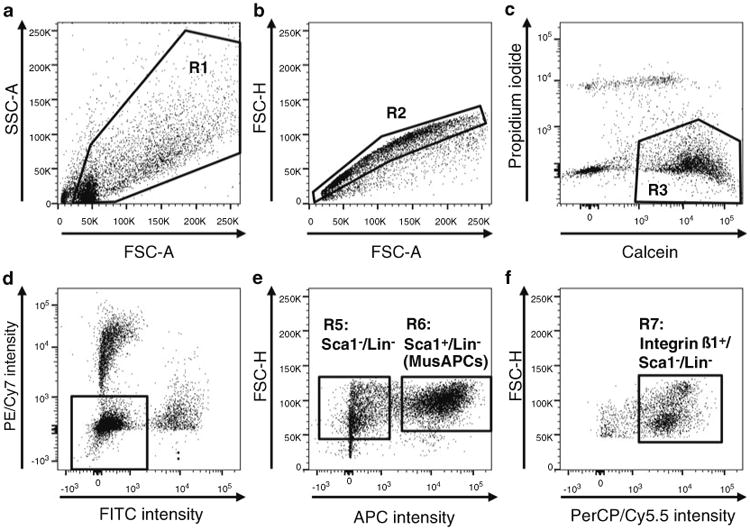
Gating strategy for FACS purification of adipogenic progenitor cells from a heterogeneous muscle cell population. (a) In the FSC vs. SSC plot, draw a polygonal region (R1) around the events that represent intact cells, thereby excluding debris. (b) Perform duplet discrimination by gating for single cells (R2) in FSC-H vs. FSC-A plot as indicated. (c) Calcein and Propidium iodide (PI) are used to discriminate between live (Calcein+/PI−, R3) and dead cells (Calcein−/PI+). (d) Exclude hematopoietic cells and endothelial cells by selecting cells that are CD11b−/CD45−/CD31− (R4: lineage negative (Lin−) cells) in the PE/Cy7 vs. FITC plot. (e) Select MusAPCs marked as the Sca1+/Lin− population (R6) for sorting. For optional analysis/collection of myogenic progenitors, define a gate for the Sca1−/Lin− population (R5). (f) To enrich satellite cells within the myogenic Sca1−/Lin− population, select the Integrin ß1 positive population (R7) and apply a specific selection marker for satellite cells (see Note 8)
3.3 Cell Culture and (Brown) Adipogenic Differentiation of MusAPCs
Centrifuge the collected Sca1 positive progenitor cells at 4 °C and 50 × g for 5 min. Resuspend in an appropriate volume of growth medium to plate approximately 50,000 cells per well on coated 24-well cell culture plates. Wash the tube with growth medium to collect and plate residual cells. Important: Use growth medium supplemented with 50 μg/mL gentamycin to prevent bacterial contamination.
After 2 days, add fresh growth medium without gentamycin.
Expand cells until they reach 90–95 % confluence. This should take approximately 1 week.
For adipogenic differentiation, seed cells into Matrigel-coated 48-well plates and leave to adhere overnight (15,000 cells per well in a 48-well plate).
Pretreatment with bone morphogenic protein 7 (BMP7) can be used to induce brown adipogenesis and UCP1 expression in the mature adipocytes [6]. Seeding 15,000–20,000 cells will allow the cells to reach confluence within a 72 h treatment with BMP7. The pretreatment with 3.3 nM BMP7 is for 72 h in basal growth medium with growths factors (termed as day 3 of the time course of differentiation; Fig. 3). BMP7 does not need to be replaced during this period (see Note 14).
For adipogenic induction, cells are treated with adipogenic induction medium without growth factors (Table 3) for 48 h followed by differentiation in growth medium without growth factors, but addition of T3 and insulin only for 7 days (Fig. 3). Cells are re-fed fresh medium every other day until cells are differentiated into mature adipocytes (see Note 15).
To harvest cells for gene expression analysis of brown adipogenic markers, add 0.5 mL Trizol reagent or similar to lyse cells. Pipette up and down several times to fully lyse cells before transferring liquid to reaction tubes.
Fig. 3.
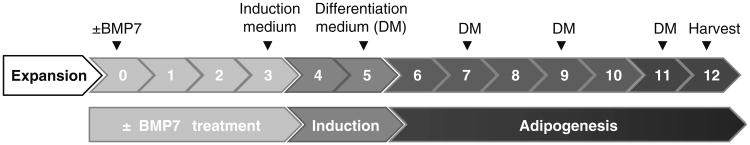
Time course of adipogenic differentiation of MusAPCs. After expansion of purified MusAPCs, cells are seeded into Matrigel-coated 48-well plates and left to adhere overnight. Before starting the differentiation procedure, a BMP7 pretreatment is performed for 72 h. Start adipogenic differentiation after removal of BMP7 and by treating the cells with adipogenic induction medium for 48 h. On day 5, cells are treated with differentiation medium and re-fed fresh medium containing only insulin and T3 every other day until they are differentiated into mature adipocytes (usually on day 12)
3.4 Cell Culture and Implantation of Brown Adipogenic Progenitors (See Note 16)
Grow and pretreat MusAPCs with BMP7 as described. Harvest cells after a 72 h exposure to 3.3 nM BMP7 by trypsinizing. Be sure not to over-trypsinize since the cells detach very quickly.
Count and spin down the cells for resuspension in an appropriate volume of F-10 medium with 2 % Matrigel. Resuspend to a final concentration of one million cells per 25 μL injection volume.
Inject cells using a cooled 25 μL Hamilton syringe into gastrocnemius muscle after removing fur from the lower hind legs, or the subcutaneous or epididymal fat pads. While the muscle can be easily reached by intramuscular injections, injection into the fat pad requires a surgical procedure to either depot.
Sacrifice animals 10 days after implantation for monitoring engraftment of cells. GFP+ cells can be identified under a fluorescence microscope prior to embedding for further analysis of the implanted cells.
4 Notes
An appropriate collagenase batch has to be determined in pilot studies. Usually, a higher enzyme activity leads to better dissociation of the tissue. However, a lower viability could be an adverse side effect.
Use no less than 10 mL of digestion buffer per animal. A lower volume will negatively affect cell viability.
To culture MusAPCs, it is recommended to use coated cell culture dishes. To avoid uneven coating by polymerization, keep Matrigel at 4 °C all the time and work quickly as pure Matrigel solidifies at room temperature. It should be kept on ice until diluted in cold media. The ready-to-use Matrigel solution in F-10 can be used for up to 2 weeks if kept cold and sterile. Only discard aliquots that have been used for coating.
Less time-consuming protocols for muscle cell isolation performing only one enzymatic digestion have been reported [9, 21, 22]. In previous tests of different methods, one step digestion results in muscle digests containing much more debris and a significantly lower yield of adipogenic progenitors.
Be careful not to destroy the muscle pieces, as this will result in loss of intact fibers.
Make sure the opening is not too wide in order to generate some mechanical strain while pipetting up and down. This will help to break open the predigested muscle pieces to release individual fibers.
Do not touch the fiber pellet as it is only lightly packed.
Including additional surface markers in the FACS analysis enables the prospective purification of multiple muscle cell populations, for instance endothelial cells (CD31+/Sca1+/CD45−) [22]. Moreover, several protocols for the isolation of satellite cells by FACS with distinct marker configurations have been published. Specifically, satellite cells could be identified by expression of Integrin β1 (CD29) and CXCR4 (C-X-C chemokine receptor type 4; CD184) [7, 19]. Similarly, the selection of Integrin α7+/CD34+ double positive cells and negative selection for CD45, CD31, CD11b, and Sca1 has been shown before [23]. We successfully incorporated the isolation of myogenic progenitors (Integrin β1+/Sca1−/CD11b−CD45−/CD31−) in a complex gating strategy (Fig. 2). In the FSC-H versus APC plot, gate for Sca1−, lineage− (Lin−) cells (R5, Fig. 2d). Myogenic progenitors are further enriched by gating for Integrin ß1 positive population (R7, see Fig. 2f). To discriminate satellite cells more rigorously, specific markers have to be included, like abovementioned CXCR4 [24], CD34 [25], or Integrin α7 [26].
The staining controls described here are sufficient for a setup for most FACS-instrument brands.
Contrary to calcein, it is not necessary to compensate for propidium iodide as PI+/dead cells are excluded in the subsequent gating strategy (Fig. 2c).
Prior to use in a FACS experiment, the antibody concentrations need to be optimized by titration to avoid unspecific binding (false positive signals). To ensure reliable FACS data acquisition, perform a titration with each antibody lot. Titration protocols have been described elsewhere [27].
Keep all the samples on ice and in the dark until FACS analysis is carried out. Vortex and filter all samples before analysis.
The strategy for color compensations and subsequent setups strongly depends on the instrument and should be performed by experienced personnel.
Other reagents that promote browning of adipogenic cells have been described in the literature and may do so in MusAPCs as well. Typical inducers are Rosiglitazone [28], Irisin [29], or FGF21 [30]. The efficiency of browning through these factors would have to be determined. Our studies indicate that a pretreatment with BMP7 is critical for induction of brown adipogenesis, whereas a full-time exposure to BMP7 throughout differentiation may not promote brown adipogenesis as efficiently. Our findings suggest that BMP7 indeed acts as a lineage commitment factor that switches an adipogenic cell between a white adipocyte differentiation program and a brown adipocyte- like differentiation program.
Importantly, and depending on the mouse strain used, the cells will not normally express high levels of brown adipocyte markers without an inductive cue, such as BMP7. Moreover, high levels of brown adipogenesis are strongly dependent on differentiation time and serum batch. In order to achieve high level of brown adipocyte-marker expression, several batches of FBS may need to be tested. We typically obtain induction of UCP1 expression after pretreatment with BMP7 ranging from 10- to 30-fold.
MusAPCs can be transplanted into skeletal muscle or adipose tissue of recipient mice. We typically use cells pretreated with BMP7 to achieve good implantation and subsequent differentiation into adipocytes. To allow identification of the engrafted cells after implantation, we suggest using cells from GFP-transgenic animals or a similar genetic intervention that permanently labels the donor cells. Our studies show that implanted cells do not survive prolonged periods of time in healthy, immunocompetent mice, which is consistent with previous reports showing that adipogenic progenitors expressing our described set of markers do not persist in normal mice, likely due to the presence of a large population of competing adipogenic progenitors already present within the host [31].
Acknowledgments
This work was supported by grants from the German Research Foundation (DFG; grant # SCHU 2445/2-1) and the European Research Council (grant # ERC-StG-2012-311082) to TJS.
References
- 1.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol. 2001;91:534–551. doi: 10.1152/jappl.2001.91.2.534. [DOI] [PubMed] [Google Scholar]
- 3.Zammit PS, Partridge TA, Yablonka-Reuveni Z. The skeletal muscle satellite cell: the stem cell that came in from the cold. J Histochem Cytochem. 2006;54:1177–1191. doi: 10.1369/jhc.6R6995.2006. [DOI] [PubMed] [Google Scholar]
- 4.Uezumi A, Fukada S, Yamamoto N, et al. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010;12:143–152. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- 5.Joe AWB, Yi L, Natarajan A, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12:153–U144. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schulz TJ, Huang TL, Tran TT, et al. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc Natl Acad Sci U S A. 2011;108:143–148. doi: 10.1073/pnas.1010929108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerletti M, Jurga S, Witczak CA, et al. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell. 2008;134:37–47. doi: 10.1016/j.cell.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heredia JE, Mukundan L, Chen FM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153:376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mozzetta C, Consalvi S, Saccone V, et al. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol Med. 2013;5:626–639. doi: 10.1002/emmm.201202096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Almind K, Manieri M, Sivitz WI, et al. Ectopic brown adipose tissue in muscle provides a mechanism for differences in risk of metabolic syndrome in mice. Proc Natl Acad Sci U S A. 2007;104:2366–2371. doi: 10.1073/pnas.0610416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qu Z, Balkir L, van Deutekom JC, et al. Development of approaches to improve cell survival in myoblast transfer therapy. J Cell Biol. 1998;142:1257–1267. doi: 10.1083/jcb.142.5.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richler C, Yaffe D. The in vitro cultivation and differentiation capacities of myogenic cell lines. Dev Biol. 1970;23:1–22. doi: 10.1016/s0012-1606(70)80004-5. [DOI] [PubMed] [Google Scholar]
- 14.Arsic N, Mamaeva D, Lamb NJ, et al. Muscle-derived stem cells isolated as non-adherent population give rise to cardiac, skeletal muscle and neural lineages. Exp Cell Res. 2008;314:1266–1280. doi: 10.1016/j.yexcr.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Qu-Petersen ZQ, Deasy B, Jankowski R, et al. Identification of a novel population of muscle stem cells in mice: potential for muscle regeneration. J Cell Biol. 2002;157:851–864. doi: 10.1083/jcb.200108150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gharaibeh B, Lu A, Tebbets J, et al. Isolation of a slowly adhering cell fraction containing stem cells from murine skeletal muscle by the preplate technique. Nat Protoc. 2008;3:1501–1509. doi: 10.1038/nprot.2008.142. [DOI] [PubMed] [Google Scholar]
- 17.Jankowski RJ, Haluszczak C, Tr ucco M, et al. Flow cytometric characterization of myogenic cell populations obtained via the pre-plate technique: potential for rapid isolation of muscle-derived stem cells. Hum Gene Ther. 2001;12:619–628. doi: 10.1089/104303401300057306. [DOI] [PubMed] [Google Scholar]
- 18.Conboy IM, Conboy MJ, Smythe GM, et al. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–1577. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- 19.Sher wood RI, Christensen JL, Conboy IM, et al. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543–554. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 20.Steenhuis P, Pettway GJ, Ignelzi MA., Jr Cell surface expression of stem cell antigen-1 (Sca-1) distinguishes osteo-, chondro-, and adipoprogenitors in fetal mouse calvaria. Calcif Tissue Int. 2008;82:44–56. doi: 10.1007/s00223-007-9083-4. [DOI] [PubMed] [Google Scholar]
- 21.Pisani DF, Clement N, Loubat A, et al. Hierarchization of myogenic and adipogenic progenitors within human skeletal muscle. Stem Cells. 2010;28:2182–2194. doi: 10.1002/stem.537. [DOI] [PubMed] [Google Scholar]
- 22.Ieronimakis N, Balasundaram G, Reyes M. Direct isolation, culture and transplant of mouse skeletal muscle derived endothelial cells with angiogenic potential. PLoS One. 2008;3 doi: 10.1371/journal.pone.0001753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasut A, Oleynik P, Rudnicki MA. Isolation of muscle stem cells by fluorescence activated cell sorting cytometry. Methods Mol Biol. 2012;798:53–64. doi: 10.1007/978-1-61779-343-1_3. [DOI] [PubMed] [Google Scholar]
- 24.Ratajczak MZ, Majka M, Kucia M, et al. Expression of functional CXCR4 by muscle satellite cells and secretion of SDF-1 by muscle-derived fibroblasts is associated with the presence of both muscle progenitors in bone marrow and hematopoietic stem/progenitor cells in muscles. Stem Cells. 2003;21:363–371. doi: 10.1634/stemcells.21-3-363. [DOI] [PubMed] [Google Scholar]
- 25.Beauchamp JR, Heslop L, Yu DS, et al. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J Cell Biol. 2000;151:1221–1234. doi: 10.1083/jcb.151.6.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanco-Bose WE, Yao CC, Kramer RH, et al. Purification of mouse primary myoblasts based on alpha 7 integrin expression. Exp Cell Res. 2001;265:212–220. doi: 10.1006/excr.2001.5191. [DOI] [PubMed] [Google Scholar]
- 27.Hulspas R. Titration of fluorochrome-conjugated antibodies for labeling cell surface markers on live cells. Curr Protoc Cytom. 2010;54:6.29.1–6.29.9. doi: 10.1002/0471142956.cy0629s54. [DOI] [PubMed] [Google Scholar]
- 28.Petrovic N, Walden TB, Shabalina IG, et al. Chronic peroxisome proliferator-activated receptor gamma (PPAR gamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem. 2010;285:7153–7164. doi: 10.1074/jbc.M109.053942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bostrom P, Wu J, Jedrychowski MP, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisher FM, Kleiner S, Douris N, et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26:271–281. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135:240–249. doi: 10.1016/j.cell.2008.09.036. [DOI] [PubMed] [Google Scholar]