
Figure 1.
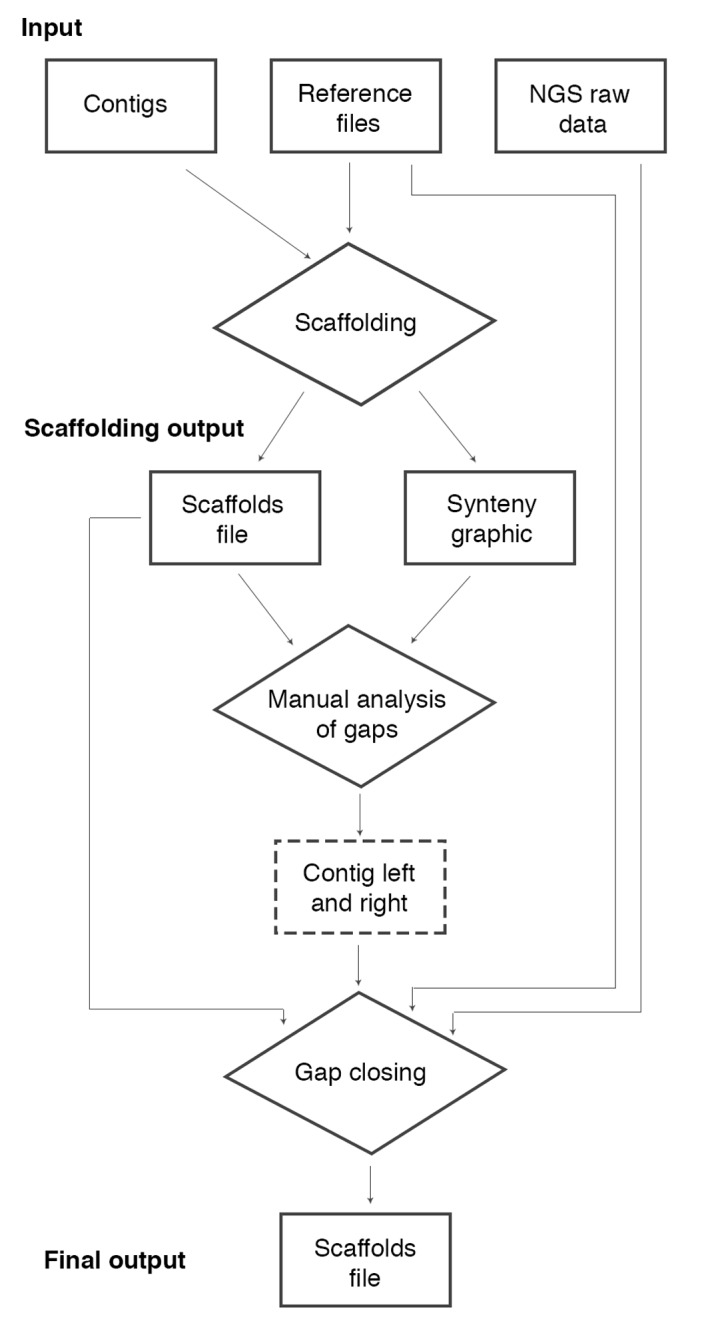
pipeline flowchart. The pipeline receives as input: contigs file, reference files (Fasta and GenBank files) and NGS raw data (reads file). The first step is the scaffolding of the contigs. This step can be realized by a modified version of the CONTIGuator software and it has as output a scaffolds file and a synteny graphic with colored targets indicating repetitive regions in the reference file and the gaps׳ positions in the scaffolds file. Using this file it is possible to conduct a manual analysis to choose two contigs׳ names as neighbors to a gap. Note that in the scaffolds file, we do not have contigs (orientated are called scaffolds), however we preserve this denomination in this flowchart to facilitate the comprehension. After this step, we developed the movednaa.py script to correct the beginning of the scaffold file for circular genomes searching the gene dnaA. We also developed the script cut_left.pl to remove barcodes on raw data, when needed. Thus, one can complete the assembly of repetitive regions based on the extraction of the consensus sequence of the mapping of raw data to the reference genome. To automate this step, we developed a software called MapRepeat. It receives as input the name of the two contigs and the path of the scaffolds file, reference Fasta file and the folder containing the NGS raw data file. MapRepeat has as output a new scaffolds file with a closed gap that was indicated in the step before. To analyze the result we developed the scripts: mcontig.py (to divided scaffold files in Multi-Fasta files breaking Ns regions) and contiginfo.py (to analyze number of gaps, length of the genome, length of larger and smaller contigs, and calculate the N50 value).