
Figure 2.
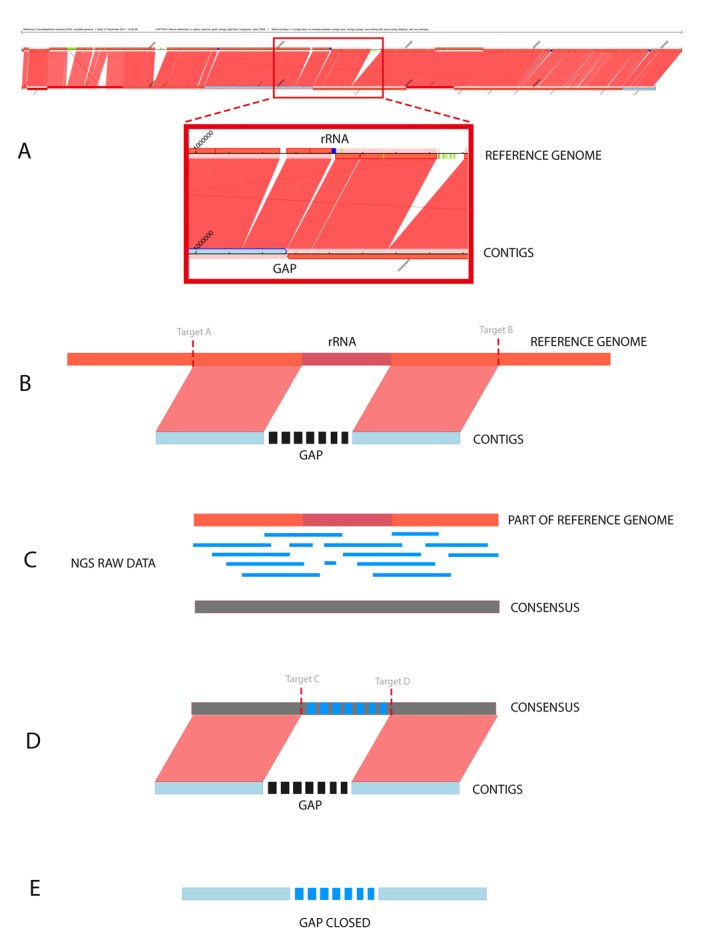
(A) Synteny graphic generated by CONTIGuator. The figure shows the reference genome on top, and below are the contigs aligned with the localization of gaps, which may be used as input parameters for MapRepeat; (B) First step of running MapRepeat. The software uses BLAST to detect whether there are similarities between two neighbors of contigs. If there are similarities, targets will be used to delimit the initial position of similarity with the left contig and the final position of similarity with the right contig. MapRepeat analyzes a region in the left contig until 3,000 pb before the gap and in the right contig until 3,000 pb after the gap; (C) The region between the targets is extracted, and then the raw data of sequencing are mapped against the extracted sequence using Mira assembler. A consensus sequence is generated based on whether there is coverage that proves the existence of this region in the reference genome and also in the genome sequenced; (D) The consensus sequence is aligned against the fragments of the two contigs. New targets (C and D) are used to identify the unknown regions from the contigs file mapped in the consensus; (E) The sequence contained between the targets C and D is extracted and used to close the gap.