
FIGURE 3.
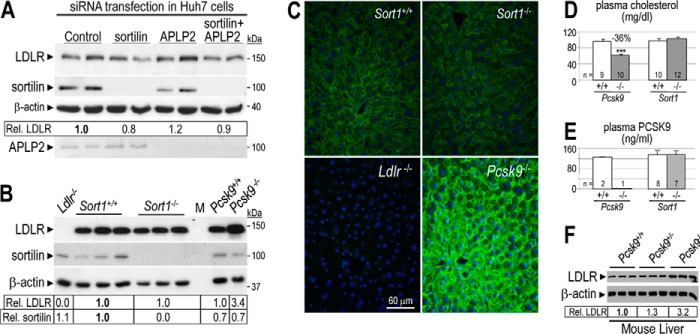
Sortilin depletion does not affect LDLR degradation by PCSK9 and has no cholesterol phenotype in mice. A, Huh7 cells were transfected with control non-target, APLP2-, and/or sortilin-specific siRNAs. After 48 h, cells were incubated in serum-free media for 24 h. Cell lysates were then subjected to Western blotting using LDLR, sortilin, APLP2, and β-actin antibodies. B, total liver extracts of Sort+/+ (n = 3), Sort1−/− (n = 3), Ldlr−/− (n = 1), Pcsk9+/+ (n = 1), and Pcsk9−/− (n = 1) mice were analyzed by Western blot analysis. Accurate quantification of LDLR and sortilin signals and their normalization to that of β-actin was obtained by LI-COR analysis of a duplicate gel. Relative LDLR/β-actin signals were normalized to that of control (A and B in bold). C, immunohistochemistry of surface LDLR (green) in the liver of Sort+/+, Sort1−/−, Ldlr−/−, and Pcsk9−/− mice. Surface LDLR levels in the liver of Sort1−/− and Sort+/+ mice were similar. Bar = 60 μm. Plasma (D) total cholesterol and (E) PCSK9 measured by ELISA, in control Pcsk9+/+ and Pcsk9−/− mice, and in Sort+/+ and Sort1−/− mice. Error bars represent S.E. ***, p < 3 × 10−5 (Student's t test). F, Western blot analysis of total liver extracts of Pcsk9+/+ (n = 3), Pcsk9+/− (n = 3), and Pcsk9−/− (n = 3) mice. Accurate quantification of LDLR and its normalization to that of β-actin was obtained by BioRad Image Lab 5.2 analysis of the same gel. Relative LDLR/β-actin signals were normalized to that of Pcsk9+/+ (in bold). These data are representative of at least two independent experiments.