

Abstract
Downstream processing of nanoplexes (viruses, virus-like particles, bacteriophages) is characterized by complexity of the starting material, number of purification methods to choose from, regulations that are setting the frame for the final product and analytical methods for upstream and downstream monitoring. This review gives an overview on the nanoplex downstream challenges and chromatography based analytical methods for efficient monitoring of the nanoplex production.
Keywords: bacteriophage, CIM monolithic column, DSP, gene therapy vector, PAT, purification, USP, vaccine, VLP, virus
Introduction
Purified virus preparations have been used for many years as vaccines and have a great potential in the future as gene therapy vectors. Currently, the number of approved gene therapy clinical trials worldwide that have been conducted or are still ongoing is larger than 1900. The most commonly used gene transfer virus vectors in clinical trials are adenovirus, retrovirus, vaccinia virus, adeno-associated virus and lentivirus.1
Bacterial viruses or bacteriophages (phages) have proven to be able to treat bacterial infections already in the previous century, but the discovery of antibiotics slowed down or restricted research toward their use in human therapeutics. Due to the growing resistance of bacteria to antibiotics the potential of phages to serve as an alternative or at least as a backup treatment for bacterial infections is now again being recognized.
The most differential feature of virus based biopharmaceuticals compared to protein based biopharmaceuticals is their size. A few orders of magnitude difference in size presented in the past (and for some viruses even today) a challenge in the downstream processing (DSP) of these huge entities. Since chromatography based tools for purification of biopharmaceuticals were optimized for purification of smaller molecules and proteins, purification of virus particles was carried out mainly by using other techniques (density gradient ultracentrifugation, precipitation, etc.). Poor scalability of these processes, sometimes inadequate purity of the product and/or poor economics together with the recognized potential of gene therapy and higher regulatory requirements regarding the purity of virus preparations used for vaccination led to the development of new and advanced technologies in the field of chromatography media. The structure and characteristics of these new media enabled the shift toward chromatography based DSPs of nanoparticles.
The main goal of chromatography media designed for nanoparticle purification was the creation of larger surface area accessible for nanoparticles. In case of particle based packed columns larger surface area was achieved with porous/perfusion particles in the early nineties.2 Although large surface area inside the pores is accessible for nanoparticles, high diffusional resistance of nanoparticles allows only low flow rates to be used in order to give nanoparticles enough time to diffuse in and out of the particle pores. However, relatively small pore sizes still prevent viruses to enter and reach the active surface area inside the pores3-5 leading to smaller dynamic binding capacity of these resins for nanoparticles.6 Wu et al. exposed POROS® HS 50 to 100 nm VLPs in batch and flow conditions and analyzed the beads with transmission electron microscopy (TEM) after exposure. TEM analysis revealed the formation of a distinct VLP layer at the particle outer surface indicating that bound VLPs block access to the underlying pore network for other VLPs.3 Similarly, Trilisky and Lenhoff4 studied the adsorption of 80 nm adenovirus type 5 (Ad5) on PL-SAX 4000 A resins that has one of the widest available pores (400 nm in diameter) among beaded resins. Although relatively large, these pores do not allow substantial penetration of Ad5, limiting the use of the internal surface area of the wide-pore resin and giving the internal surface area little practical utility in preparative separations.4 These results suggest that matrices with larger pores are needed to accommodate nanoparticles and to take advantage of the effects of perfusion for their adsorption.
In parallel to porous/perfusion particles different membrane- and monolith-based media were developed. The main advantage of these media over packed porous particles is that the mass transfer is predominantly based on convection rather than diffusion, which enables high flow rates to be used (10-20 times higher compared to packed porous particles) without any loss of dynamic binding capacity (DBC) and resolution. This results in short separation times and leads to higher productivity of chromatographic processes which can be increased by at least one order of magnitude as compared to traditional chromatographic columns packed with porous particles.7 Figure 1 is a schematical illustration of the ability of virus particles to reach active binding sites in columns packed with porous particles and CIM monolithic columns.
Figure 1.
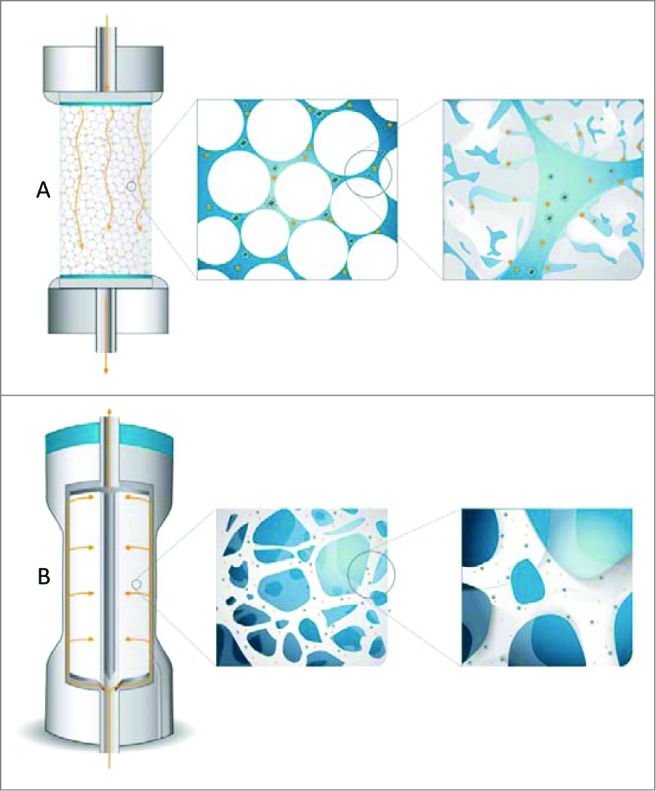
Scheme illustrating the ability of virus particles to reach active binding sites in columns packed with porous particles (A) and CIM monolithic columns (B).
High DBC (for target molecule and binding impurities) of a chromatography media is a prerequisite for an efficient capture step. Monoliths and membranes have active sites located at the surface of large channel structure/membrane and they are easily accessible also for large nanoparticles resulting in high dynamic binding capacity for large molecules which increases the productivity of the purification process. However, for some DSP steps (e.g. polishing step) good resolution is as important as high DBC, because this enables efficient removal of impurities. Due to the large void volume of membranes compared to monoliths, the resolution, regardless of the flow rate used, is superior with monoliths. This advantage of monoliths is especially important and has already been recognized in different DSP steps of nanoparticles.6,8,9-11 Excellent resolution of monoliths was demonstrated by Lock's group9 who developed a method for separation of 2 very similar viral particles (viral capsids with and without genetic material) using a CIM monolithic column and a very shallow linear gradient.
In addition to high DBC and good resolution the most important characteristic of any chromatography resin used in the DSP of any biomolecule is the ability to provide high yield of the product and thus enable good economics of the DSP. Superiority of monolithic columns in this respect was previously shown for different nanoparticles.6,10,11 Burden et al.6 performed a comparative study and showed that the dynamic binding capacity of CIM OH monolithic column for the HB-VLPs is approximately 3 to 4 times larger than that on Butyl-S 6 Sepharose FF with equivalent 90% recovery of the VLP. Similarly, Bandeira et al.10 showed 3-fold higher lentivirus recovery with monoliths when compared to membrane based adsorbers.
A very important part of a nanoparticle production represents the set-up of appropriate analytical methods for downstream and upstream monitoring of the process and for the final product analysis. Reproducible DSP and final products are possible only when appropriate analytical assays are in place. There is currently a variety of traditional biological analytical methods available, however, due to their relatively high variability and different information that they provide, usually a combination of several are needed to assure adequate product characterization. In addition, most of these methods are time consuming, which represents a significant bottleneck in the DSP of a new vaccine. Since 2004 when FDA started with the Process Analytical Technology (PAT) initiative,12 emphasizing the importance of in-depth monitoring of the complete production process and not only final products, rapid methods for in-process and final product control became highly desired. As a consequence chromatographic HPLC based methods started to emerge in the field of nanoparticle production and started to gain their importance due to their speed and precision, providing information about product purity and quantity in terms of minutes. Chromatographic methods applicable for PAT can be roughly divided into 2 types of methods; quantification methods and fingerprinting methods. Quantification methods are methods used for quantifying viral particles. A pure viral standard is needed for calibration curve preparation, the method needs to be validated, and parameters like quantification limits, repeatability, reproducibility and precision need to be determined. Fingerprinting methods are fast methods, providing almost real-time information about a particular step in the DSP. A fingerprinting method does not necessarily provide information about the quantity of a particular virus but gives relevant information to make timely decisions for subsequent purification steps within the DSP. Results provided by fingerprinting methods can be included in the decision criteria such as optimization of purification runs, duration of fermentation, nuclease treatment steps, etc. Their most beneficial feature compared to traditional analytical methods is time efficiency.
Chromatographic methods can be developed on different types of chromatographic media, each having their advantages and disadvantages for a certain molecule of choice. Monolithic columns exhibit some quite advantageous properties for separation of viruses and VLPs, compared to the particle based columns. For analytical applications, their largest advantage is the flow unaffected resolution, which is a consequence of convective mass transfer between the mobile and the stationary phase (Fig. 2). Thus chromatographic methods become even shorter with chromatographic runs lasting just a few minutes, providing almost real-time in-process results.
Figure 2.

Flow independent resolution of CIM monolithic columns; gradient elution of the mixture of 6 proteins on CIMac SO3 Analytical ColumnTM at the flow rates 0.5, 1.0 and 2.0 mL/min. Column: CIMac SO3 Analytical ColumnTM (5.2 mm I.D. × 5.0 mm); Sample: Test protein mixture (1) myoglobin, (2) trypsinogen, (3) ribonuclease A, (4) α-chymotrypsinogen A, (5) cytochrome C, (6) lysozyme; Injection volume: 10 μl; mobile phase A: Buffer A: 20 mM Na-phosphate, pH 6.0, Mobile phase B: Buffer B: 20 mM Na-phosphate + 1.0 M NaCl, pH 6.0, Gradient: a linear gradient from 0 to 28% buffer B in 30 CV, Detection: UV at 280 nm, HPLC system: Knauer high pressure gradient HPLC system.
Virus Downstream Processing
The starting virus material which needs to be purified before it is used as a pharmaceutical can be produced in different production/expression systems like bacteria cells (propagation of phages), eggs (e.g., influenza virus for vaccine), or now days more and more in different cell lines. When the virus/phage/VLP is harvested at the end of the upstream process (UPS) the resulting virus material contains apart from virus particles also processing reagents (media components), cell debris, host cell related molecules (host cell proteins, DNA, endotoxins) and potentially also host cell derived viral agents (e.g. retroviruses and endogenous viruses). All these impurities present a health risk and therefore need to be removed to a certain extent which is set by regulatory authorities before virus preparation is used as a pharmaceutical.
The potential risk of residual cellular DNA in vaccines is transmission of latent viruses and other agents and incorporation of cellular DNA into host genetic material. These theoretical safety issues are considered by regulatory authorities during vaccine registration/approval process, especially if vaccine is produced in a new cell substrate/line.14 Recommendations for acceptable levels of residual cellular DNA in the final product are determined in regard to the characteristics of the cell substrate used, the intended use of the vaccine and the effect of the manufacturing process on the size, quantity and biological activity of the residual cellular DNA fragments15 and are product specific.16
Current recommendations regarding the level of residual cell-substrate DNA for vaccines produced in cell lines derived from human tumors are ≤10 ng per dose and a median DNA size of 200 bp or lower.17 Similar FDA expectations were reported for adenoviral vector gene transfer products.18
Regulators have also specified that the amount of host cell proteins (HCP) needs to be qualitatively and quantitatively analyzed in the final vaccine bulk17,19 and gene therapy products.20 Namely, there is a potential for HCP to induce immune response and/or cause side effects.
In the past and still today methods like density gradient ultracentrifugation, selective precipitation and ultrafiltration were/are used to purify virus preparations. Ultracentrifugation of viruses can be carried out in different density gradients like caesium chloride,21 sucrose22 or iodixanol.23,24 In general the method enables efficient purification and concentration of virus particles in one step, but requires expensive equipment, process times are long (with subsequent step of density gradient material removal by dialysis or size exclusion chromatography) and sometimes several cycles are needed21,25 to reach the required virus purity. In addition, some density gradients are not suitable for certain viruses. For example, high viscosity and hyper-osmotic property of sucrose can cause damage to the extremely labile virus24 leading to loss of virus infectivity and unsatisfactory infectious virus yields.25 Another problem is extensive virus aggregation during gradient ultracentrifugation which was observed by Peng et al.23 when trying to purify recombinant adeno vectors in CsCl density gradient.
Selective precipitation uses different chemical agents like ammonium sulfate and polyethylene glycol25 to precipitate either the virus or the impurities. However, this method is not very suitable for preparative scale virus purification/production; it is especially challenging to carry out this method in a batch mode under cGMP conditions. Both, density gradient ultracentrifugation and selective precipitation are difficult to scale up and can suffer from low virus yields.
Ultrafiltration, a filtration method based on molecular weight differences between the target compound and impurities can be designed as a very robust method resulting in high virus yields. The scale up of the method to the industrial level is relatively straightforward and if coupled with diafiltration also provides a buffer exchange step. However, precipitation and ultrafiltration alone cannot deliver a product of sufficient purity and need to be combined with other techniques.
Chromatography was recognized as a method which can meet the challenges regarding virus preparation purity. It proved to be very efficient for protein based pharmaceuticals and has an advantage of easy scale up from laboratory to the preparative scale. However, traditional chromatography media were not designed and optimized for purification of large entities like virus particles, while the new generation chromatography media, CIM monolithic columns, as already previously described, were. The superiority of CIM monolithic columns in terms of virus recovery compared to particle based and/or membrane based resin for purification of different viruses like lentivirus and influenza virus10,13 was already presented.
Apart from high virus yield and efficient removal of impurities, also high dynamic binding capacity (DBC) of the chromatographic supports used for virus purification is of great importance. The higher the DBC of the chromatography column, the better the economics of the process is. If column capacity for a particular virus material is high, the size of the column needed to produce sufficient amount of pharmaceutical is smaller, which consequently leads toward smaller costs for the column itself and to smaller footprint of the column in the production plant. Although the DBC of a resin for a particular virus material depends on the purification steps applied prior to chromatography step, the superiority of CIM monolithic columns compared to other resins in terms of virus binding capacity was already demonstrated for different viruses.8,10 Segura et al.8 compared CIM DEAE monolithic column and Fractogel DEAE resin for purification of canine adenovirus vector and showed that CIM DEAE monolithic column has 16 times higher DBC. Dynamic binding capacity for a certain virus material can also depend on the chemistry selected for virus purification11 although the same conditions for virus binding are used. Therefore DBC of the resin for a particular virus material can serve as additional criteria when searching the optimal resin/column chemistry. Dynamic binding capacities up to 8.2E + 13 pfu/ml for different phage materials were previously reported26-28 and very high dynamic binding capacity of CIM monolithic columns for adenovirus was presented by Seigneuret et al.29 The authors developed a single step chromatography purification process based on a CIM monolithic column on a small scale and scaled up the process to 80 ml CIM column. They reported that further scale up of the process would be able to produce up to 1E + 15 recombinant adenovirus particles in a single batch. Table 1 summarizes dynamic binding capacities and virus recoveries achieved with different chromatography supports for different virus (-like) materials.
Table 1.
Dynamic binding capacity and virus recovery on different chromatography supports for different virus (-like) materials
| Nanoparticle | Type | Capacity | Recovery | Reference |
|---|---|---|---|---|
| Influenza | CIM QA monolith | 2E + 10 TCID₅₀/mL | 70%–87% 42% |
11 12 |
| Mustang Q | 2E + 10 TCID₅₀/mL | 40% | 12 | |
| Q Sepharose XL | 1E + 9 TCID₅₀/mL | 24% | 12 | |
| Adenovirus 5 | Q Sepharose XL | 4E + 13 VP/mL | n/d | 4 |
| Sartobind Q | 1.5E + 13 VP/mL | 60% | 73 | |
| Canine Ad 2 (CAV2) | Fractogel EMD propyl (capture) | 4.5E + 10 VGCN/mL | 77%–100% | 8 |
| CIM DEAE monolith (polishing) | 7E + 11 viral VGCN/mL | 58%–77% | 8 | |
| Adenovirus 3 VLP | CIM QA monolith | 1.3E + 16 VP/mL | 52% | 42 |
| Lentivirus | CIM DEAE monolith | >8E + 7 IP/mL | 80% | 10 |
| Q membrane | n/d | 70% | 74 | |
| Retroviral vectors | DEAE Sepharose FF | >3E + 7 cfu/mL | 53% | 75 |
| Flavivirus | CIM QA monolith | 4.5E + 9 FFU/mL | 72% | 76 |
| Mustang Q membrane | n/d | 36% | 76 | |
| Capto Q resin | n/d | 37% | 76 | |
| Baculovirus | CIM QA monolith (capture) | 2.4E + 11 pfu/ml | up to 99% | 77 |
| Sartobind D membrane | 7.7E + 10 TP/mL | 65% | 78 | |
| Capto DEAE resin | n/d | 33% | 78 | |
| Rotavirus | CIM QA monolith | 4E + 12 VP/mL | n/d | 79 |
| Rotavirus like particle | Resource Q | n/d | 50% | 80 |
| Rotavirus like particle | Sartobind D membrane (capture) | n/d | 55% | 81 |
| Rotavirus like particle | Sartobind D membrane (concentration) | n/d | 97% | 81 |
| T7 bacteriophage | CIM QA monolith | 1E + 13 pfu/mL | 100% | 25 |
| M13 bacteriophage | CIM QA monolith | 4.5E + 13 pfu/mL | 100% | 25 |
| Lambda bacteriophage | CIM QA monolith | 1E + 13 pfu/mL | 100% | 25 |
| PRD1 bacteriophage | CIM DEAE/QA monolith | 6E + 13 pfu/ml | 60%–80% | 27 |
Although new generation chromatography media are very efficient, chromatography based purification of viruses intended for human use still represents only a part of the virus downstream processing (DSP) scheme which usually consists of several purification steps and needs to deliver a virus preparation of high purity and efficacy (e.g., ratio between virus particles and infectious units (VP: IU) in case of adenovirus products) for a specific biopharmaceutical. General scheme of virus production process with emphasis on DSP part is illustrated in Figure 3.
Figure 3.
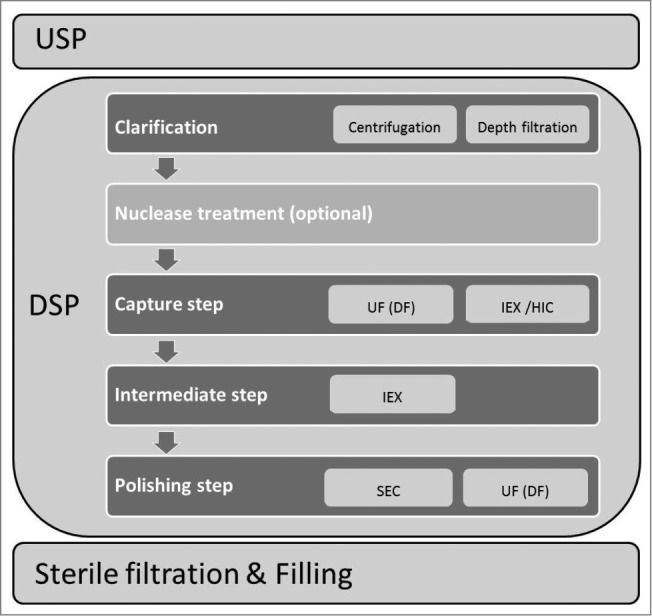
General scheme of virus production process with emphasis on DSP part. Abbreviations: UF-ultrafiltration; DF-diafiltration; IEX-ion exchange; HIC-hydrophobic interaction mode; SEC-size exclusion chromatography.
The first step in virus particle DSP is usually clarification, providing cellular debris removal. It can be carried out with centrifugation (not widely used in the preparative scale) or depth filtration. Optimized clarification step can result in virus yields up to 90% as previously reported for lentivirus10 or even 100% as described for canine Ad vectors.8
The second step in the DSP scheme can incorporate a concentration step which is very important especially for low titer virus productions and can be performed with precipitation or ultrafiltration (UF; e.g. tangential flow filtration), and can be coupled with diafiltration (DF) for buffer exchange. Diafiltration is very important since it is usually followed by a chromatography step and enables the introduction of the virus into the appropriate buffer system and pH environment. In some cases the concentration step is placed also after the chromatography based purification step.10
Chromatography part of the DSP can consist of one or several steps, using different chromatography modes/chemistries depending on purity requirements of the final product and the type of nanoparticle applied. Purification with chromatography methods can be either based on size (size exclusion chromatography; SEC)-usually used as a polishing step in the virus DSP, hydrophobicity (hydrophobic interaction chromatography, HIC), (pseudo) affinity30-33 or charge (anion/cation exchange chromatography).
The most widely used chromatography mode for the capture step of industrial scale virus DSP is charge specific separation where viruses are separated from impurities on the basis of the difference in their surface charge distribution. Since majority of viruses have isoelectric points below 6, they efficiently bind to anion exchange matrixes at neutral pH. However, also majority of contaminants bind to anion exchangers at neutral pH, therefore the separation of viruses from impurities is performed by selective elution. Elution is carried out by changing the pH of the buffer (not widely used as viruses are pH sensitive) or by increasing the ionic strength of the buffer (salt concentration in the buffer). Viruses bind to cation exchangers when low pH buffers (below their isoelectric point) are used, but for human viruses, as they are sensitive to low pH, this usually is not an option. However, certain phages are more stable in low pH environment34 and in these cases using a cation exchanger is of great advantage, because host cell DNA (a very important impurity needed to be removed) does not bind to the cation exchanger and is therefore removed very efficiently.
Some viruses bind to both anion and cation exchange matrixes at neutral pH,11 although their isoelectric point is below 6. This phenomenon can be explained by the fact that viruses are large macromolecular assemblies with non-uniform surface charge distribution. Clusters of positively/negatively charged moieties on the viral surface enable the binding of these large entities on 2 differently charged matrixes under the same conditions.35,36 In such cases impurity depletion is excellent, as cation exchanger efficiently removes host cell DNA, while anion exchanger removes host cell proteins.
New generation chromatography media, CIM monolithic columns were used in DSPs of different viruses like influenza11,13,37 adenovirus,8,29 adeno-associated virus,9,38 lentivirus,10,39 rubella,40 different phages26-28,41,42 and different VLPs6,43 with good virus/phage/VLP recoveries while reaching high dynamic binding capacities at the same time.
When host cell DNA (HC DNA) concentration after chromatography purification of virus material is above the limit set by regulatory authorities or the size of DNA fragments is above the specifications additional steps to lower the amount of DNA and decrease its size need to be incorporated into the DSP. Such a step (deoxiribonuclease treatment) can be performed before or after chromatography purification step; by placing this step early in the DSP scheme, for example prior to UF/DF, the latter enables also partial removal of DNA fragments that result from the deoxiribonuclease treatment.
However, if the chromatography step enables efficient HC DNA removal it is preferred that no deoxiribonuclease is used in the DSP as DNA fragmentation and size reduction can increase DNA content in the final virus pool after chromatography step. Additionally, deoxiribonuclease has to be removed from the final product intended for human use.13
The incorporation of DNA degradation step into the downstream process of phages was studied by Smrekar et al.44 They have investigated the influence of DNase on the purification process of lytic phages and reported that addition of DNase lowers process productivity because of long degradation time and due to the risk of non-completed DNA degradation resulting in co-elution of the phage and DNA fragments. However, the success of DNA degradation step depends on the elution profile of the phage; while complete DNA degradation approach might be an option for T4 phage, elution profile of T7 phage allows complete separation of phage from host cell DNA only when DNA is still intact.44 Chromatography step in the DSP should also, if possible, enable separation of infectious particles from non-infectious particles. The ratio between virus particles to infectious units (VP: IU) is a well-accepted and required component for quality control of any adenoviral vector and it is also an indicator of product potency.45 The VP:IU ratio of 30:1 is recommended by the Food and Drug Administration (FDA).45 The capacity of CIM monolithic columns to separate empty from full capsids was already demonstrated by Lock et al. for adeno-associated virus.9
The last step in the DSP (polishing step) should provide final impurity removal, additional concentration of the active substance (nanoparticle) and buffer exchange into final formulation. This final step can be performed by applying size exclusion chromatography (SEC) or UF/DF. SEC is usually performed in a group mode separation where the virus elutes in the void volume, while all remaining low molecular weight impurities elute later on. Optimized SEC method can result in high virus yields and it is very efficient in simultaneous removal of residual impurities and buffer exchange.13 Bandeira et al.10 increased lentivirus yield with optimization of SEC as a polishing step from 27% to 70% and up to 100% virus yields were reported for Influenza virus purification with SEC.13,37 However, SEC does not enable virus concentration and there are limitations regarding the flow rates of the mobile phases and the loading volume of the sample; usually up to a few percentages of the SEC column volume can be loaded in a single run. On the other hand UF/DF does not have these limitations and also enables final concentration of the virus and it is therefore used in many virus DSPs.
At the end of the virus DSP additional sterile filtration step can be introduced; UF concentrate or SEC eluate is filtered through 0.2 μm filter for final removal of bioburden.
Chromatographic Analytical Methods in Virus Production
The final viral product intended for gene therapy and vaccine applications needs to be well characterized and of proper quantity, purity and potency. This can be achieved only if adequate characterization methods are in place and the viral production process is well established, understood and reproducible. Sample control and product characterization are not important only in the final product but also during different steps within the production process. In depth knowledge of both up- and downstream processing is crucial since the up-stream production greatly affects the DSP process and sometimes even slight changes in the USP can result in less efficient DSP. The USP therefore needs to be monitored and the harvest material well characterized before it enters the DSP. The DSP consists of several subsequent steps where monitoring of viral concentration and sample components is important as predefined final products are needed. Since the Process Analytical Technology (PAT) initiative was presented in 2004 by the FDA,12 a lot of emphasis has been put on the in depth understanding of the production processes itself. The initiative encouraged pharmaceutical companies to increase research and use of new analytical technologies to perform timely measurements of the critical quality attributes of raw materials and intermediates allowing for better process understanding and control. The desired goal of PAT is to design and develop well understood processes that will ensure a predefined quality of the product at the end of the manufacturing process. A satisfactory level of understanding the manufacturing process and assuring control of the final viral product can only be achieved when accurate and reproducible characterization assays for monitoring are in place. In the production process of viruses thorough product evaluation is needed to determine viral potency, identity, quantity, residuals (i.e., Triton and deoxiribonuclease), aggregation, empty capsids, protein content and safety. There are different characterization assays available and their applicability depends on the type of virus purified as well as the expression system used in the USP.
Infectious titer assays are considered to be representative for quantifying active, infectious viral particles. Standard plaque assay or tissue culture infectious dose (TCID50) assays using complementing cell lines can be employed to assess infectious titers. These are laborious and time consuming methods that lack accuracy and precision.
The quantity of viruses can be measured using a variety of different methods based e.g., on determining the viral genetic material (quantitative Polymerase Chain Reaction, qPCR), the ability of viruses to agglutinate red-blood cells (Haemagglutination Assay, HA) protein-antibody interactions (Enzyme-Linked ImmunoSorbent Assay, ELISA and Single Radial Immunodiffusion SRID) or counting viral particles (Transmission Electron Microscopy, TEM and Dynamic Light Scattering, DLS, new technologies such as ViroCyt and Nanosight devices) as well as measuring the optical density (OD) of pure virus preparations. Purity is determined via Polyacrylamide Gel Electrophoresis (SDS-PAGE) using different types of staining, Western blot is used for identification of specific viral proteins, DNA and protein specific tests are used for host cell residuals.
As already mentioned most of the methods described above are laborious and time consuming, have questionable accuracy and are not precise. As an alternative, chromatographic methods, based on absorbance or fluorescence measurement of viral products and impurities started to emerge for process monitoring (quantification of viruses, detection of impurities). Chromatographic methods are rapid, reproducible and accurate. They cannot be applied for measuring infective viral particles, but are a good alternative to traditional methods when it comes to total virus particle quantitation since the resolving power of high-performance liquid chromatography (HPLC) permits separation of intact virus particles from other cellular contaminants or virus particle fragments. HPLC methods can therefore be applied for monitoring the impurity/purity profiles of viral products in DSP as well as the USP. They can be used as fingerprinting methods where the purpose is to take a snap-shot of a certain process step, and rapidly proceed to the next step in the process, without having to wait for days for all the standard analytics to come through in order to make further decisions on process development. Fingerprinting methods provide information that is needed for reaching a decision point within a certain process, e.g. when is the right time to stop the fermentation or when to stop collecting a sample in a chromatographic run but do not necessarily provide information about virus quantity.
When purified virus preparations that can be used as standards are available, HPLC method can be applied for virus quantification yielding results that are far more precise (RDS values usually vary from 1–10%) than in the case of biological assays mentioned above.46-49 However results obtained from HPLC analytics are accurate and precise when chromatographic columns are performing properly. Opposed to preparative chromatographic columns that are often single use columns or used just for a few times several thousand injections of different samples can be performed on a single analytical column. Column performance and lifetime of the columns must therefore be monitored to assure reliable results and a type of system suitability test is greatly advised. Various system suitability tests can be applied for examining column performance such as pulse response tests, backpressure monitoring, capacity tests or the ability of the column to separate a mixture of 2 or more different compounds (e.g., proteins). The column performance must be examined at point zero (before any viral sample is injected on the column–a reference point for determining proper column performance) and after a certain number of viral sample injections. The lifetime of every column depends on the complexity/purity of the injected virus samples, the type of virus itself and the pretreatment of the sample prior to injection (e.g., filtration, addition of detergents, organic solvents, etc). The lifetime of the column can be prolonged with proper cleaning in place procedures that are usually provided by column producers as well as proper storage of the column.
Due to their features, monolithic columns offer great advantages when it comes to HPLC analytics of viruses, VLPs and phages. Active binding sites for molecules are situated in large flow-through channels and are fully accessible; as a result the resolution power is extremely high. Due to the convective mass transfer between the mobile and stationary phase, column performance is not affected by flow-rate, and fast chromatographic methods can be developed. Since the channels within the monoliths are large (1–2 um), processing of complex samples is not an issue and they can be applied for monitoring harvest, intermediate and final product samples in the DSP process as well as in the USP part of virus production.
The effective use of HPLC analytics in the up-stream production of bacteriophages was reported by Smrekar et al.44 as they demonstrated the importance of up-stream optimization for further downstream processing of bacteriophages. An indirect analytical method for on-line monitoring of phage titer was developed and applied for the termination of lytic phage cultivation. In order to achieve high phage purity and quantity at the end of phage production process determination of phage cultivation proved to be essential. If optimal cultivation termination was missed, the DSP could not have been done properly and the phage fraction in the DSP process was contaminated with DNA released from E.coli cells. Using a rapid 5 minutes long analytical method on a CIM DEAE column and a Tris buffering system, fermentation of phages was monitored. Crude T4 and T7 lysate samples were injected on the column and their chromatographic profiles were examined (Fig. 4). A correlation curve between the amount of the released E.coli genomic DNA peak area and phage concentration obtained from a plaque assay was created. The correlation curve was derived already during the process development phase and implemented for fast monitoring of phage titer during its manufacturing as long as cultivation conditions were constant. While it gave no direct information about the phage titer its main advantage was that information on when to perform the harvest was obtained within few minutes and could therefore be used to decide if and how to proceed with the phage purification. DNA peak area and phage titer correlated closely until maximal phage titer was achieved. After that point, due to activities of DNases present in E.coli lysate, DNA concentration started to decrease while phage titer remained constant. The appearance of DNA fragments affected the phage DSP, since DNA fragments co-eluted with the phage fraction and as a consequence the final phage product was contaminated with fragmented host cell DNA. Cultivation monitoring and determination of cultivation termination using an HPLC analytical method was essential for achieving high phage quantity and pure final phage product. The same analytical method was applied for measuring the purity of the final phage product as well.44
Figure 4.
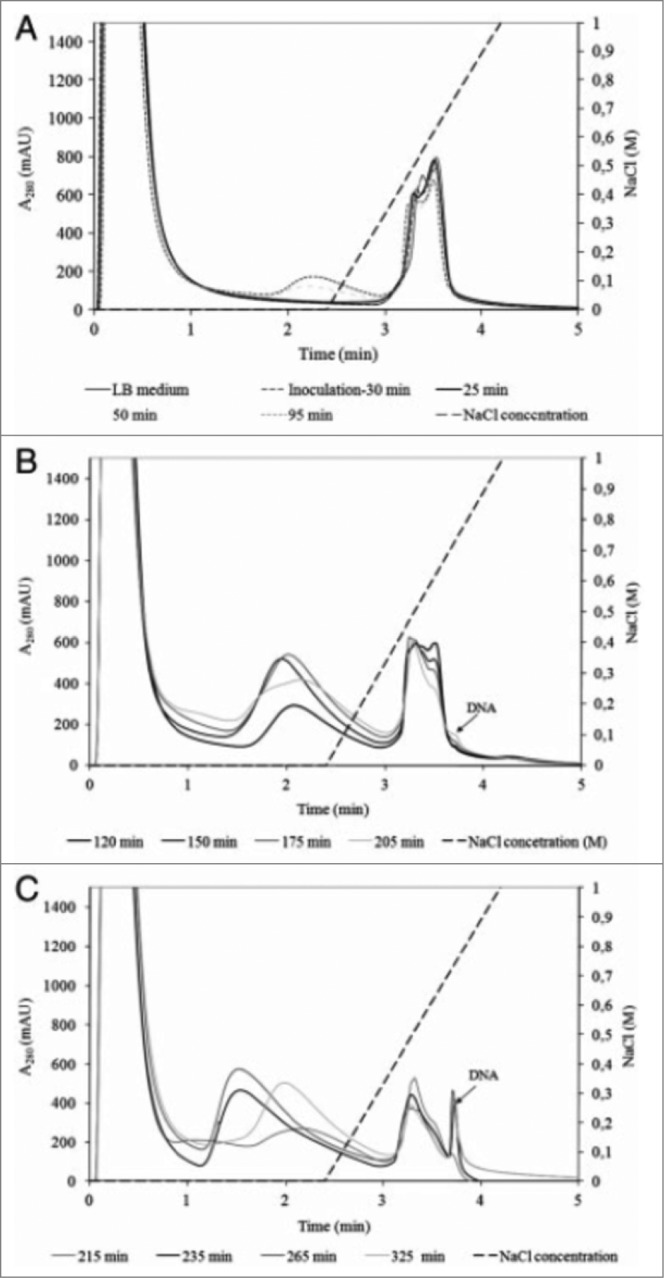
Analysis of crude T4 lysate collected during fermentation: (A) between 0 and 95 min; (B) between 120 and 205 min; (C) between 215 and 325 min. Conditions: Stationary phase: 0.34 mL CIM DEAE disk monolithic column; Mobile phase: Buffer A: 20 mM Tris, pH 7.5. Buffer B: 20 mM Tris, 1.0 M NaCl, pH 7.5; Gradient: linear gradient from 0 to 100% buffer B in 1.5 min; Flow rate: 4 mL/min; Injection volume: 1 mL; Detection: UV at 280 nm; reused from Smrekar et. al.44.
HPLC analytical methods can also be used as fingerprinting methods during DSP development providing rapid information on the efficiency of a particular process step and greatly contributing to faster DSP development. DSP process consists of several purification steps. After each step an aliquot of processed viral material needs to be collected and injected on an HPLC analytical column (Fig. 5). The elution profile provides a snap-shot of a particular purification step and a rough estimation on the virus content as well as the impurity profile of components present in the processed sample (e.g., DNA, host cell proteins). Urbas at al.43 used a CIMac analytical QA column to monitor the purity profiles of Ad3 VLPs after different steps of Ad3 VLPs purification. Estimation on host cell DNA, protein and VLPs presence in the material was obtained in terms of minutes.
Figure 5.
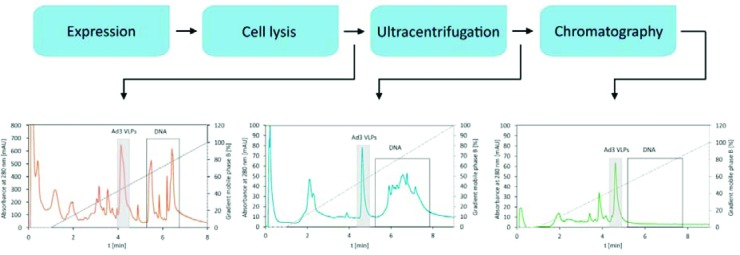
Fingerprint HPLC elution profiles of Ad3 VLPs purification process. Elution profiles depict the purity/impurity profiles of samples containing Ad3 VLPs after particular DSP steps.
When purified viral material (virus standard) is available, a quantitation method can be developed and used for determining the concentration of a particular virus, together with impurity profile monitoring. Provided that the sensitivity and resolution power of the method are adequate, in-process as well as final product quantitation can be performed.
Method using a monolithic column that enabled rapid process decisions as well as virus quantification during development of Ad5 DSP scheme for production of Ad5 particles intended for clinical applications was developed by Whitfield et al.46 using a Bio-Monolith QA HPLC analytical column. An example of Bio-Monolith QA analytical column application was an evaluation of centrifugation versus depth filtration for material clarification. Cellular lysate clarified by both methods was independently injected on the column. The peak profile of centrifuged material was different to that of material clarified by depth filtration. Analysis of centrifuged material demonstrated a mixture of species indicative of adenovirus and host cell components. By comparison, analysis of the same volume of material clarified by depth filtration displayed an appreciable reduction in the signal of all eluted species. This may have been due to filter fouling, although operational parameters did not indicate sub-optimal processing. As this stage was at the start of the purification process, the only other suitable analytical method to evaluate this process step would have been via an infectivity assay. As discussed, up to 7 days may be required to perform this analysis, over which duration the stability of the intermediate Ad5 particles is unknown. Application of the method, however, allowed a rapid decision to be made in favor of centrifugation rather than depth filtration in this instance. The method was also applied to ease the decision making in other stages during development of a purification platform. Once developed, the method was applied as an in-process assay to evaluate product purity. With the Bio-Monolith column it could be seen that he majority of protein or nucleic acid contaminants were removed by the primary capture chromatography step. The remaining contaminating proteins, detected in the flow through or during the first hold step, were removed during the final, polishing chromatography step. Product purity was demonstrated, as Ad5 particles were detected as a single peak after concentration and exchange into the final formulation buffer. Using a purified Ad5 standard the same method was qualified to enable quantification of Ad5 in the final product. Excellent repeatability (<1% RSD) was demonstrated for peak area, height and retention time of the virus peak at 260 nm. Linearity was determined by plotting the Ad5 concentration as a function of peak area. The working linear range was determined as 7.5 × 108 to at least 2.4 × 1010 VPs. A method for similar purposes was developed by Urbas et al.50 for monitoring the production and making fast process decisions for DSP of adenoviruses using a CIMac Adeno column. By changing the detection from UV to fluorescence the limit of quantitation was lowered for one order of magnitude reaching 7.5 × 107 VP. The same as in the case of Whitfied,46 the time of analysis was very short, and together with equilibration of the column lasted only 10 minutes. Before the introduction of monoliths, several chromatography based analytical methods have been developed for different viruses using particle based supports. The first HPLC quantification method for adenoviral vectors was developed on a Source 15Q column51 followed by a method on a Q Sepharose XL52 and Resource Q.49 Beside particle based supports methods were also developed on a continuous bed matrix support–UNO Q.49,47 It was however noticed that UNO Q was prone to fouling which was observed by increase of the pressure on the column. All of the described methods have similar detection limits and linear range but are almost 2 times longer than the methods using monolithic analytical supports.
Beside adenoviruses different HPLC methods for analytics of the influenza viruses have been developed, most of them based on the detection of haemagglutinin, the structural protein of the influenza virus and involve quite some preparation prior to HPLC analysis. Methods have been developed on different types of supports using different types of column chemistries. Phelan and Cohen53 managed to separate the major proteins of the detergent-disrupted influenza virus by RP-HPLC on conventional porous media, however, individual protein recoveries were low, as was the case with the ion-exchange chromatography.54 Size exclusion chromatography showed higher potential to separate viral proteins with recoveries >90%.54 A 2D HPLC method was reported for quantitative detection of virus proteins in vaccines, utilizing SEC and a non-porous silica RP column.55 Non-porous C18 silica-based column gave satisfactory results in terms of sensitivity, protein recovery and sensibility as opposed to conventional porous silica particle based C4 column.56 Kapteyn et al. developed an RP-HPLC method using a perfusion particle- based POROS column. The method is based on measuring the peak area of HA1, the hydrophilic subunit of HA, which is proportional to the amount of the analyzed HA.57,58 A similar method was developed on monoliths.59 Lorbetskie et al.60 improved the already established method55 from García-Cañas by detection of the virus via native fluorescence and gradient elution optimization, which resulted in 10 fold increase in sensitivity and excellent separation of different HA1 viral strains. Creskey at al developed61 a method for vaccine analysis using a BEH130 C18 RP-HPLC column that enabled simultaneous quantification of HA and NA levels much more rapidly than conventional HA quantification techniques, while providing additional valuable information on the total protein content. Enzymatically digested vaccine proteins were analyzed by LC–MSE, a mass spectrometric technology that allows absolute quantification of analytes, including the HA and NA antigens, other structural influenza proteins and chicken egg proteins associated with the manufacturing process. This method has potential application for increasing the accuracy of reference antigen standards and for validating label claims for HA content in formulated vaccines. It can also be used to monitor NA and chicken egg protein content in order to monitor manufacturing consistency.
The methods described above are all appropriate for final vaccine characterization and quantification, provided that appropriate viral standards are available. However, due to quite extensive sample preparation they are mostly not most suitable for in-process monitoring for the DSP.
Just recently Transfiguracion at al.62 developed an ion-exchange method for the quantification of influenza particles using a CIMac QA Analytical column using native fluorescence for detection. No significant sample preparation except for filtration prior to injection on the HPLC column is needed since the method is used for quantification of whole viral particles. The method was applied for quantification of 5 different strains of influenza particles and it is able to separate, identify and detect the influenza virus not only in ultracentrifugation purified samples but in cell culture supernatant samples as well making the method applicable for in-process analysis of influenza virus production in real time.Beside adenoviruses and influenza HPLC methods using different supports methods have been developed also for human Reovirus type 3 (UNOQ polishing column- in various stages of Reovirus purification,63,48 murine leukemia virus derived retroviral vectors,64 baculovirus particles,65 Chikungunya VLPs,66 Recombinant Hepatitis B Surface Antigen,67 polioviral proteins69,70 and proteins from human papilloma virus.71
Conclusion Remarks
Differences between viruses (virus strains), different upstream processes (expression systems) and different applications where these viruses are used make the development of a platform process for a DSP of a certain virus very challenging. However, similarities between different protocols for a particular virus can be observed4,11 and a general scheme or a so called semi-platform process can be proposed taking into account that fine tuning for each process step needs to be performed.
Once DSP is developed the reproducibility needs to be verified. According to the study conducted by TriMark DSP accounts for 75% to 80% of biopharmaceutical manufacturing operating costs.72 Semi-platform processes, together with rapid analytics that support process monitoring, enhance process reproducibility and enable production of pre-defined final products would definitely have positive effects on the process economics.
Disclosure of Potential Conflicts of Interest
The authors work for BIA Separations (Ajdovščina, Slovenia), the company that develops and manufactures CIM monolithic columns. However, this review presents different options for virus downstream processing (DSP) and process analytical technology (PAT) and advantages/disadvantages of different techniques are presented and referenced with published papers in a non-biased manner.
References
- 1. Wiley and Sons Gene Therapy Clinical trials Worldwide. Provided by the Journal of Gene Medicine. Updated June 2014, cited September 2014 Available from: http://www.abedia.com/wiley/vectors.php [Google Scholar]
- 2. Afeyan NB, Gordon NF, Mazsaroff I, Varady L, Fulton SP, Yang JB, Regnier FE. Flow-through particles for the high-performance liquid chromatographic separation of biomolecules: perfusion chromatography. J Chromatogr A 1990; 519 (1):1-29; http://dx.doi.org/ 10.1016/0021-9673(90)85132-F [DOI] [PubMed] [Google Scholar]
- 3. Wu Y, Simons J, Hooson S, Abraham D, Carta G. Protein and virus-like particle adsorption on perfusion chromatography media. J Chromatogr A 2013; 1297:96-105; PMID:23726244; http://dx.doi.org/ 10.1016/j.chroma.2013.04.062 [DOI] [PubMed] [Google Scholar]
- 4. Trilisky EI, Lenhoff AM. Sorption processes in ion-exchange chromatography of viruses. J Chromatogr A 2007; 1142:2-12; PMID:17240385; http://dx.doi.org/ 10.1016/j.chroma.2006.12.094 [DOI] [PubMed] [Google Scholar]
- 5. Mengran Y, Yan L, Songping Z, Xiunan L, Yanli Y, Yi C, Guanghui M, Zhiguo S. Improving stability of virus-like particles by ion-exchange chromatographic supports with large pore size: advantages of gigaporous media beyond enhanced binding capacity. J Chromatogr A 2014; 1331:69-79; PMID:24485037; http://dx.doi.org/ 10.1016/j.chroma.2014.01.027 [DOI] [PubMed] [Google Scholar]
- 6. Burden CS, Jin J, Podgornik A, Bracewell DG. A monolith purification process for virus-like particles from yeast homogenate. J Chromatogr B 2012; 880:82-9; http://dx.doi.org/ 10.1016/j.jchromb.2011.10.044 [DOI] [PubMed] [Google Scholar]
- 7. Štrancar A, Podgornik A, Barut M, Necina R. Short monolithic columns as stationary phases for biochromatography. In: Freitag R, editor. Modern advances in chromatography (Advances in Biochemical Engineering/Biotechnology). Berlin: Springer; 2002. 49-85 [DOI] [PubMed] [Google Scholar]
- 8. Segura MM, Puig M, Monfar M, Chillon M. Chromatography purification of canine adenoviral vectors. Hum Gene Ther Meth 2012; 23:182-97; http://dx.doi.org/ 10.1089/hgtb.2012.058 [DOI] [PubMed] [Google Scholar]
- 9. Lock M, Alvira MR, Wilson JM. Analysis of particle content of recombinant adeno-associated virus serotype 8 vectors by ion-exchange chromatography. Hum Gene Ther Methods 2012; 23:56-64; PMID:22428980; http://dx.doi.org/ 10.1089/hgtb.2011.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bandeira V, Peixoto C, Rodrigues AF, Cruz PE, Alves PM, Coroadinha AS, Carrondo MJ. Holmes. Downstream processing of lentiviral vectors: releasing bottlenecks. Hum Gene Ther Methods 2012; 23(4):255-63; PMID:22934827; http://dx.doi.org/ 10.1089/hgtb.2012.059 [DOI] [PubMed] [Google Scholar]
- 11. Banjac M, Roethl E, Gelhart F, Kramberger P, Lah Jarc B, Jarc M, Štrancar A, Muster T, Peterka M. Purification of Vero cell derived live replication deficient influenza A and B virus by ion exchange monolith chromatography. Vaccine 2014; 32:2487-92; PMID:24631091; http://dx.doi.org/ 10.1016/j.vaccine.2014.02.086 [DOI] [PubMed] [Google Scholar]
- 12. United States Food and Drug Administration (FDA) , Guidance for industry PAT—A Framework for Innovative Pharmaceutical Manufacturing and Quality Assurance, September 2004. [Google Scholar]
- 13. Peterka M, Štrancar A, Banjac M, Kramberger P, Maurer E, Muster T, inventors; von Kreisler Selting Werner, representative. Method for influenza virus purification. Chinese State Intellectual Property Office, CN101490248 B, 2013 May 1. [Google Scholar]
- 14. WHO: Weekly Epidemiological Record [Internet] Switzerland, WHO Geneva. No. 1, 2005;; 80: 1–8. Available from: http://www.who.int/wer [Google Scholar]
- 15. Grachev V, Magrath D, Griffiths E. Requirements for the use of animal cells as in vitro substrates for the production of biologicals. WHO: WHO Technical Report Series; 1998 56p. No. 878, Annex 1: 19-56. [DOI] [PubMed] [Google Scholar]
- 16. Petricciani J, Sheets R, Stacey G, Bar P. and Knezevic I. Recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological medicinal products and for the characterization of cell banks. WHO, October 2010 94p. Proposed replacement of TRS 878, Annex 1. [Google Scholar]
- 17. FDA Briefing Document Vaccines and Related Biological Products Advisory Committee Meeting. Cell Lines Derived from Human Tumors for Vaccine Manufacture. September 19, 2012.
- 18. Bauer SR, Pilaro AM, Weiss KD. Chapter 21—testing of adenoviral vector gene transfer products: FDA expectations. Adenoviral Vectors Gene Ther 2002; 615-54; http://dx.doi.org/ 10.1016/B978-012199504-1/50022-5 [DOI] [Google Scholar]
- 19. European Medicines Agency (EMA), Committee for Medicinal Product for Human Use (CHMP) Guideline on quality, non-clinical and clinical aspects of live recombinant viral vectored vaccines. EMA/CHMP/VWP/141697/2009. 2010 June 24, 14. [Google Scholar]
- 20. Food and Drug Administration (FDA) , Guidance for Industry: Guidance for Human Somatic Cell Therapy and Gene Therapy. US. Department of Health and Human Services, Food and Drug Administration Center for Biologics Evaluation and Research, March 1998 [Google Scholar]
- 21. Woo YJ, Zhang JCL, Taylor MD, Cohen JE, Hsu VM, Sweeney HL. One year transgene expression with adeno-associated virus cardiac gene transfer. Int J Cardiol 2005; 100(3):421-26; PMID:15837086; http://dx.doi.org/ 10.1016/j.ijcard.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 22. Mbiguino A, Menezes J. Purification of human respiratory syncytial virus: superiority of sucrose gradient over percoll, renografin, and metrizamide gradients. J Virol Methods 1991; 31(2-3):161-70; PMID:1650782; http://dx.doi.org/ 10.1016/0166-0934(91)90154-R [DOI] [PubMed] [Google Scholar]
- 23. Peng HH, Wu S, Davis JJ, Wang L, Roth JA, Marini FC, Fang B. A rapid and efficient method for purification of recombinant adenovirus with arginine–glycine–aspartic acid-modified fibers. Anal Biochem 2006; 354(1):140-7; PMID:16707084; http://dx.doi.org/ 10.1016/j.ab.2006.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gias E, Nielsen SU, Morgan LAF, Toms GL. Purification of human respiratory syncytial virus by ultracentrifugation in iodixanol density gradient. J Virol Methods 2008; 147(2):328-32; PMID:18029032; http://dx.doi.org/ 10.1016/j.jviromet.2007.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trépanier P, Payment P, Trudel M. Concentration of human respiratory syncytial virus using ammonium sulfate, polyethylene glycol or hollow fiber ultrafiltration. J Virol Methods 1981; 3(4):201-11; http://dx.doi.org/ 10.1016/0166-0934(81)90071-9 [DOI] [PubMed] [Google Scholar]
- 26. Smrekar F, Ciringer M, Štrancar A, Podgornik A. Characterisation of methacrylate monoliths for bacteriophage purification. J Chromatogr A 2011; 1218:2438-44; PMID:21238969; http://dx.doi.org/ 10.1016/j.chroma.2010.12.083 [DOI] [PubMed] [Google Scholar]
- 27. Adriaenssens EM, Lehman SM, Vandersteegen K, Vandenheuvel D, Philippe DL, Cornelissen A, Clokie MRJ, García AJ, Proft M, Maes M, et al. . CIM® monolithic anion-exchange chromatography as a useful alternative to CsCl gradient purification of bacteriophage particles. Virology 2012; 434(2):265-70; PMID: 23079104; http://dx.doi.org/ 10.1016/j.virol.2012.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oksanen HM, Domanska A, Bamford DH. Monolithic ion exchange chromatographic methods for virus purification. Virology 2012; 434:271-7; PMID: 23089255; http://dx.doi.org/ 10.1016/j.virol.2012.09.019 [DOI] [PubMed] [Google Scholar]
- 29. Seigneuret F, Enga M, Roblin A, Pennors M, Toublanc E, Douthe S, Darmon C, Moullier P, Mariau J, Brument N. CIM Monolithic matrix as an efficient and polyvalent tool to prepare and analyze a cGMP compliant batch of recombinant Adenovirus serotype 5. Poster session presented at: 5th Monolith Summer School & Symposium; 2012 June 1-6; Portorož, Slovenia. [Google Scholar]
- 30. Njayou M, Quash G. Purification of measles virus by affinity chromatography and by ultracentrifugation: a comparative study. J Virol Methods 1991; 32(1):67-77; PMID:1648573; http://dx.doi.org/ 10.1016/0166-0934(91)90186-4 [DOI] [PubMed] [Google Scholar]
- 31. Opitz L, Hohlweg J, Reichl U, Wolff MW. Purification of cell culture-derived influenza virus A/Puerto Rico/8/34 by membrane-based immobilized metal affinity chromatography. J Virol Methods 2009; 161(2):312-6; PMID:19591872; http://dx.doi.org/ 10.1016/j.jviromet.2009.06.025 [DOI] [PubMed] [Google Scholar]
- 32. Hu J, Ni Y, Dryman BA, Meng XJ, Zhang C. Purification of porcine reproductive and respiratory syndrome virus from cell culture using ultrafiltration and heparin affinity chromatography. J Chromatogr A 2010; 1217(21):3489-93; PMID:20371065; http://dx.doi.org/ 10.1016/j.chroma.2010.03.023 [DOI] [PubMed] [Google Scholar]
- 33. Ohtaki N, Takahashi H, Kaneko K, Gomi Y, Ishikawa T, Higashi Y, Todokoro M, Kurata T, Sata T, Kojima A. Purification and concentration of non-infectious West Nile virus-like particles and infectious virions using a pseudo-affinity Cellufine Sulfate column. J Virol Methods 2011; 174(1-2):131-5; PMID: 21440004; http://dx.doi.org/ 10.1016/j.jviromet.2011.03.021 [DOI] [PubMed] [Google Scholar]
- 34. Holmes DH. The Effects of Various Physical and Chemical Agents on a Staphylococcus Bacteriophage. Butler University Botanical Studies 1956; 13: Article 8. [Google Scholar]
- 35. Gagnon P. Purification Tools for Monoclonal Antibodies. Validated Biosystems, Inc., Tuscon, AZ, ZDA, 1996, 1-254. [Google Scholar]
- 36. Carta G., Jungbauer A, Downstream Processing of Biotechnology Products. In Protein Chromatography: Process Development and Scale-Up. Wiley-VCH, Weinheim, Germany, 2010; 1-55. [Google Scholar]
- 37. Maurer E, Peterka M, Gassner M, Seper H, Gelhart F, Banjac M, Jarc M, Lah B, Kramberger P, Štrancar A, et al. . Influenza virus purification platform. Poster session presented at: 3rd Monolith Summer School & Symposium; June 2008; Portorož, Slovenia. [Google Scholar]
- 38. Flotte TR, Trapnell BC, Humphries M, Carey B, Calcedo R, Rouhani F, Campbell-Thompson M, Yachnis AT, Sandhaus RA, McElvaney NG, et al. . Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing a1-antitrypsin: interim results. Hum Gene Ther 2011; 22:1239-47; PMID:21609134; http://dx.doi.org/ 10.1089/hum.2011.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lesch HP, Laitinen A, Peixoto C, Vicente T, Makkonen K-E, Laitinen L, Pikkarainen JT, Samaranayake H, Alves PM, Carrondo MJT, et al. . Production and purification of lentiviral vectors generated in 293T suspension cells with baculoviral vectors. Gene Ther 2011, 1-8; http://dx.doi.org/ 10.1038/gt.2010.162 [DOI] [PubMed] [Google Scholar]
- 40. Forcic D, Brgles M, Ivancic-Jelecki J, Šantak M, Halassy B, Barut M, Jug R, Markušić M, Štrancar A. Concentration and purification of rubella virus using monolithic chromatographic support. J Chromatogr B 2011; 879(13-14):981-6; http://dx.doi.org/ 10.1016/j.jchromb.2011.03.012 [DOI] [PubMed] [Google Scholar]
- 41. Smrekar F, Ciringer M, Peterka M, Podgornik A, Štrancar A. Purification and concentration of bacteriophage T4 using monolithic chromatographic supports. J Chromatogr B 2008; 861(2):177-80; http://dx.doi.org/ 10.1016/j.jchromb.2007.05.048 [DOI] [PubMed] [Google Scholar]
- 42. Kramberger P, Honour RC, Herman RE, Smrekar F, Peterka M. Purification of the Staphylococcus aureus bacteriophages VDX-10 on methacrylate monoliths. J Virol Methods 2010; 166:60-4; PMID:20188758; http://dx.doi.org/ 10.1016/j.jviromet.2010.02.020 [DOI] [PubMed] [Google Scholar]
- 43. Urbas L, Lah Jarc B, Barut M, Zochowska M, Chroboczek J, Pihlar B, Szolajska E. Purification of recombinant adenovirus type 3 dodecahedric virus-like particles for biomedical applications using short monolithic columns. J Chromatogr A 2011; 1218:2451-9; PMID:21295784; http://dx.doi.org/ 10.1016/j.chroma.2011.01.032 [DOI] [PubMed] [Google Scholar]
- 44. Smrekar F, Ciringer M, Jančar J, Raspor P, Štrancar A, Podgornik A. Optimization of lytic phage manufacturing in bioreactor using monolithic supports. J Sep Sci 2011; 34:2152-8. [DOI] [PubMed] [Google Scholar]
- 45. McIntyre M. Development of viral vectors for use in gene transfer trials; product characterization and quality concerns. Presented at: 8th Annual Meeting: Viral Vectors & Vaccines; November 2001; Lake Tahoe, Nevada. [Google Scholar]
- 46. Whitfield RJ, Battom SE, Barut M, Gilham DE, Ball PD. Rapid high-performance liquid chromatographic analysis of adenovirus type 5 particles with a prototype anion-exchange analytical monolith column. J Chromatogr A 2009; 1216:2725-9; PMID:19041094; http://dx.doi.org/ 10.1016/j.chroma.2008.11.010 [DOI] [PubMed] [Google Scholar]
- 47. Transfiguracion J, Bernier A, Arcand N, Chahal P, Kamen A. Validation of a high-performance liquid chromatographic assay for the quantification of adenovirus type 5 particles. J Chromatogr B 2001; 761:187-94; http://dx.doi.org/ 10.1016/S0378-4347(01)00330-9 [DOI] [PubMed] [Google Scholar]
- 48. Transfiguracion J, Berniera A, Voyera R, Coelho H, Coffey M, Kamen A. Rapid and reliable quantification of reovirus type 3 by high performance liquid chromatography during manufacturing of Reolysin®. J Pharm Biomed Anal 2008; 48:598-605; PMID:18632239; http://dx.doi.org/ 10.1016/j.jpba.2008.05.038 [DOI] [PubMed] [Google Scholar]
- 49. Klyushnichenko V, Bernier A, Kamen A, Harmsen E. Improved high-performance liquid chromatographic method in the analysis of adenovirus particles. J Chromatogr B 2001; 755:27-36; http://dx.doi.org/ 10.1016/S0378-4347(00)00597-1 [DOI] [PubMed] [Google Scholar]
- 50. Urbas L, Mersich C, Paril C, Barut M, Štrancar A. Monolithic columns for the purification, in-process and final control of chimpanzee adenoviruses. Presented at: 5th Monolith Summer School & Symposium; 2012 June 1-6; Portorož, Slovenia. [Google Scholar]
- 51. Shabram PW, Giroux DD, Goudreau AM, Gregory RJ, Hörn MT, Huyghe BG, Liu X, Nunnally MH, Sugarman BJ, Sutjipto S. Analytical anion-exchange HPLC of recombinant type-5 adenoviral particles. Hum Gene Ther 1997; 453-65; PMID:9054520; http://dx.doi.org/ 10.1089/hum.1997.8.4-453 [DOI] [PubMed] [Google Scholar]
- 52. Blanche F, Cameron B, Barbot A, Ferrero L, Guillemin T, Guyot S, Somarriba S, Bisch D. An improved anion-exchange HPLC method for the detection and purification of adenoviral particles. Gene Ther 2000; 7:1055-62; PMID:10871755; http://dx.doi.org/ 10.1038/sj.gt.3301190 [DOI] [PubMed] [Google Scholar]
- 53. Phelan MA, Cohen KA. Gradient optimization principles in reversed-phase high-performance liquid chromatography and the separation of influenza virus components. J Chromatogr A 1983; 266:55-66; http://dx.doi.org/ 10.1016/S0021-9673(01)90879-9 [DOI] [PubMed] [Google Scholar]
- 54. Calam DH, Davidson J. Isolation of influenza viral proteins by size-exclusion and ion-exchange high-performance liquid chromatography: the influence of conditions on separations. J Chromatogr 1984; 296:285-92; PMID:6480744; http://dx.doi.org/ 10.1016/S0021-9673(01)96422-2 [DOI] [PubMed] [Google Scholar]
- 55. García-Cañas V, Lorbetskie B, Bertrand D, Cyr TD, Girard M. Selective and quantitative detection of influenza virus proteins in commercial vaccines using two-dimensional high-performance liquid chromatography and fluorescence detection. Anal Chem 2007; 79:3164-72; http://dx.doi.org/ 10.1021/ac0621120 [DOI] [PubMed] [Google Scholar]
- 56. García-Cañas V, Lorbetskie B, Girard M. Rapid and selective characterisation of influenza virus constituents in monovalent and multivalent preparations using non-porous reversed-phase high performance liquid chromatography columns. J Chromatogr A 2006; 1123:225-32; http://dx.doi.org/ 10.1016/j.chroma.2006.04.003 [DOI] [PubMed] [Google Scholar]
- 57. Kapteyn JC, Saidi MD, Dijkstra R, Kars C, Tjon JC, Weverling GJ, de Vocht ML, Kompier R, van Montfort BA, Guichoux JY, et al. . Haemagglutinin quantification and identification of influenza A&B strains propagated in PER.C6® cells: a novel RP-HPLC method. Vaccine 2006; 24:3137-44; PMID:16490287; http://dx.doi.org/ 10.1016/j.vaccine.2006.01.046 [DOI] [PubMed] [Google Scholar]
- 58. Kapteyn JC, Porre AM, de Rond EJP, Hessel WB, Tijms MA, Kessen H, Slotboom AME, Oerlemans MA, Smit D, van der Linden J, et al. . HPLC-based quantification of haemagglutinin in the production of egg- and MDCK cell-derived influenza virus seasonal and pandemic vaccines. Vaccine 2009; 27:1468-77; PMID:19110022; http://dx.doi.org/ 10.1016/j.vaccine.2008.11.113 [DOI] [PubMed] [Google Scholar]
- 59. Urbas L, Košir B, Peterka M, Pihlar B, Štrancar A, Barut M. Reversed phase monolithic analytical columns for the determination of HA1 subunit of influenza virus haemagglutinin. J Chromatogr A 2011; 1218:2432-7; PMID:21251658; http://dx.doi.org/ 10.1016/j.chroma.2010.12.082 [DOI] [PubMed] [Google Scholar]
- 60. Lorbetskie B, Wang J, Gravel C, Allen C, Walsh M, Rinfret A, Li X, Girard M. Optimization and qualification of a quantitative reversed-phase HPLC method for hemagglutinin in influenza preparations and its comparative evaluation with biochemical assays. Vaccine 2011; 29:3377-89; PMID:21397719; http://dx.doi.org/ 10.1016/j.vaccine.2011.02.090 [DOI] [PubMed] [Google Scholar]
- 61. Creskey MC, Li C, Wang J, Girard M, Lorbetskie B, Gravel C, Farnsworth A, Li X, Smith DGS, Cyr TD. Simultaneous quantification of the viral antigens hemagglutinin and neuraminidase in influenza vaccines by LC–MSE. Vaccine 2012; 30:4762-70; PMID:22643214; http://dx.doi.org/ 10.1016/j.vaccine.2012.05.036 [DOI] [PubMed] [Google Scholar]
- 62. Transfiguracion J, Manceur AP, Petiot E, Thompson CM, Kamen A. Particle quantification of influenza viruses by high performance liquid chromatography. Vaccine 2014; 33(1):78-84; PMID:25448111; http://dx.doi.org/ 10.1016/j.vaccine.2014.11.027 [DOI] [PubMed] [Google Scholar]
- 63. Chahal PS, Transfiguracion J, Bernier A, Voyer R, Coffey M, Kamen A. Validation of a high-performance liquid chromatographic assay for the quantification of Reovirus particles type 3. J Pharm Biomed Anal 2007; 45:417-21; PMID:17692493; http://dx.doi.org/ 10.1016/j.jpba.2007.06.025 [DOI] [PubMed] [Google Scholar]
- 64. Transfiguracion J, Coelho H, Kamen A. High-performance liquid chromatographic total particles quantification of retroviral vectors pseudotyped with vesicular stomatitis virus-G glycoprotein. J Chromatogr B 2004; 813:167-73; http://dx.doi.org/ 10.1016/j.jchromb.2004.09.034 [DOI] [PubMed] [Google Scholar]
- 65. Transfiguracion J, Mena JA, Aucoin MG, Kamen A. Development and validation of a HPLC method for the quantification of baculovirus particles. J Chromatogr B 2011; 879:61-8; http://dx.doi.org/ 10.1016/j.jchromb.2010.11.011 [DOI] [PubMed] [Google Scholar]
- 66. Shytuhina A, Pristatsky P, He J, Casimiro DR, Schwartz RM, Hoang VM, Ha S. Development and application of a reversed-phase high-performance liquid chromatographic method for quantitation and characterization of a chikungunya virus-like particle vaccine. J Chromatogr A. Forthcoming 2014. http://dx.doi.org/ 10.1016/j.chroma.2014.05.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. O`Keefe DO, Paiva AM. Assay for Recombinant hepatitis B surface antigen using reversed-phase high-performance liquid chromatography. Anal Biochem 1995; 230:48-54; PMID:8585629; http://dx.doi.org/ 10.1006/abio.1995.1436 [DOI] [PubMed] [Google Scholar]
- 68. Urbas L, Brument N, Ruščić J, Marc D. CIMac analytical columns for in-process control of adenoviruses. Presented at: Engineering Conferences International, 2014 June 8-13; Paradisus Playa del Carmen, Mexico. [Google Scholar]
- 69. Heukeshoven J, Dernick R. Reversed-phase high-performance liquid chromatography of virus proteins and other large hydrophobic proteins in formic acid containing solvents. J Chromatogr 1982; 252:241-54; PMID:6304128; http://dx.doi.org/ 10.1016/S0021-9673(01)88415-6 [DOI] [PubMed] [Google Scholar]
- 70. Heukeshoven J, Dernick R. Reverse-phase high-performance liquid chromatography of virus proteins and other hydrophobic proteins. Chromatographia 1984; 19:95-100; http://dx.doi.org/ 10.1007/BF02687724 [DOI] [PubMed] [Google Scholar]
- 71. Yuan Y, Shane E, Oliver C. N. Reversed-phase high-performance liquid chromatography of virus-like particles. J Chromatogr A 1998; 816:21-8; PMID:9741097; http://dx.doi.org/ 10.1016/S0021-9673(98)00065-X [DOI] [PubMed] [Google Scholar]
- 72. TriMark Publications, LLC Bioseparation systems for global biopharmaceutical markets August 2013, TMRBIOS13-0801. [Google Scholar]
- 73. Peixoto C, Ferreira TB, Sousa MFQ, Carrondo MJT, Alves PM. Towards purification of adenoviral vectors based on membrane technology. Biotechnol Prog 2008; 24:1290-6; PMID:19194943; http://dx.doi.org/ 10.1002/btpr.25 [DOI] [PubMed] [Google Scholar]
- 74. Slepushkin V, Chang N, Cohen R, et al. . Large-scale purification of a lentiviral vector by size exclusion chromatography or Mustang Q ion exchange capsule. Bioproc J 2003; 2(5):89-95. [Google Scholar]
- 75. Rodrigues T, Carvalho A, Roldao A, Carrondo MJT, Alves PM, Cruz PE. Screening anion-exchange chromatographic matrices for isolation of onco-retroviral vectors. J Chromatogr B 2006; 837:59-68; http://dx.doi.org/ 10.1016/j.jchromb.2006.03.061 [DOI] [PubMed] [Google Scholar]
- 76. Mundle S, Anderson S, inventors; Michaud SM, agent Purification of flaviviruses. World Intellectual Property Organisation, WO 2013/106337 A1, 2013 July 18. [Google Scholar]
- 77. Gerster P, Kopecky EM, Hammerschmidt N, Klausberger M, Krammer F, Grabherr R, Mersich C, Urbas L, Kramberger P, Paril T, et al. . Purification of infective baculoviruses by monoliths. J Chromatogr A 2013; 1290:36- 45; PMID:23587319; http://dx.doi.org/ 10.1016/j.chroma.2013.03.047 [DOI] [PubMed] [Google Scholar]
- 78. Vicente T, Peixoto C, Carrondo MJT, Alves PM. Purification of recombinant baculoviruses for gene therapy using membrane processes. Gene Ther 2009; 16:766-75; PMID:19340018; http://dx.doi.org/ 10.1038/gt.2009.33 [DOI] [PubMed] [Google Scholar]
- 79. Gutiérrez-Aguirrea I, Banjac M, Steyer A, Poljšak-Prijatelj M, Peterka M, Štrancar A, Ravnikar M. Concentrating rotaviruses from water samples using monolithic chromatographic supports. J Chromatogr A 2009; 1216:2700-4; http://dx.doi.org/ 10.1016/j.chroma.2008.10.106 [DOI] [PubMed] [Google Scholar]
- 80. Peixoto C, Sousa MFQ, Silva AC, Carrondo MJT, Alves PM. Downstream processing of triple layered rotavirus like particles. J Biotech 2007; 127:452-61; http://dx.doi.org/ 10.1016/j.jbiotec.2006.08.002 [DOI] [PubMed] [Google Scholar]
- 81. Vicente T, Sousa MFQ, Peixoto C, Mota JPB, Alves PM, Carrondo MJT. Anion-exchange membrane chromatography for purification of rotavirus-like particles. J Membrane Sci 2008; 311:270-83; http://dx.doi.org/ 10.1016/j.memsci.2007.12.021 [DOI] [Google Scholar]