

Abstract
Ups1 forms a complex with Mdm35 and is critical for the transport of phosphatidic acid (PA) from the mitochondrial outer membrane to the inner membrane. We report the crystal structure of the Ups1-Mdm35-PA complex and the functional characterization of Ups1-Mdm35 in PA binding and transfer. Ups1 features a barrel-like structure consisting of an antiparallel β-sheet and three α-helices. Mdm35 adopts a three-helical clamp-like structure to wrap around Ups1 to form a stable complex. The β-sheet and α-helices of Ups1 form a long tunnel-like pocket to accommodate the substrate PA, and a short helix α2 acts as a lid to cover the pocket. The hydrophobic residues lining the pocket and helix α2 are critical for PA binding and transfer. In addition, a hydrophilic patch on the surface of Ups1 near the PA phosphate-binding site also plays an important role in the function of Ups1-Mdm35. Our study reveals the molecular basis of the function of Ups1-Mdm35 and sheds new light on the mechanism of intramitochondrial phospholipid transport by the MSF1/PRELI family proteins.
Keywords: cardiolipin synthesis, lipid transfer protein, mitochondrial morphology, phosphatidic acid transport
Introduction
Cellular membranes are largely comprised of lipids, and each membrane has its own characteristic lipid composition to ensure the cell normal function 1,2. Whereas lipids are predominantly synthesized in the endoplasmic reticulum (ER), they are non-randomly distributed to cellular membranes including subcellular organelles as well as leaflets of organelle membranes 1. Lipid redistribution from its synthesis site to destination could be accomplished by either vesicular or non-vesicular transport, and the non-vesicular transport, prevailing in cells, is mediated through lipid transfer proteins (LTPs) 3,4. There are many LTPs in cells, which can act alone or form complexes to extract specific lipids from donor membranes, accommodate the lipids in a hydrophobic pocket, and then transfer them through aqueous phase to the target acceptor membranes. Elucidation of the underlying molecular mechanisms of the functions of LTPs is critical for understanding the cell normal function and thus has attracted extensive studies 5,6,7,8,9,10.
Cardiolipins (CLs) are featured lipids of the inner membrane of mitochondria (MIM) and are required for its proper function 11,12. The majority of CLs are synthesized at the MIM using phosphatidic acids (PAs) as precursors, while the latter are mainly produced at the ER membrane which can be transported to the outer membrane (MOM) and further to the MIM of mitochondria by LTPs. Recently, a highly conserved protein complex Ups1-Mdm35 was identified in yeast to mediate the PA transport from the MOM to the MIM 13. Ups1 was initially identified as a member of the conserved MSF1/PRELI family proteins 14. Cells lacking Ups1 exhibit defective cell growth and decreased ratio of normal tubular mitochondria 15. Yeast has three Ups proteins, Ups1, Ups2, and Ups3, which share ∼25% sequence identity and are all located in mitochondrial inter-membrane space (IMS). Like Ups1, Ups2 and Ups3 may also form stable complexes with Mdm35 to function in metabolic regulation of mitochondrial phospholipids 13,16,17. In human, there are four Ups1 homologues named SLMO1 (slowmo homology 1), SLMO2, PRELI1 (Protein of Relevant Evolutionary and Lymphoid Interest 1), and PRELI2; and PRELI1 can rescue the growth defects of yeast Ups1Δ cells 14,15. Mdm35 is another IMS-localized protein that contains two Cx9C sequence motifs; and cells lacking Mdm35 show abnormal mitochondrial morphology, while the cell growth is not affected 17,18,19. At molecular level, Ups1-Mdm35 could serve as a PA-transfer protein complex in the IMS by presenting PA from the MOM to the CL synthetic cascade in the MIM 20,21. In addition, high concentration of CL can bind to Ups1 tightly to prevent its dissociation from MIM, which attenuates the PA transport and the CL accumulation. In human, PRELI was also found to bind to TRIAP1, a homologue of Mdm35, to form a complex which can transport PA in vitro; and loss of PRELI or TRIAP1 could impair the accumulation of CL in mitochondria and facilitate the release of cytochrome c, leading to cell apoptosis 22.
Here, we report the crystal structure of Ups1-Mdm35 bound with a PA molecule at 2.0 Å resolution. Structural and functional analyses reveal the molecular basis of Ups1-Mdm35-mediated PA binding and transfer and shed new lights on the mechanism of intramitochondrial lipid transport by the MSF1/PRELI family proteins.
Results
Structure of the Ups1-Mdm35-PA complex
Numerous attempts for expression and purification of soluble Mdm35 or Ups1 independently failed due to protein aggregation or formation of inclusion body. The homogenous Ups1-Mdm35 complex was purified in a 1:1 molar ratio when the two proteins were co-expressed together with a 6× His tag at the N-terminus of Ups1 (Fig1A). Structure of the Ups1-Mdm35 complex was solved by single-wavelength anomalous dispersion (SAD) method using a dataset (3.1 Å) collected from a native crystal soaked with sodium iodide (Table1). There are two Ups1-Mdm35 complexes containing eight iodide atoms per asymmetric unit (Fig EV1). The structure of Ups1-Mdm35 was refined against a 2.55-Å dataset. After model building and refinement, evident yet discontinuous electron density peaks were found at the putative lipid-binding site of Ups1 (Fig EV2), indicating that there is partial binding of lipid(s). To characterize the type of lipid(s) bound to Ups1-Mdm35, we extracted lipids from the protein complex and analyzed the lipid composition by LC-MS. The results show that there is indeed PA binding to Ups1-Mdm35, with 32:1 (16:0-16:1) PA and 34:1 (16:0-18:1) PA as the highest contents (Fig EV3). Since we did not add lipids in the purification system, the lipids bound with Ups1 are apparently from the expression system. To improve the occupancy of PA and get a PA-bound complex structure, we tried co-crystallization of Ups1-Mdm35 with several PAs of various lengths, including di 16:0, di 18:1, and 16:0–18:1 PAs, but only obtained co-crystals of the Ups1-Mdm35-PA (di 16:0) complex. The structure of Ups1-Mdm35-PA was solved to 2.0 Å resolution by molecular replacement using the Ups1-Mdm35 structure as a search model (Fig1B). The statistics of diffraction data collection and model refinement of Ups1-Mdm35 and Ups1-Mdm35-PA are summarized in Table1.
Figure 1.
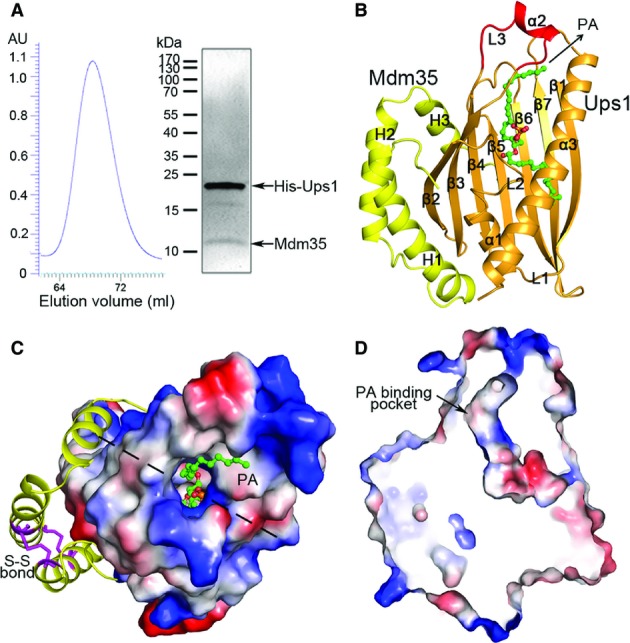
- Purification of the Ups1-Mdm35 complex. Left panel: gel filtration chromatography profile of Ups1-Mdm35; right panel: SDS–PAGE of the purified complex stained with Coomassie blue.
- Overall structure of Ups1-Mdm35-PA in ribbon cartoon. Ups1 is colored in orange and Mdm35 in yellow; the bound di 16:0 PA is shown with a ball-and-stick model in green.
- Structure of Ups1-Mdm35-PA viewed from the top. Ups1 is shown with electrostatic potential surface (blue and red colors represent positive and negative charges, respectively) with the L3 loop and the α2 helix omitted to show the PA-binding pocket more clearly. Mdm35 is shown with ribbon cartoon with the disulfide bonds shown with magenta sticks. The dotted lines indicate the cross section position of (D).
- A cross section drawing of the complex showing the PA-binding pocket in Ups1.
Data information: Structural data have been deposited with the Protein Data Bank (PDB) under accession numbers 4XHR for Ups1-Mdm35 and 4XIZ for Ups1-Mdm35-PA.
Table 1.
Statistics of data collection and structure refinement
| Ups1-Mdm35 | Ups1-Mdm35-PA | Soak_I | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.9791 | 0.9793 | 1.7000 |
| Space group | P21 | P21 | P21 |
| Cell dimensions | |||
| a, b, c (Å) | 43.0, 74.9, 87.6 | 43.0, 74.1, 87.9 | 42.9, 72.8, 87.5 |
| β (°) | 95.3 | 95.1 | 95.2 |
| Resolution (Å) | 50.0–2.55 (2.64–2.55)a | 50.0–2.00 (2.07–2.00) | 50.0–3.10 (3.21–3.10) |
| Unique reflections | 17,575 | 36,723 | 10,079 |
| Redundancy | 3.5 | 4.1 | 5.7 |
| I/sigma (I) | 12.0 (4.6) | 11.2 (4.9) | 21.7(13.4) |
| Completeness (%) | 97.2 (98.8) | 99.2 (97.8) | 99.0 (91.1) |
| Refinement | |||
| Rwork/Rfree (%)b | 23.0/27.7 | 19.6/24.0 | |
| Number of atoms | 3,887 | 4,328 | |
| Ligand | 92 | ||
| Water | 11 | 380 | |
| Protein residues | 484 | 481 | |
| r.m.s. deviations | |||
| Bond lengths (Å) | 0.026 | 0.009 | |
| Bond angles (°) | 1.4 | 1.3 | |
| Ramachandran | |||
| Favored (%) | 97 | 99 | |
| Allowed (%) | 100 | 100 | |
| Average B factor | 100.4 | 26.5 | |
| Ligand | 63.4 | ||
| Water | 69.4 | 31.7 | |
| Protein | 100.5 | 25.2 | |
Numbers in parentheses represent the highest resolution shell.
R = Σhkl||Fo| − |Fc||/Σhkl|Fo|.
Structure comparison of Ups1-Mdm35 and Ups1-Mdm35-PA reveals no significant conformational differences (rmsd = 0.39 Å); thus, we use the Ups1-Mdm35-PA complex for structure description and analyses. The structure model of the Ups1-Mdm35-PA complex comprises of residues 1–170 of Ups1, residues 6–75 of Mdm35, and one di 16:0 PA (Fig1B and C). Ups1 consists of an antiparallel β-sheet and three α-helices (α1–α3). The β-sheet is formed by seven β-strands with a connectivity of 2345671; strands β1 and β2 are intervened by helix α1 and connecting loops L1 and L2, and strands β3 and β4 are connected by a 16-residue loop L3, in the middle of which five residues form a short α-helix (α2). Strands β5, β6, and β7 follow β4 to complete the β-sheet. A long α-helix (α3) flanks the C-terminus of the β-sheet and is oriented diagonally across the β-sheet. There is a long tunnel-like pocket inside Ups1 to bind the PA molecule. Mdm35 is composed of three α-helices (H1–H3), and the relative conformation of H1 and H2 is defined by two disulfide bonds formed between Cys13 and Cys52, and Cys23 and Cys42. Mdm35 acts like a clamp to wrap around one side of the β-sheet of Ups1 (Fig1C).
The lipid-binding pocket
The lipid-binding pocket is formed by the concave β-sheet, the L2 loop, the L3 loop, and the helices α1-α3 of Ups1. The α1 helix seals the pocket from the bottom, and the small α2 helix acts like a lid to cover the pocket from the top. The upper part of the pocket has a cylinder shape of ∼8 Å in diameter and ∼28 Å in length and is hydrophobic in nature; the bottom part of the pocket has a bowel-like shape of ∼12 Å in both width and depth and is hydrophilic in nature (Fig1B and D). There is a small opening of ∼4 Å in diameter at the joint region of the upper and bottom parts of the pocket, which is also hydrophilic in nature. Comparing with the structure of Ups1-Mdm35 complex, the electron density of PA (di 16:0) in the structure of Ups1-Mdm35-PA complex is much improved (Figs EV2 and EV4). The upper part of the pocket has well-defined electron density which was modeled as sn-1 acyl chain of PA, and the joint region has fairly defined density which was interpreted as the phosphate group of PA. However, the bottom part of the pocket has fragmentary electron density peaks which did not allow us to model the sn-2 acyl chain of PA. We made extensive efforts including co-crystallization and soaking the crystals with PA liposome to further improve the occupancy and electron density of PA, but were unsuccessful so far. This might be due to unfavorable interactions between the hydrophobic sn-2 acyl chain of PA and the hydrophilic bottom part of the pocket (Figs1D and 2A and B).
Figure 2.

- Chemical structure of di 16:0 PA.
- The conformation of PA and its interactions with the surrounding residues inside the pocket. PA is shown with a green stick model, and the interacting residues are shown with side chains.
- Isothermal titration calorimetry analyses of the binding of PA with the Ups1-Mdm35 complex containing wild-type (WT) or mutant Ups1.
- Yeast (Ups1Δ) growth assay complemented with wild-type (WT) or mutant Ups1. Growth of the yeast cells was observed under five concentrations of 10-fold dilutions.
- Mitochondria morphology of the yeast cells (Ups1Δ) complemented with wild-type or mutant Ups1 was examined using confocal microscope. Left: differential interference contrast (DIC); right: fluorescence (Su9-GFP). Two different patterns of mitochondria were recorded in cells expressing wild-type Ups1 or Ups1Δ plus empty vector. Scale bars: 1 μm.
- Quantitation of the results of (E). The cells containing tubular mitochondria were scored. Data are presented as the means ± standard deviation (SD) of three independent experiments.
The sn-1 acyl chain of PA assumes an extended conformation and lies in the upper part of the pocket formed by a number of hydrophobic residues (Fig2B). C2-C6 of the sn-1 acyl chain are stabilized via hydrophobic interactions by residues Ile78 and Val106 that act like a clip to hold the acyl chain; C6-C8 interact with residue Asn97; and C11-C16 are stabilized by residues Trp144 and Met104. There is a ∼120-degree bend at C10 of the sn-1 acyl chain. Then, we analyzed the effects of mutations of these residues on the binding of PA using isothermal titration calorimetry (ITC) since Ups1 proteins containing V106E or I78D mutation could be co-purified with Mdm35 (Fig EV5). The results show that binding affinity of these two mutants with PA was substantially decreased compared with that of the wild-type (Fig2C). Previous studies showed that Ups1 regulates the sorting of Mgm1p that is required for fusion, inheritance, and morphology of yeast mitochondria and the deletion of Ups1 (Ups1Δ) in yeast could severely affect the cell viability and mitochondrial morphology 15,23,24,25,26, and we thus used complementary experiments to verify the functional importance of these residues. As expected, the Ups1Δ yeast cells transformed with empty vector show growth defect and abnormal mitochondrial morphology with aggregated or fragmented tubule (Fig2D–F). These abnormalities can be rescued by the wild-type Ups1 but cannot be rescued by the I78D, N97A, and V106E single mutants or the M104E/W144E double mutant. As a negative control, mutation of Ile103 (which is distant from the PA-binding pocket) to Glu has no effect on the growth and mitochondrial morphology. These results indicate that the residues constituting the lipid-binding pocket are important for the function of Ups1-Mdm35.
In the Ups1-Mdm35-PA structure, the small helix α2 of Ups1 covers the top of the lipid-binding pocket; therefore, the bound PA is barely accessible from the top. This implies that during substrate uptake or release, helix α2 and possibly other structural elements have to undergo conformational changes. Structural analyses reveal that helix α2 and the connecting L3 loop have much higher B factors than other regions, indicative of high flexibility (Fig3A). These properties of α2 are reminiscent of the functional lid in the yeast lipid transfer protein Osh4 6. To test whether helix α2 plays a similar functional role, we purified the Mdm35-Ups1 complex containing α2 deletion (residues 62–73 are deleted, and refer to as α2-Del), or Ala substitution (residues 62–73 are substituted with Ala, and refer to as α2-Ala) and carried out functional analyses (Fig EV5). The results show that compared with the wild-type protein, the binding ability of α2-Del or α2-Ala with PA is almost abolished (Figs2C and 3B), suggesting that α2 is essential for PA binding. Consistently, the in vitro lipid transfer activity of α2-Del or α2-Ala is dramatically decreased (Fig3C). Furthermore, both α2-Del and α2-Ala mutants cannot rescue the growth defect of Ups1Δ cells (Fig3D). Ups1Δ cells co-transformed with the α2-Del or α2-Ala mutant have only 2% of normal tubular mitochondria compared to 93% with the wild-type (Fig3E and F). Collectively, these results suggest that helix α2 may function as a lid of the lipid-binding pocket and plays important roles in PA binding and transfer.
Figure 3.
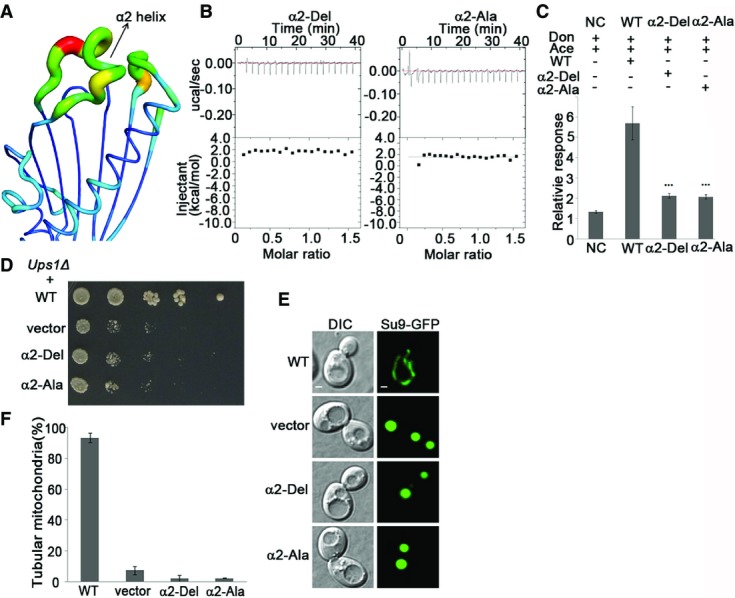
- A ribbon model of Ups1 is represented by the B factors of the residues to reflect their relative flexibilities. The increase of the B factors is represented by blue to red color. The α2 helix has the highest B factor.
- The binding ability of PA to α2-Del or α2-Ala Ups1 mutant as determined by ITC.
- The PA transfer ability of Ups1-Mdm35-containing wild-type (WT) or mutant Ups1 as analyzed by LC-MS. Each experiment was repeated five times. The stars above the columns indicate the significant difference of PA transfer ability between wide type and mutations (one-way ANOVA test). Values are mean ± SD.
- Yeast (Ups1Δ) growth assay complemented with wild-type (WT) or mutant Ups1.
- Mitochondrial morphology of the yeast cells (Ups1Δ) complemented with wild-type or mutant Ups1 was examined using confocal microscopy. Left: differential interference contrast (DIC); right: fluorescence (Su9-GFP). Scale bars: 1 μm.
- Quantitation of the results of (E). Values are the means ± SD (three independent experiments of (E) were performed and quantified).
The interaction interface between Ups1 and Mdm35
Unlike most other lipid transfer proteins, which function as monomer or homodimer, Ups1 needs to form a heterodimer with Mdm35 to transfer lipid. In the Ups1-Mdm35-PA structure, the interaction interface between Ups1 and Mdm35 buries 3,856 Å2 or 30% of the total surface area. The interactions involve helix α1 and strands β2–β5 of Ups1 and helices H1–H3 of Mdm35 and can be divided into three major regions (Fig4A and B). In region I, the guanidine group of Arg96 from β5-Ups1 forms three hydrogen bonds directly or via a water molecule with Ala65, Glu68, and Glu72 from H3-Mdm35. In region II, the side chain of Trp77 of β4-Ups1 makes hydrophobic contacts with Phe9 from H1 and Ile62 from H3 of Mdm35 and forms hydrogen bonds with Gln60 from H2 and Ile62 from H3 of Mdm35 through a water molecule. Interactions in region III are more extensive than those in regions I and II and involve H1 of Mdm35 and strands β2–β4 and helix α1 of Ups1. Specifically, Arg42 of β2-Ups1 forms hydrogen bonds with Tyr20 and Asp21 (directly or via a water molecule) and forms cation-π interactions with Phe24 of H1-Mdm35. In addition, Phe23, Leu50, and Vla84 of Ups1 form hydrophobic interactions with Phe24, Trp27, Tyr28, Phe32, and Leu33 of H1-Mdm35. Through these three interaction regions, Mdm35 acts like a clamp to wrap around Ups1 to form a stable Ups1-Mdm35 complex.
Figure 4.
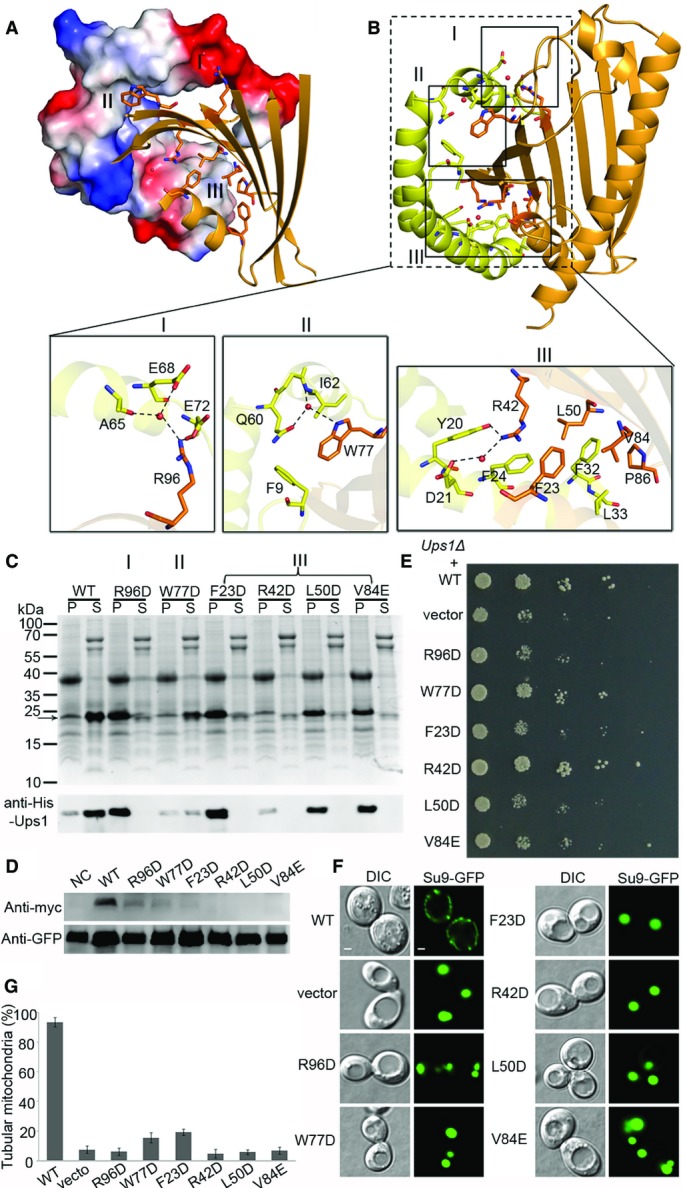
- Overview of the interaction interface of Ups1 and Mdm35. Mdm35 and Ups1 are shown with electrostatic potential surface and ribbon cartoon, respectively.
- Residues constituting the interaction interface (regions I, II, and III) are shown with side chains. The close-up views show the detailed interactions in regions I, II, and III.
- Purification of the Ups1-Mdm35 complex containing different Ups1 mutants. The upper panel shows the SDS–PAGE stained with Coomassie blue; the lower panel shows the Western blot results of Ups1 with anti-His antibody (TIANGEN, Cat No. AB102). Molecular markers are shown on the left. P: pellets of centrifugation of the lysed cells; S: elution samples after Ni2+-chelating purification.
- Detection of the protein level of Ups1 wild-type and mutants in mitochondria using Western blot. NC, negative control.
- Yeast (Ups1Δ) growth assay complemented with wild-type (WT) or mutant Ups1 containing mutations at the interaction interface with Mdm35.
- Mitochondrial morphology of the yeast (Ups1Δ) complemented with wild-type and mutant Ups1 was examined using confocal microscopy. Left: differential interference contrast (DIC); right: fluorescence (Su9-GFP). Scale bars: 1 μm.
- Quantitation of the results of (F). Values are the means ± SD (three independent experiments of (F) were performed and quantified).
As Ups1 and Mdm35 could not be individually expressed and purified and the soluble Ups1-Mdm35 complex could be obtained only by co-expression of the two proteins, apparently Mdm35 is required to stabilize Ups1 and facilitate its function in lipid binding and transfer. Thus, we used this feature to analyze the functional roles of the residues involved in the interactions between Ups1 and Mdm35. The results show that mutation W77D or R42D in Ups1 leads to substantially decreased amount of soluble Ups1-Mdm35 complex; and mutation R96D, F23D, L50D, or V84E in Ups1 leads to very little amount of soluble Ups1-Mdm35 complex (Fig4C). We also detected the protein level of wild-type and mutant Ups1 in the mitochondria via Western blot using anti-Myc antibody (Ups1 contains a C-terminal Myc tag). The results suggest that these mutations also lead to decrease of the Ups1 level in vivo (Fig4D). Then, we further analyzed the effects of these mutations on the function of Ups1-Mdm35 in vivo. As shown in Fig4E, the W77D and R42D Ups1 mutants can partially rescue the growth defect of Ups1Δ yeast cells, while all other mutations tested fail to rescue the growth defect. Consistently, the percentage of tubular mitochondria is reduced to below 20% in Ups1Δ cells containing the F23D Ups1 mutant and is decreased to background level in Ups1Δ cells containing the R42D, L50D, W77D, V84E, or R96D Ups1 mutant (Fig4F and G). These results together suggest that the residues at the interaction interface play critical roles in stabilization of the Ups1-Mdm35 complex, and their mutations may impair the formation and proper function of the complex.
A functionally important hydrophilic patch on Ups1
As discussed above, the lipid-binding pocket is connected to the outside through a small opening near the PA phosphate-binding site. Structural analysis shows that at the periphery of the opening, there are a number of basic residues including His33, Lys58, Lys61, Lys148, and Lys155, forming a large positively charged surface patch (Fig5A). To analyze whether this hydrophilic patch is of biological relevance, we constructed a series of mutations on this patch and tested their biological functions in vivo. The results demonstrate that the Ups1 mutant containing H33E, K58E, and K61E triple mutations (Ups1-3E) can partially rescue the growth defect of Ups1Δ cells; the Ups1 mutant containing H33E, K58E, K61E, K148E, and K155E quintuple mutations (Ups1-5E) cannot rescue the growth defect; and the effect of the Ups1 mutant containing H33E, K58E, K61E, and K148E quadruple mutations (Ups1-4E) lies in between. In addition, the Ups1 mutant containing H33A, K58A, K61A, K148A, and K155A mutations (Ups1-5A) can also partially rescue the growth defect of Ups1Δ cells (Fig5B). The effects of the above mutants on the mitochondrial morphology are consistent with the growth assay results: the normal tubular mitochondria of Ups1Δ cells containing the Ups1-5A mutant occupy about 50%, while those of Ups1Δ cells containing the Ups1-3E, Ups1-4E, and Ups1-5E mutants exhibit gradually decreased percentages with the Ups1-5E mutant being less than 6% (Fig5C and D). These data suggest that the hydrophilic patch plays an important role in the function of Ups1-Mdm35. It is possible that the positive charges of this patch might facilitate the Ups1-Mdm35 complex to interact with the phosphate group of PA or other lipids in the membrane.
Figure 5.
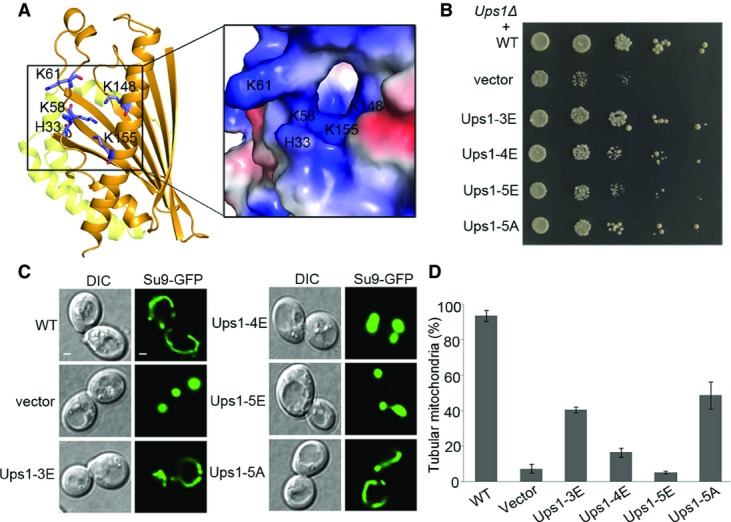
- Location of the hydrophilic patch in the Ups1-Mdm35-PA structure. A close-up view of the electrostatic potential surface of the patch is shown on the right.
- Yeast (Ups1Δ) growth assay complemented with wild-type (WT) and mutant Ups1 containing mutations in the hydrophilic patch. Ups1-3E: Ups1 mutant containing H33E, K58E, and K61E mutations; Ups1-4E: Ups1 mutant containing H33E, K58E, K61E, and K148E mutations; Ups1-5E: Ups1 mutant containing H33E, K58E, K61E, K148E, and K155E mutations; and Ups1-5A: Ups1 mutant containing H33A, K58A, K61A, K148A, and K155A mutations.
- Mitochondrial morphology of the yeast cells (Ups1Δ) complemented by wild-type and mutant Ups1 was examined using confocal microscopy. Left: differential interference contrast (DIC); right: fluorescence (Su9-GFP). Scale bars: 1 μm.
- Quantitation of the results of (C). Values are the means ± SD (three independent experiments of (C) were performed and quantified).
Discussion
Previous studies showed that Ups1 can bind with several negatively charged phospholipids including PA and CL, and CL can prevent the dissociation of Ups1 from the membrane 20. We tried to obtain the structure of Ups1-Mdm35 bound with CL, but have been unsuccessful so far. Thus, we docked a CL molecule into the Ups1-Mdm35 complex to analyze the potential interactions. The docking experiments using both Autodock and Haddock 27,28 converged to a similar model showing that two acyl chains of CL are bound in the lipid-binding pocket of Ups1, while the other two acyl chains go through the small opening to reside in a shallow groove on the surface patch downside the opening (Fig EV6). The phosphate groups lie along the edge of the opening and form hydrophilic interactions with the basic residues constituting the hydrophilic patch. This structure model is consistent with the observation that the size of the lipid-binding pocket in Ups1 can only accommodate PA but not CL. These results imply that when Ups1-Mdm35 binds to the CL-containing membrane, the lipid-binding pocket may bind only to two acyl chains of CL, while the other two acyl chains may remain in the membrane, thus anchoring the Ups1-Mdm35 complex to the membrane. This notion could explain the previous data showing that high concentration of CL can prevent Ups1 dissociation from the membrane.
As a representative of the MSF1/PRELI protein family, the Ups1 structure features a lipid-binding pocket that accommodates a PA molecule and the small α2 helix that acts as a lid to cover the top of the pocket, and forms a stable complex with Mdm35 through three interaction regions. Sequence alignment of the MSF1/PRELI family proteins shows that the residues constituting the interaction regions II and III of Ups1-Mdm35 and the hydrophobic residues constituting the lipid-binding pocket of Ups1 are conserved, suggesting that other members of the MSF1/PRELI protein family may form similar complexes with Mdm35/TRIAP and have similar lipid-binding mode (Fig EV7). In this issue, Miliara et al report another structure of MSF1/PRELI family protein complex-TRIAP1-SLMO1, which shares a similar conformation with Ups1-Mdm35 29. Our structural modeling studies of yeast Ups2, Ups3, and mammalian PRELI1 using the Ups1 structure as a template (Fig EV8) demonstrate that Ups2, Ups3, and PRELI1 may adopt similar overall structures except that Ups2 and PRELI1 have two extra α-helices at the C-terminus to form the LEA (late embryogenesis abundant) domain 30. Both the lipid-binding pocket and the α2 helix are found in the structure models; however, the volume and shape of the pockets are different. Intriguingly, the putative lipid-binding pocket of Ups2 in the structure model seems open to the outside. Although the substrates of these proteins remain unknown, our structure models suggest that they may have different substrate specificity. The characteristics of these predicted lipid-binding pockets may provide clues to identify the substrates of these proteins.
Cardiolipins are featured phospholipids of mitochondria and play essential roles in the normal mitochondrial morphogenesis, and thus, the processes of synthesis and regulation of CL are extremely important 31,32,33. As the precursor of CL, proper amount of PA should be transported from MOM to MIM through Ups1-Mdm35 complex 20. While the import of the “twin Cx9C” protein Mdm35 relies on the mitochondrial disulfide relay system 19,34, the translocation and stability of Ups1/2 depend on Mdm35 that forms complex with Ups1/2 in IMS 13,17,35. To initiate the PA trafficking, Ups1 may attach to the MOM through charge–charge interactions (i.e., via the hydrophilic patch around the lipid-binding pocket), and the opening of the lid α2 may allow Ups1 to capture PA molecule through the lipid-binding pocket. Once PA molecule binds into the pocket, helix α2 may undergo conformational change to close the pocket, and then, Mdm35 binds with Ups1 to form Ups1-Mdm35-PA complex and dissociates from MOM and translocates PA across the IMS to the MIM to release PA. This process may be dynamic and reversible. Since the synthesis of CL using PA occurs at MIM, the concentration of PA at MIM is usually lower than that at MOM, and Ups1-Mdm35 continues translocating PA from MOM to MIM; once the amount of CL at MIM is sufficient, CL may capture Ups1-Mdm35 complex at MIM to feedback-regulate PA trafficking (Fig EV9). In a word, the structural and functional analyses of the Ups1-Mdm35-PA complex provide the molecular basis for how Ups1 and Mdm35 form a stable complex to bind and transfer PA, and shed new lights on the feedback regulation of Ups1-Mdm35-dependent PA trafficking by CL at the mitochondrial inner membrane.
Materials and Methods
Cloning, expression, and purification of the Ups1-Mdm35 complex
Genes encoding Ups1 and Mdm35 were amplified by PCR from the genomic DNA of S. cerevisiae and were inserted into pETDuet vector. The constructs containing point mutations were generated by PCR and verified by DNA sequencing. Plasmid was transformed into E. coli Rosette Origami (DE3) strain for protein expression. The bacterial cells were grown at 37°C until OD600 reached to 1.2, and then, the protein expression was induced by adding 0.5 mM IPTG (isopropyl β-D-thiogalactoside) for 6 h. The cells were collected, resuspended in buffer A (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM PMSF, and 40 mM imidazole), and then lysed by high-pressure cell disruptor. The lysate was centrifuged, and the super-natant was loaded through a Ni-NTA column. After washing with buffer A, the protein was eluted with a linear gradient of imidazole. The peak fractions were collected and further purified by gel filtration using a Superdex G75 16/60 column (GE Healthcare) in buffer B (50 mM Tris/HCl, pH 8.0, and 100 mM NaCl). The proteins were collected and concentrated to about 20 mg/ml and stored at −80°C for further studies.
Crystallization, diffraction data collection, and structure determination
Crystallization was performed using the sitting-drop vapor-diffusion method at 4°C. Equal volumes (0.4 μl) of the protein solution (12 mg/ml) and the reservoir solution were mixed and equilibrated against 100 μl of the reservoir solution. Crystals of the Ups1-Mdm35 complex were grown in drops containing the reservoir solution of 2% (v/v) Tacsimate (pH 6.0), 0.1 M Bis-Tris (pH 6.5), and 20% (w/v) PEG 3350. To obtain crystals of the Ups1-Mdm35-PA complex, the purified protein was treated with 10-fold excess amount of PA-containing liposomes at 4°C in a buffer containing 50 mM Tris–HCl (pH 8.0), 100 mM NaCl, and 1 mM DTT. After incubation, the mixture was centrifuged at 18,000 g and the supernatant was used for crystallization. To determine the phases, the I− derivative crystals were prepared by soaking the native crystals in the crystallization solution containing 500 mM NaI for 1 min. Single-wavelength anomalous dispersion (SAD) diffraction data were collected to 3.1 Å resolution, and diffraction data for Ups1-Mdm35 and Ups1-Mdm35-PA were collected to 2.55 Å and 2.0 Å resolution, respectively. The diffraction data were processed with HKL2000 36.
The structure of Ups1-Mdm35 was solved by the SAD method implemented in Phenix 37 at 3.1 Å resolution and refined to 2.55 Å resolution. The structure of Ups1-Mdm35-PA was solved by the molecular replacement (MR) method implemented in Phenix 37 using the Ups1-Mdm35 structure as the template. The structure models were refined with Phenix and manually built with Coot 38. The statistics of the structure refinement and the structure models are summarized in Table1.
Isothermal titration calorimetry analysis
ITC experiments were performed with a MicroCal ITC200 system (Malvern) at 20°C in a buffer consisting of 50 mM Tris–HCl (pH 8.0) and 100 mM NaCl. Proteins were purified as described above; liposomes were prepared with 10% PA and 90% PC in molar ratio, and liposomes in the negative control were prepared with PC only. The syringe was filled with 280 μM of the protein complex, and the sample cell was filled with the liposome solution at a concentration of 40 μM. The protein solution was added to the liposome solution by sequential injections of 2-μl aliquots followed by 120 s of equilibration after each injection, and there were 20 injections in total. For analysis, the heat released by each injection was integrated, and the background was subtracted with the negative control. The data were fit to the Wiseman isotherm with the Origin ITC analysis package. The experiments were performed at least twice for each sample with similar results.
Yeast growth assay and confocal analysis
The open reading frame (ORF) of yeast Ups1 plus promoter region (1,000 bp away from the initial condon) and terminator region (500 bp away from the stop condon) was amplified from the S. cerevisiae genomic DNA and was cloned into pRS316 (CEN-Ura) to yield the pRS316-Ups1 plasmid. The Su9-EGFP gene and the ADH1 promoter were amplified from pYX232-mtGFP (Add Gene) and S. cerevisiae genomic DNA, respectively, and were inserted into pRS313 vector to yield the pRS313-Su9-GFP plasmid 39.
Saccharomyces cerevisiae Ups1Δ strain used in this study was derived from BY4742 (MATΔ his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0). To carry out the growth assay, pRS316 empty vector and pRS316-Ups1 (wild-type or mutants) were transformed into the Ups1Δ strain and grown on the plate containing synthetic-defined (S.D.) medium (2% w/v glucose, 0.17% w/v yeast nitrogen base without amino acid or ammonium sulfate, 0.5% w/v ammonium sulfate, 0.09% amino acid dropout mix, 1% succinic acid, and 0.6% w/v sodium hydroxide) lacking uracil plus 2% agar. To examine the mitochondrial morphology, pRS316-Ups1 (wild-type or mutants) and pRS313-Su9-GFP were co-transformed into the Ups1Δ strain and selected on S.D. medium lacking uracil and histidine. The resulting cells were grown to log phase and then harvested and suspended in 0.5% low-melt agarose for confocal analyses. The cells were examined using an Olympus X81 confocal microscope with an 100×/1.3NA objective.
Protein level detection of Ups1 and its mutants in yeast mitochondria
pRS313-Su9-GFP was co-transformed with pRS316-Ups1-Myc and its mutants (containing C-terminal Myc tag) to the S. cerevisiae Ups1Δ strain. Transformed yeast was cultured overnight and then harvested. Mitochondria were purified according to a previously reported protocol 40. The purified mitochondria were lysed and analyzed by SDS–PAGE and immunoblotting using anti-Myc and anti-GFP antibodies.
Liposome preparation
Lipids in stock solutions in chloroform were mixed at the desired molar ratio, and the solvent was evaporated under nitrogen to form lipid film. Large multilamellar liposomes were formed by rehydrating the lipid film in a rehydration buffer (50 mM Tris–HCl, pH 7.5, 100 mM NaCl, and 1 mM DTT) at 42°C for 1 h. Liposomes were extruded through a polycarbonate filter (400 nm or 100 nm) for 21 times using a mini-extruder (Avanti Polar Lipids).
In vitro lipid transfer assay
The lipid transfer assay was carried out using a method as described previously 7. The donor liposomes (400 nm extruded) were made of PC plus 10% PA (molar ratio) and rehydrated in the rehydration buffer containing 25% sucrose. Liposomes were collected by centrifugation at 16,000 g for 20 min and then washed with the rehydration buffer. The acceptor liposomes (100 nm extruded) were prepared with PC. The purified Ups1-Mdm35 complex (1 μM) was incubated with the donor and acceptor liposomes (2 mM total lipids) in 300 μl assay buffer (10 mM HEPES, pH 7.4, and 250 mM NaCl) at 25°C for 30 min. The supernatant (250 μl) was pooled after centrifugation at 16,000 g for 30 min. The lipids in the supernatant were extracted and quantified by mass spectrometry.
Lipid extraction and mass spectrometric analysis
Lipids were extracted from the purified protein sample or assay sample with a single-phase chloroform/methanol/H2O system 41. The extracted lipids were dissolved in 10 mM ammonium acetate in methanol and analyzed with HPLC-MS. Mass spectrometric analysis was done on a 5500 QTrap triple quadrupole mass spectrometer (AB Sciex, Germany). PA measurement was performed in positive ion mode. The internal standard 17:0-14:1 PA (Avanti, USA) was used for correction and relative quantification of PA in the samples according to peak intensity. Mass spectra were processed using the LipidView software version 1.1 (AB Sciex, Germany) for identification and quantification of lipids as described previously 20. Lipid amounts (relative ratio) were corrected for response differences between internal standard and lipid PA.
Data deposition
Structural data have been deposited with the Protein Data Bank (PDB) under accession numbers 4XHR for Ups1-Mdm35 and 4XIZ for Ups1-Mdm35-PA.
Acknowledgments
We thank the staff members at BL17U of Shanghai Synchrotron Radiation Facility (SSRF) and BL19U of National Center for Protein Science Shanghai (NCPSS) for technical assistance in data collection. We thank Dr. Jinqiu Zhou for providing us the yeast strain and vectors used in the growth assay. This work was supported by grants from the National Natural Science Foundation of China (31400650 to FY, 31322016 to PZ, and 31130060 to HX) and the Chinese Academy of Sciences (XDB08010302 to JD, and 2012OHTP to PZ), and funding from the National Key Lab of Plant Molecular Genetics, Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, CAS.
Author contributions
FY and FH performed the gene cloning, protein expression, purification, crystallization, ITC analysis, yeast growth assay, cell imaging, and lipid transfer assay; and CW, FH, and JW carried out the diffraction data collection, structure determination, and modeling overseen by PZ and JD. HY did the LC-MS lipid quantification experiment overseen by HX. JL and XQ contributed to the data analysis and discussion. All authors contributed to the experimental design, data analysis, and discussion. PZ, JD, and HX conceived the project and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Expanded View Figures PDF
Review Process File
References
- 1.Holthuis JC, Levine TP. Lipid traffic: floppy drives and a superhighway. Nat Rev Mol Cell Biol. 2005;6:209–220. doi: 10.1038/nrm1591. [DOI] [PubMed] [Google Scholar]
- 2.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lev S. Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat Rev Mol Cell Biol. 2010;11:739–750. doi: 10.1038/nrm2971. [DOI] [PubMed] [Google Scholar]
- 4.Prinz WA. Lipid trafficking sans vesicles: where, why, how? Cell. 2010;143:870–874. doi: 10.1016/j.cell.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ile KE, Schaaf G, Bankaitis VA. Phosphatidylinositol transfer proteins and cellular nanoreactors for lipid signaling. Nat Chem Biol. 2006;2:576–583. doi: 10.1038/nchembio835. [DOI] [PubMed] [Google Scholar]
- 6.Im YJ, Raychaudhuri S, Prinz WA, Hurley JH. Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature. 2005;437:154–158. doi: 10.1038/nature03923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M, Gavin A-C. Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature. 2013;501:257–261. doi: 10.1038/nature12430. [DOI] [PubMed] [Google Scholar]
- 8.Raychaudhuri S, Im YJ, Hurley JH, Prinz WA. Nonvesicular sterol movement from plasma membrane to ER requires oxysterol-binding protein-related proteins and phosphoinositides. J Cell Biol. 2006;173:107–119. doi: 10.1083/jcb.200510084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roderick SL, Chan WW, Agate DS, Olsen LR, Vetting MW, Rajashankar KR, Cohen DE. Structure of human phosphatidylcholine transfer protein in complex with its ligand. Nat Struct Biol. 2002;9:507–511. doi: 10.1038/nsb812. [DOI] [PubMed] [Google Scholar]
- 10.Simanshu DK, Kamlekar RK, Wijesinghe DS, Zou X, Zhai X, Mishra SK, Molotkovsky JG, Malinina L, Hinchcliffe EH, Chalfant CE, et al. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature. 2013;500:463–467. doi: 10.1038/nature12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santucci R, Sinibaldi F, Polticelli F, Fiorucci L. Role of cardiolipin in mitochondrial diseases and apoptosis. Curr Med Chem. 2014;21:2702–2714. doi: 10.2174/0929867321666140414112156. [DOI] [PubMed] [Google Scholar]
- 13.Potting C, Wilmes C, Engmann T, Osman C, Langer T. Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 2010;29:2888–2898. doi: 10.1038/emboj.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dee CT, Moffat KG. A novel family of mitochondrial proteins is represented by the Drosophila genes slmo, preli-like and real-time. Dev Genes Evol. 2005;215:248–254. doi: 10.1007/s00427-005-0470-4. [DOI] [PubMed] [Google Scholar]
- 15.Sesaki H, Dunn CD, Iijima M, Shepard KA, Yaffe MP, Machamer CE, Jensen RE. Ups1p, a conserved intermembrane space protein, regulates mitochondrial shape and alternative topogenesis of Mgm1p. J Cell Biol. 2006;173:651–658. doi: 10.1083/jcb.200603092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tamura Y, Endo T, Iijima M, Sesaki H. Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J Cell Biol. 2009;185:1029–1045. doi: 10.1083/jcb.200812018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamura Y, Iijima M, Sesaki H. Mdm35p imports Ups proteins into the mitochondrial intermembrane space by functional complex formation. EMBO J. 2010;29:2875–2887. doi: 10.1038/emboj.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herrmann JM, Riemer J. Mitochondrial disulfide relay: redox-regulated protein import into the intermembrane space. J Biol Chem. 2012;287:4426–4433. doi: 10.1074/jbc.R111.270678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longen S, Bien M, Bihlmaier K, Kloeppel C, Kauff F, Hammermeister M, Westermann B, Herrmann JM, Riemer J. Systematic analysis of the twin cx(9)c protein family. J Mol Biol. 2009;393:356–368. doi: 10.1016/j.jmb.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 20.Connerth M, Tatsuta T, Haag M, Klecker T, Westermann B, Langer T. Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science. 2012;338:815–818. doi: 10.1126/science.1225625. [DOI] [PubMed] [Google Scholar]
- 21.Tatsuta T, Scharwey M, Langer T. Mitochondrial lipid trafficking. Trends Cell Biol. 2014;24:44–52. doi: 10.1016/j.tcb.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 22.Potting C, Tatsuta T, Konig T, Haag M, Wai T, Aaltonen MJ, Langer T. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 2013;18:287–295. doi: 10.1016/j.cmet.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 23.Jones BA, Fangman WL. Mitochondrial DNA maintenance in yeast requires a protein containing a region related to the GTP-binding domain of dynamin. Genes Dev. 1992;6:380–389. doi: 10.1101/gad.6.3.380. [DOI] [PubMed] [Google Scholar]
- 24.Shepard KA, Yaffe MP. The yeast dynamin-like protein, mgm1p, functions on the mitochondrial outer membrane to mediate mitochondrial inheritance. J Cell Biol. 1999;144:711–720. doi: 10.1083/jcb.144.4.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sesaki H, Southard SM, Yaffe MP, Jensen RE. Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell. 2003;14:2342–2356. doi: 10.1091/mbc.E02-12-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong ED, Wagner JA, Scott SV, Okreglak V, Holewinske TJ, Cassidy-Stone A, Nunnari J. The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol. 2003;160:303–311. doi: 10.1083/jcb.200209015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AM. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- 28.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miliara X, Garnett JA, Tatsuta T, Abid Ali F, Baldie H, Pérez-Dorado I, Simpson P, Yague E, Langer T, Matthews S. Structural insight into the TRIAP1/PRELI-like domain family of mitochondrial phospholipid transfer complexes. EMBO Rep. 2015;16:824–835. doi: 10.15252/embr.201540229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall BM, Owens KM, Singh KK. Distinct functions of evolutionary conserved MSF1 and late embryogenesis abundant (LEA)-like domains in mitochondria. J Biol Chem. 2011;286:39141–39152. doi: 10.1074/jbc.M111.259853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren M, Phoon CK, Schlame M. Metabolism and function of mitochondrial cardiolipin. Prog Lipid Res. 2014;55:1–16. doi: 10.1016/j.plipres.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Baile MG, Lu YW, Claypool SM. The topology and regulation of cardiolipin biosynthesis and remodeling in yeast. Chem Phys Lipids. 2014;179:25–31. doi: 10.1016/j.chemphyslip.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlattner U, Tokarska-Schlattner M, Rousseau D, Boissan M, Mannella C, Epand R, Lacombe ML. Mitochondrial cardiolipin/phospholipid trafficking: the role of membrane contact site complexes and lipid transfer proteins. Chem Phys Lipids. 2014;179:32–41. doi: 10.1016/j.chemphyslip.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Gabriel K, Milenkovic D, Chacinska A, Muller J, Guiard B, Pfanner N, Meisinger C. Novel mitochondrial intermembrane space proteins as substrates of the MIA import pathway. J Mol Biol. 2007;365:612–620. doi: 10.1016/j.jmb.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 35.Herrmann JM. Ups delivery to the intermembrane space of mitochondria: a novel affinity-driven protein import pathway. EMBO J. 2010;29:2859–2860. doi: 10.1038/emboj.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr A. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr A. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Westermann B, Neupert W. Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast. 2000;16:1421–1427. doi: 10.1002/1097-0061(200011)16:15<1421::AID-YEA624>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 40.Daum G, Bohni PC, Schatzg G. Import of proteins into mitochondria. J Biol Chem. 1982;257:13028–13033. [PubMed] [Google Scholar]
- 41.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File