

Abstract
During nuclear DNA replication, proofreading-deficient DNA polymerase α (Pol α) initiates Okazaki fragment synthesis with lower fidelity than bulk replication by proofreading-proficient Pol δ or Pol ε. Here, we provide evidence that the exonuclease activity of mammalian flap endonuclease (FEN1) excises Pol α replication errors in a MutSα-dependent, MutLα-independent mismatch repair process we call Pol α-segment error editing (AEE). We show that MSH2 interacts with FEN1 and facilitates its nuclease activity to remove mismatches near the 5′ ends of DNA substrates. Mouse cells and mice encoding FEN1 mutations display AEE deficiency, a strong mutator phenotype, enhanced cellular transformation, and increased cancer susceptibility. The results identify a novel role for FEN1 in a specialized mismatch repair pathway and a new cancer etiological mechanism.
Keywords: DNA mismatch repair, flap endonuclease 1, MutSα, Okazaki fragment maturation, α-segment error editing
Introduction
During replication of the eukaryotic nuclear genome, most of the nascent leading and lagging strands are synthesized by DNA polymerase ε (Pol ε) and Pol δ, respectively. These replicases are highly accurate, partly because their intrinsic 3′ exonucleases can proofread the rare mismatches they generate. In contrast, the third nuclear replicase, Pol α, lacks an intrinsic proofreading exonuclease (EXO) activity and is tenfold to 100-fold less accurate (Kunkel, 2009). Pol α's role in replication is to initiate Okazaki fragments by extending primers synthesized by its associated RNA primase. Thus, any mismatches made by Pol α will be near the 5′ ends of Okazaki fragments, in what can be called the α-segment. Genetic evidence suggests that some mismatches generated by Pol α are proofread by the EXO activity of Pol δ (Pavlov et al, 2006). Theoretically, mismatches generated by Pol α that escape proofreading could be removed during Pol δ-catalyzed strand displacement synthesis associated with normal Okazaki fragment maturation (OFM) (Burgers, 2009; Zheng & Shen, 2011; Balakrishnan & Bambara, 2013). However, studies of yeast strains encoding variant alleles of Pol α (Niimi et al, 2004; Nick McElhinny et al, 2008, 2010) indicate that OFM alone does not remove all mismatches from the α-segment. In fact, the most recent studies indicate that despite Okazaki fragment processing, DNA synthesized by Pol α is retained in vivo and DNA-binding proteins including histones and transcription factors that rapidly re-associate, post-replication, act as partial barriers to Pol-δ-mediated displacement of the α-segment, resulting in increased mutation rates in the region (Clausen et al, 2015; Daigaku et al, 2015; Koh et al, 2015; Reijns et al, 2015). Pol α variant strains have mild mutator phenotypes, resulting from DNA replication errors generated by Pol α, and their mutation rates are synergistically increased by loss of MSH2-dependent mismatch repair (MMR). Thus, MSH2-dependent MMR in yeast plays a major role in correcting mismatches in the α-segment.
How is this done? A recent study (Liberti et al, 2013) indicated that some mismatches generated by yeast Pol α are excised by Exonuclease 1 (EXO1), a 5′ exonuclease involved in MMR (Tishkoff et al, 1997b; Wei et al, 2003). Interestingly, the loss of MMR resulting from deleting EXO1 is mild compared to the loss of MMR from deleting MSH2, implying the existence of a MSH2-dependent but EXO1-independent mechanism that removes mismatches from the α-segment. This mechanism could be the same as the mechanism used to repair more internal mismatches generated by Pols ε and δ. In addition, a non-exclusive possibility was previously suggested for mismatches generated by Pol α near the 5′ end of an Okazaki fragment, namely FEN1-dependent mismatch removal (Nick McElhinny et al, 2010). This possibility is supported by an earlier genetic study (Johnson et al, 1995), suggesting that yeast FEN1 participates in MMR in yeast, and it is consistent with a biochemical study of mammalian MMR in vitro, providing evidence for EXO1-independent MMR via strand displacement synthesis by Pol δ (Kadyrov et al, 2009). The present study investigates a role for FEN1 in MSH2-dependent removal of mismatches from the α-segment. We show that FEN1 and MSH2 interact and that FEN1 can remove mismatches from the α-segment in an in vitro reaction, referred to as the α-segment error editing (AEE) assay. This action is strongly stimulated by MutSα; FEN1 mutants are defective in the AEE reaction, and these mutations result in a mutator phenotype and increased cellular transformation in mouse cells and increased cancer susceptibility in mice.
Results
Pol α error editing during Okazaki fragment maturation
To identify the protein machinery responsible for editing the α-segment during OFM, we designed five sets of DNA substrates that mimic the α-segment (Supplementary Fig S1). The first set (Supplementary Fig S1A) is a pair of gapped-flap substrates starting with an “A” in the three or four nucleotide (nt) single-stranded (ss) DNA region and with or without a “C/T” mismatch in the downstream duplex. C is unique in the entire downstream template sequence. Incorporation of radiolabeled T in the matched substrate is used to monitor normal flap removal, gap filling, and ligation, referred to here as RNA primer removal (RPR). Incorporation of radiolabeled G in the mismatched substrate is used to monitor removal of the flap containing the mismatch, gap filling, and ligation, referred to here as AEE. The second pair of substrates uses the same principle, but contains a T in the ssDNA region and a G/T mismatch in the downstream duplex, with the G also being unique in the entire downstream template sequence. We obtained similar results with both pairs of substrates and use them interchangeably in the experiments below, as indicated in the text, figures, and Figure legends. In the second set of substrates (Supplementary Fig S1B), we varied the distance between the first paired nt of the downstream duplex and mismatched nts (indicated as X nt), where X nt is the 3rd, 6th, 12th, or 18th nt. The third set of substrates was designed to test the nuclease activities of EXO1, DNA2 and wild-type (WT) and mutant FEN1. These substrates include one with a 3′ single nt flap and a 40-nt-long 5′ flap, two without a 3′ flap but with different lengths of 5′ flaps (long 5′ flap = 40 nt and short 5′ flap = 5 nt), and one with a nicked duplex DNA without a flap (Supplementary Fig S1C). Set 4 (D1, 2, 3, 4) contains gapped substrates with or without an upstream 3′ flap, and nicked substrates with or without an upstream 3′ flap, used to elucidate the best substrate for the FEN1/MSH2 complex in the removal of Pol α errors (Supplementary Fig S1D). Finally, set five contains substrates that are similar to A1 and A2 but contain an RNA flap (Supplementary Fig S1E). The oligonucleotides used to construct these substrates are listed in Supplementary Table S1.
We performed the RPR assay using the first pair of substrates with or without a downstream mismatch, following a procedure that was previously described (Turchi et al, 1994; Zheng et al, 2007a, 2008). This procedure involves incubating nuclear extracts (NEs) with the indicated DNA substrates and nts. In the RPR assays, the gapped-flap substrate was designed in such a way that the first ss DNA nt is an A. In the reaction mixture, we have included radiolabeled dTTP and the other three non-labeled nts. Therefore, if the flap is cleaved, the gap is filled by polymerization and the nick is sealed to generate an 80-nt-long radiolabeled DNA product; then, we know that all of the functional components necessary for OFM are intact in NEs. Indeed, in the first experiment described in Fig1, we observed a full length product from the 80 nt DNA fragment and 39–42 nt intermediate products incorporating [α-32P] dTTP, indicating that the NEs had the functional enzymatic components needed for flap removal, gap filling, and nick sealing. When incubated with WT NEs, substrates with or without Pol α errors produced similar amounts of non-ligated (39–42 nt) and ligated product (80 nt) (Fig1A). However, when we incubated the NEs with the same substrates in the presence of [α-32P] dGTP instead of [α-32P] dTTP, the substrate with the C/T mismatch showed significantly more ligated product compared with the reaction product from the substrate without a mismatch (Fig1B). Because the C is unique in the entire downstream template sequence, the 80-nt-long product is only generated when the first portion of the downstream nts, up to the mismatched nt, T, is removed and radiolabeled G is incorporated. This experiment was repeated with two similar substrates that contained RNA in the 5′ flap to mimic the RNA primer. We obtained very similar results using the RNA and DNA substrates, as we have previously described (Qiu et al, 1999b) (Supplementary Fig S2). Therefore, in the subsequent in vitro biochemical experiments, we used the DNA flap substrates.
Figure 1.
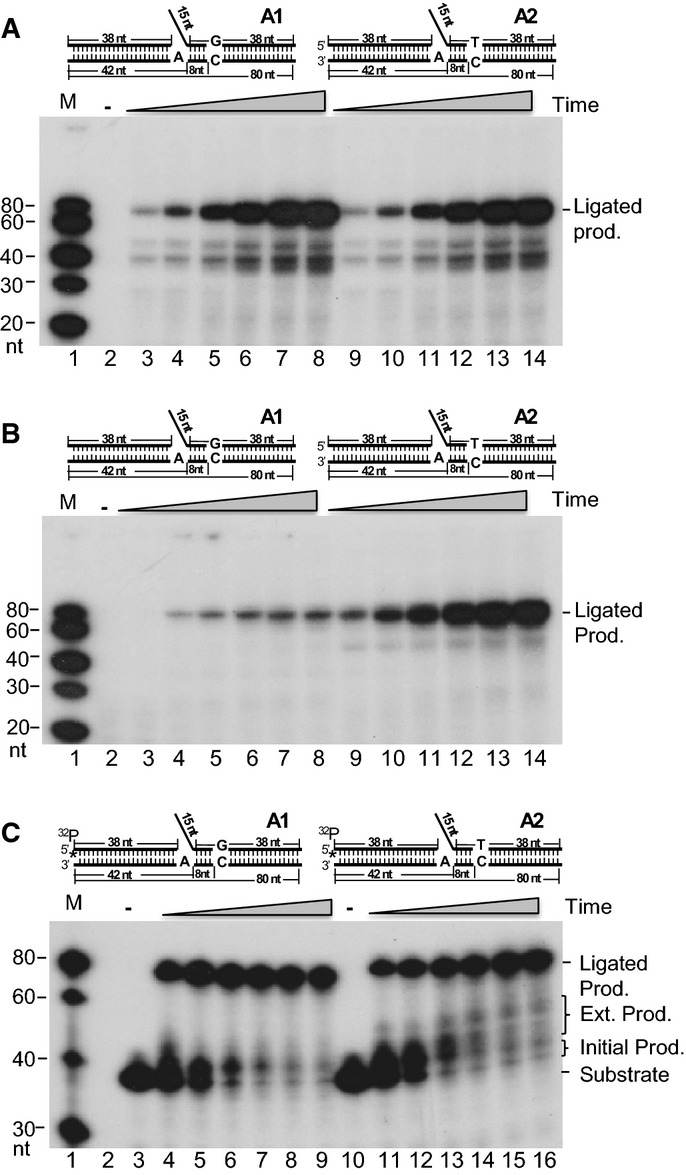
- RPR efficiency without (A1) or with (A2) a mismatch in the downstream duplex DNA region using NEs. One microgram of the NE was incubated with 500 fmol gap substrates containing a 15-nucleotide (nt) DNA flap at 37°C for 5, 10, 20, 40, 60, and 80 min. All reactions were carried out in a buffer containing 5 μCi [α-32P] dTTP and 50 μM dGTP, dCTP, and dTTP. Lanes 3–8 show substrate A1; lanes 9–14 show substrate A2; lane 1 shows molecular weight markers; lane 2 shows the reaction with A1 without enzyme.
- AEE efficiency without (A1) or with (A2) a mismatch in the downstream duplex DNA region using NEs. One microgram of the NE was incubated with 500 fmol of the gap substrates containing a 15-nt DNA flap at 37°C for 5, 10, 20, 40, 60, and 80 min. All reactions were carried out in a buffer containing 5 μCi [α-32P] dGTP and 50 μM dATP, dCTP, and dTTP. Lanes 3–8 show substrate A1; lanes 9–14 show substrate A2; lane 1 shows molecular weight markers; lane 2 shows the reaction with A1 without enzyme.
- AEE assay to show the extended gap-filling products beyond the mismatch. One microgram of the MEF NE was incubated with 500 fmol of the 5′ end upstream primer and 32P-labeled flap DNA substrates without (A1) or with (A2) a mismatch in the downstream DNA duplex. Reactions were carried out at 37°C for 10, 20, 40, 60, 80, and 100 min. The designed substrate is illustrated on the top of the panels. See also Supplementary Fig S1 and Supplementary Table S1. Lanes 4–9 show substrate A1; lanes 11–16 show substrate A2; lane 1 shows molecular weight markers; lane 2 is blank, lanes 3 and 10 show the reactions with A1 and A2, respectively, without enzyme.
The result in Fig1B indicates that there is an error editing mechanism triggered by a mismatch. To further test this hypothesis, we radiolabeled the two substrates at the 5′ end of the upstream primer. This allowed visualization of extended “nick translation” products beyond the mismatch. Similar amounts of the ligated product were generated with both of the substrates as long as the RPR reactions were completed (Fig1C). However, extended gap-filling products beyond the ss DNA region (38–52 nt) appeared in a time-dependent manner for the mismatch substrate only (Fig1C). This confirms the presence of a mechanism for mismatch editing during OFM.
How far can editing proceed beyond the ssDNA region or the gap? We designed a series of substrates with a “T/C” mismatch at the 3rd, 6th, 12th, and 18th nt from the gap in the downstream duplex, simulating Pol α errors at different positions in the α-segment. After incubating WT NEs with the four DNA substrates shown in Supplementary Fig S1B in the presence of [α-32P] dATP, we found that overall RPR efficiency was similar (Fig2A). However, when we incubated NEs with [α-32P] dCTP to assay 5′ end error editing efficiency, there were very large differences among these four substrates in the amount of product generated (Fig2B). The substrate with a mismatch at the 3rd nt was edited out most efficiently (Fig2B, lanes 2–5). As the mismatch was located farther from the gap, the editing efficiency decreased (Fig2B, lanes 6–9) such that little signal was observed when the distance was 12 nts or longer (Fig2B, lanes 10–13). These results indicated that the OFM machinery primarily edits mismatches within 1–12 nt of the gap.
Figure 2.
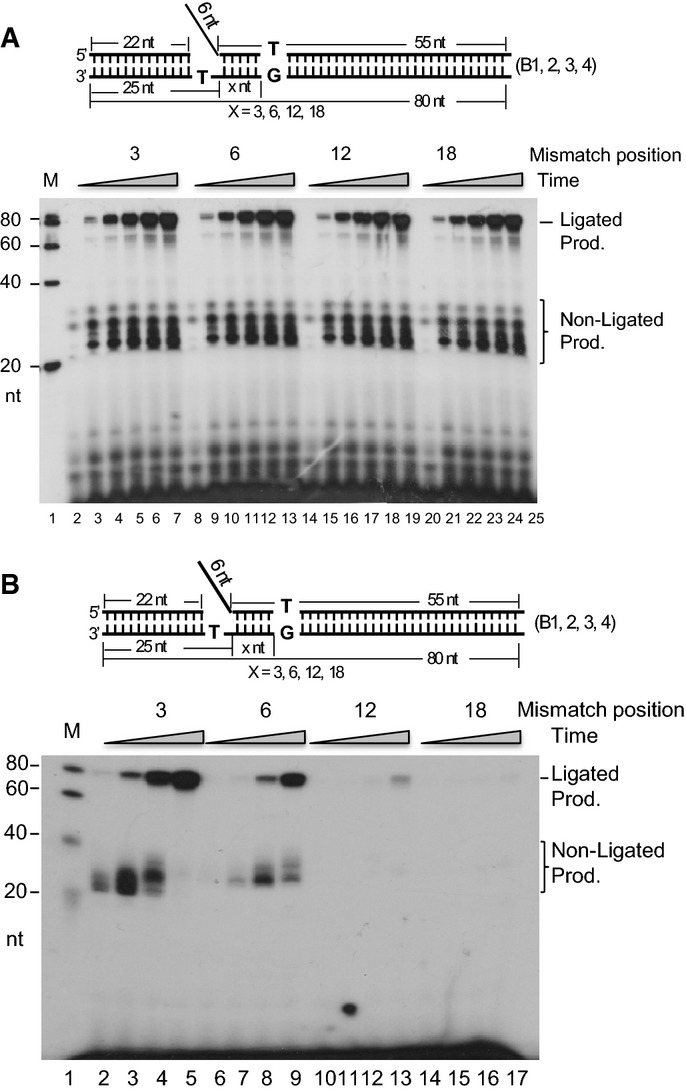
- RPR efficiency with a mismatch at different locations in the downstream duplex DNA. One microgram of the NE was incubated with 500 fmol of the gap substrates containing a 6-nt DNA flap and a variable-length spacer region (B1–4 corresponds to 3, 6, 12, and 18 nt, respectively) at 37°C for 0, 5, 10, 20, 40, and 60 min. All reactions were carried out in a buffer containing 5 μCi [α-32P] dATP and 50 μM dGTP, dCTP, and dTTP. Lanes 3–7 show substrate B1; lanes 9–13 show substrate B2; lanes 15–19 show substrate B3; lanes 21–25 show substrate B4; lane 1 shows molecular weight markers; lanes 2, 8, 14, and 20 show the reactions with B1, B2, B3, and B4, respectively, without enzyme.
- AEE efficiency with a base mismatch at different locations on the downstream duplex DNA. One microgram of the NE was incubated with 500 fmol of the gap substrates containing a 6-nt DNA flap and a variable-length spacer region (B1–4 corresponds to 3, 6, 12, and 18 nt, respectively) at 37°C for 10, 20, 40, and 60 min. All reactions were carried out in a buffer containing 5 μCi [α-32P] dCTP and 50 μM dATP, dCTP, and dTTP. Lanes 2–5 show substrate B1; lanes 6–9 show substrate B2; lanes 10–13 show substrate B3; lanes 14–17 show substrate B4; lane 1 shows molecular weight markers.
The concerted flap endonuclease and exonuclease actions of FEN1 are critical for α-segment error editing
Several nucleases, including FEN1, DNA2, and EXO1, have been proposed to play a role in RPR during OFM in yeast and mammalian cells (Waga & Stillman, 1994; Qiu et al, 1999a; Bae et al, 2001a), but the nuclease responsible for AEE has not been identified. Using the established RPR and AEE assays with purified proteins (Supplementary Fig S3), we have determined the efficiency of each of the nucleases in the RPR and AEE reactions (Fig3). We found that DNA2 was not able to complete the RPR reaction to produce the ligated products, as it was not able to completely remove the ss flap. With the residual flap left over, the ligase was not able to complete the ligation portion of the RPR reaction (Fig3A lane 5 and Fig3B lanes 7–10). In addition, DNA2 was not able to remove the embedded mismatches due to its lack of EXO activity on the double-stranded DNA duplex. It completely failed to perform the AEE reaction (Fig3C lane 5 and Fig3D lanes 7–10). On the other hand, the major function of EXO1 is to remove the mismatches from the DNA duplex, but it has little capacity to remove the flap. Therefore, as expected, EXO1 failed to produce any RPR or AEE products (Fig3A and C, lane 6, Fig3B and D, lanes 11–14). FEN1 has both flap endonuclease (FEN) and EXO activities and was able to efficiently generate both the RPR and AEE products (Fig3A and C, lane 4, Fig3B and D, lanes 3–6). We used nuclease activity assays to show that all three enzymes are active with their standard substrates (Supplementary Fig S1C and Supplementary Fig S4).
Figure 3.
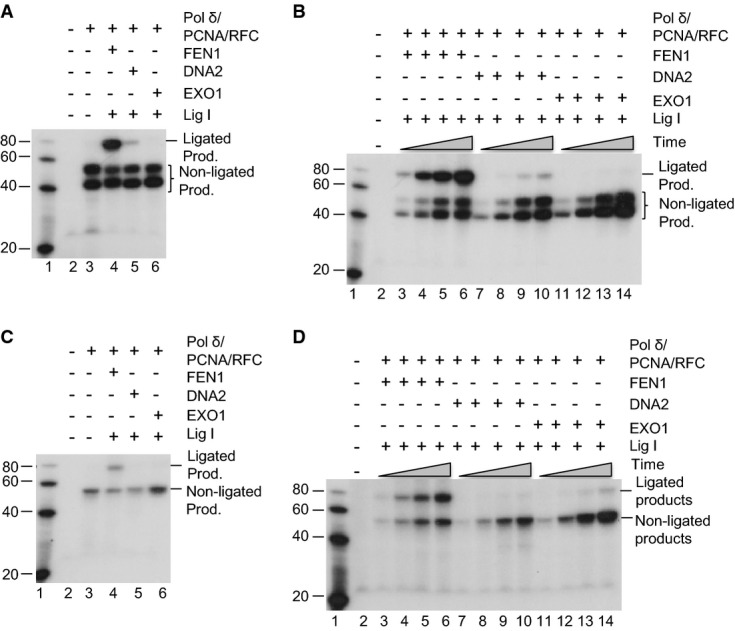
- RPR in a reconstitution assay with the purified individual nucleases: FEN1, EXO1, or DNA2 and [α-32P] dTTP. 100 fmol of Pol δ, 300 fmol of PCNA, 300 fmol of RFC, and 240 fmol of Lig I were mixed with 500 fmol of hFEN1, hDNA2, or EXO1, as indicated. The mixture was then incubated with 50 fmol of substrate in the reaction buffer containing 5 µCi [α-32P] dTTP and 50 µM of each of the other three nucleotides. The reactions were carried out at 37°C for 60 min.
- The same reactions as (A) were carried out for 10 min (lanes 3, 7, and 11), 30 min (lanes 4, 8, and 12), 60 min (lanes 5, 9, and 13), and 90 min (lanes 6, 10, and 14).
- AEE in a reconstitution assay with the individual nucleases: FEN1, EXO1, or DNA2 and [α-32P] dGTP. The reactions were carried out at 37°C for 60 min. [α-32P] dGTP, instead of [α-32P] dTTP, was included in the reactions. All other conditions are the same as in (A, B).
- The same reactions as in (C) were carried out for 10 min (lanes 3, 7, and 11), 30 min (lanes 4, 8, and 12), 60 min (lanes 5, 9, and 13), and 90 min (lanes 6, 10, and 14).
To determine the role and molecular mechanism of FEN1 in AEE, we employed two FEN1 mutants previously identified in cancer cells, E160D and A159V (Zheng et al, 2007b). We characterized their nuclease activity profiles using four standard substrates designed based on a series of publications regarding the FEN1 substrates (Kao et al, 2002) (Supplementary Figs S1C and S5). These substrates included a duplex double-flap DNA with a 3′ single nt flap and a 40-nt-long 5′ flap, two substrates without the 3′ flap but with different lengths of 5′ flaps (long 5′ flap = 40 nt and short 5′ flap = 5 nt), and one nicked duplex DNA without any flap. The E160D mutant retained 100% of WT double-flap endonuclease activity (Supplementary Fig S5A). However, its activity was approximately 50% and 45%, respectively, for cleaving the single-flap substrate with a ss flap of 40 and 5 nts in length (Supplementary Fig S5B and C), and no activity was observed with a nicked DNA substrate (Supplementary Fig S5D). In contrast, the A159V mutant retained approximately 65% of WT activity with the double-flap substrate, but had no activity with the other three substrates (Supplementary Fig S5E–H).
To test whether the different degrees of defect in the mutant activity profiles affect α-segment repair efficiency and disease susceptibility in vivo, we generated a mouse model carrying the FEN1 point mutation A159V. We constructed the FEN1 A159V mouse using a gene targeting approach (Supplementary Fig S6A), as previously described for creation of the FEN1 E160D mouse line (Zheng et al, 2007b). The genotype of the A159V mouse was confirmed by Southern blotting and DNA sequence analysis (Supplementary Fig S6B and C). No live homozygous FEN1 A159V mice were obtained. All homozygous FEN1 A159V embryos died before the E9.5 stage. Heterozygous FEN1 A159V mutant mice were viable. In the RPR assays, WT and WT/A159V NEs from mouse embryonic fibroblast (MEF) cells were incubated with the substrate with a downstream mismatch in the presence of [α-32P] dTTP for the RPR assay. The amount of ligated product for A159V was similar to that of WT or E160D, indicating that the A159V mutation did not affect the RPR efficiency (Fig4A). However, when we incubated the WT and WT/A159V NEs with the substrate in the presence of [α-32P] dGTP (AEE), a condition where the radioactivity would only be incorporated when the mismatched nt is excised, the WT/A159V NE only produced about half of the ligated product compared with the WT or E160D NEs (Fig4B). In addition, we reconstituted the RPR and AEE assays with the purified WT and mutant FEN1 enzymes and found that E160D was able to complete the RPR process by efficiently cleaving the 5′ flap, whereas A159V failed to do so (Fig4C). On the other hand, neither A159V nor E160D was able to complete the AEE reactions (Fig4D), indicating that it is the concerted actions of the flap endonuclease and EXO activities that complete the AEE functional pathway.
Figure 4.

- RPR efficiency with the A2 substrate, [α-32P] dTTP, and NEs prepared from MEFs with the indicated genotypes. One microgram NE was incubated with 500 fmol of the substrate A2 and 5 μCi [α-32P] dTTP at 37°C for 10, 20, 40, 60, and 80 min.
- AEE efficiency with the substrate A2, [α-32P] dGTP, and NEs prepared from the MEFs of the indicated genotypes. One microgram of the NE was incubated with 500 fmol of the substrate A2 and 5 μCi [α-32P] dGTP at 37°C for 10, 20, 40, 60, and 80 min.
- RPR efficiency in the reconstitution assay with the substrates A1 and A2, WT and mutant FEN1 enzymes, and [α-32P] dTTP at 3°C for 80 min.
- AEE efficiency in the reconstitution assay with WT and mutant FEN1 enzymes and [α-32P] dGTP at 37°C for 80 min. All reactions were carried out in a buffer containing 5 μCi [α-32P] dTTP or [α-32P] dGTP and 50 μM of the other three nucleotides.
MSH2 physically interacts with FEN1 and stimulates its α-segment error editing function
To identify the protein interaction partners for FEN1 in AEE, we previously pulled down FEN1 and its associated proteins from synchronized S-phase HeLa cells. Using mass spectrometry analyses, we identified that one of the dominant bands in the silver-stained gel as MSH2 (Guo et al, 2012). This was consistent with the finding that the Pol α-dependent mutation rate is strongly elevated when MSH2 is deleted in yeast (Kunkel, 2004; Niimi et al, 2004; Li, 2008; Liberti et al, 2013) and indicates that the MutSα complex may be involved in AEE in vivo. To determine whether FEN1 plays a role in the editing pathway through the recruitment of the MutSα complex, we pulled down FEN1 with an antibody against FEN1 and detected MSH2 by Western blotting analysis of HeLa cell extracts. MSH2 and its known associated proteins such as MSH6 were specifically pulled down with FEN1, using an antibody against FEN1, but not using a non-specific mouse IgG (Fig5A). In addition, we co-expressed Myc-FEN1 and FLAG-MSH2 in 293T cell lines and immunoprecipitated (IPed) using an anti-FLAG antibody and found that Myc-FEN1 was co-IPed only from cells overexpressing FLAG-MSH2 (Fig5B). To validate whether FEN1 interacts directly with MSH2, purified recombinant FEN1 or MSH2 was individually immobilized onto CNBR-Sepharose 4B beads (Guo et al, 2008). The purified MSH2 or FEN1 or BSA was reciprocally applied to the columns. We were able to detect the presence of MSH2 in the eluate from the FEN1 column, and we detected FEN1 in the eluate from the MSH2 column (Fig5C and D), suggesting a direct interaction between FEN1 and MSH2.
Figure 5.
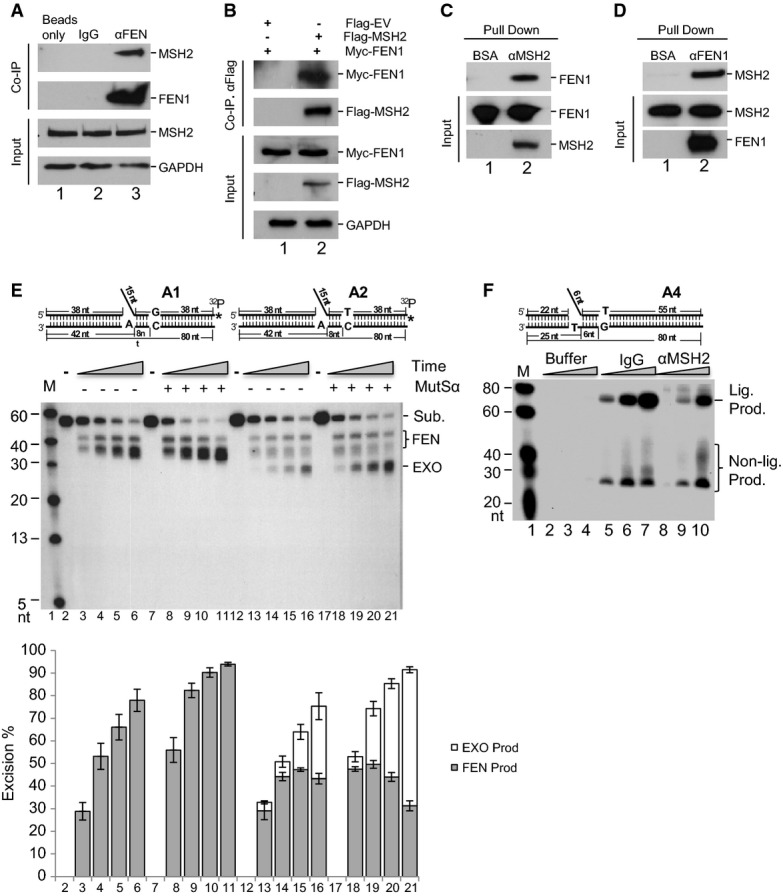
- A Co-immunoprecipitation (IP) of endogenous FEN1 with MSH2 in 293T cells. GAPDH was used as the internal control.
- B Co-IP of FLAG-MSH2 and Myc-FEN1 in 293T cells. GAPDH was used as the internal control.
- C, D Detection of FEN1 after pull-down with MSH2 (C) and detection of MSH2 after pull-down with FEN1 (D). Both FEN1 and MSH2 used in the experiments are the purified recombinant tagged proteins.
- E MutSα stimulates FEN1-extended exonuclease (EXO) activity in the presence of a mismatch-containing substrate. A total of 500 fmol of FEN1 was incubated with 1 pmol of 3′-labeled flap DNA substrate with or without 100 fmol of MutSα. Reactions were carried out at 37°C for 5 (lanes 3, 8, 13, and 18), 10 (lanes 4, 9, 14, and 19), 20 (lanes 5, 10, 15, and 20), and 30 min (lanes 6, 11, 16, and 21). Lanes 3–6 show the reactions with substrate A1 and FEN1 without MutSα; lanes 8–11 show the reactions with substrate A1, FEN1, and MutSα; lanes 13–16 show the reactions with substrate A2 and FEN1 without MutSα; lanes 18–21 show the reactions with substrate A2, FEN1, and MutSα; lane 1 shows the molecular weight markers; lanes 2, 7, 12, and 17 show the reactions without enzyme. The bottom panel shows the quantification of the cleavage products from three independent experiments. Values are means ± SD.
- F AEE assay with the substrate A4 and [α-32P] dGTP incorporation to show AEE by buffer alone, control NEs incubated with IgG (IgG), and NEs depleted of MSH2 (αMSH2). One microgram of the NE was incubated with 500 fmol of the gap substrates containing a 6-nt DNA flap at 37°C for 20 (lanes 2, 5, and 8), 40 (lanes 3, 6, and 9), and 60 (lanes 4, 7, and 10) min. All reactions were carried out in a buffer containing 5 μCi [α-32P] dGTP and 50 μM dATP, dCTP, and dTTP. Numbers at the bottom of the panels are the lane numbers for the various reactions. Lig. Prod.: ligated product.
To further investigate the role of MutSα in the error editing pathway, we radiolabeled the gapped-flap substrates with or without a mismatch in the downstream duplex at the 3′ end of the flap strand. This allowed visualization of products remaining after cleavage. Incubation of FEN1 with substrates with or without the mismatch generated similar amounts of product (38 nt) (Fig5E). In the presence of MutSα, the cleavage reaction proceeded slightly faster. However, with the mismatch-containing substrate at the 8th position in the downstream duplex region, extended exonuclease cleavage products (20–32 nt) were observed. Those products were not in the reactions with the mismatch-free substrate or in the absence of MutSα (Fig5E). The presence of MutSα increased the production of the extended exonuclease products by twofold to threefold. These results may indicate that MutSα recognizes the mismatch and facilitates the processivity of the mismatch removal reaction. To further examine the contribution of MutSα to the editing pathway, we immunodepleted MSH2 from HeLa NEs as previously described (Zheng et al, 2008). The success of immunodepletion was confirmed by Western blotting analysis. We assayed the RPR and AEE efficiency using the mismatch-containing flap substrate and the depleted NEs. We found that MSH2 depletion did not affect the RPR reaction efficiency. However, it significantly decreased the efficiency of AEE (Fig5F). Furthermore, when we reconstituted the RPR and AEE reactions with purified MutSα (Fig6), MutSα was found to increase the AEE reaction efficiency by at least 25-fold, but did not affect the RPR reaction efficiency (Fig6A lanes 8 and 12).
Figure 6.

- RPR assays with the purified recombinant proteins of Pol δ, PCNA, RFC, FEN1, the MutSα complex, Ligase 1 (Lig I), and the substrates A1 and A2. 100 fmol of hPol δ, 300 fmol of hPCNA, 300 fmol of hRFC, 150 fmol of hMutSα, and 240 fmol of Lig I were mixed with 500 fmol of hFEN1 as indicated. The mixture was then incubated with 50 fmol of substrate in the reaction buffer containing 5 µCi [α-32P] dTTP and 50 µM each of the other three nucleotides. The reactions were carried out at 37°C for 60 min.
- AEE assays were identical to (A) except for the use of 5 µCi [α-32P] dGTP instead of 5 µCi [α-32P] dTTP.
A previous study showed that the presence of the 3′ flap and its interaction with FEN1 can stimulate both the flap endonuclease and EXO activities by several fold (Finger et al, 2009). To further explore the mechanism by which MutSα facilitates the processivity and efficiency of the FEN1 EXO activity to remove the mismatch, we used an additional set of the substrates (Supplementary Fig S1D), including gapped substrates with or without an upstream 3′ flap, and nicked substrates with or without an upstream 3′ flap. We observed that the nicked substrate with a 3′ flap was the best substrate for the MutSα stimulation (Fig7A).
Figure 7.
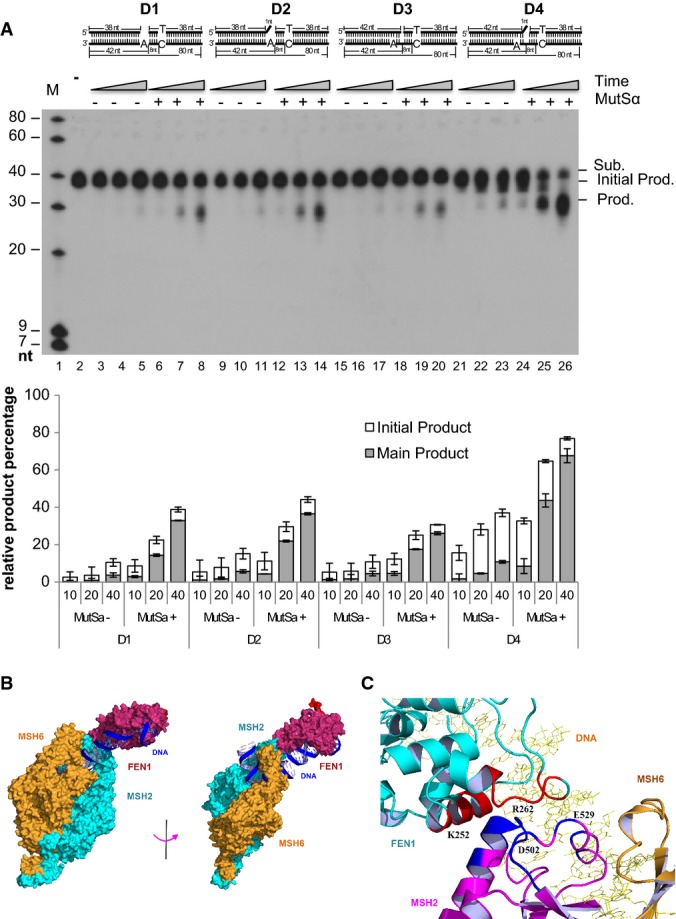
- AEE reactions with four different substrates. 500 fmol of FEN1 was incubated with 1 pmol of 3′-labeled DNA substrates with or without 100 fmol of MutSα. Reactions were carried out at 37°C for 10 (lanes 3, 6, 9, 12, 15, 18, 21, and 24), 20 (lanes 4, 7, 10, 13, 16, 19, 22, and 25), and 40 (lanes 5, 8, 11, 14, 17, 20, 23, and 26) min. Lanes 3–8 show the reactions with substrate D1 and FEN1 with or without MutSα; lanes 9–14 show the reactions with substrate D2 and FEN1 with or without MutSα; lanes 15–20 show the reactions with substrate D3 and FEN1 with or without MutSα; lanes 21–26 show the reactions with substrate D4 and FEN1 with or without MutSα; lane 1 shows molecular weight markers; lane 2 shows the reaction with substrate D1 without enzyme. Numbers at the bottom of the panels are the lane numbers for the various reactions. Sub.: substrate; Prod.: product. Values are means ± SD of three independent assays.
- The FEN1/MutSα/DNA complex model was built using ZDOCK software. The FEN1 (PDB code 3q8k) and MutSα structures (PDB code 3thx) used for protein–protein docking are from X-ray crystallographic structures in complex with DNA molecules. Two views are shown of the FEN1/MutSα/DNA complex model. The FEN1 protein is colored dark red, the DNA molecules are colored blue, MSH2 in the MutSα complex is colored cyan, and MSH6 is colored brown. FEN1 directly interacts with MSH2 according to the model. The model structure can be accessed via http://proteopedia.org/wiki/index.php/User_talk:Hongzhi_Li.
- Interaction region of FEN1 (cyan-colored) to MSH2 (magenta-colored). The residues of MSH2 that are within 4 Å of FEN1 are colored blue. The residues of FEN1 that are within 4 Å of MSH2 are shown in red. The four contacting residues, K252 and R262 in FEN1, and D502 and E529 in MSH2, are labeled at their positions. The MSH6 and DNA molecules are colored brown and yellow, respectively.
An additional question is as follows: How does FEN1 interact with the 3′ flap and remove the nearby nt as well as the mismatched nt as far as in the 8th position downstream of the 3′ flap (Fig7A)? Does MutSα facilitate such an action by looping out the single-stranded DNA in the template strand while FEN1 sequentially removes newly synthesized DNA? To address these questions, we built a model of a FEN1/MutSα/DNA complex using ZDOCK software (Pierce et al, 2014). For docking, we used the FEN1 [PDB code 3q8k (Tsutakawa et al, 2011)] and MutSα [PDB code 3thx (Warren et al, 2007; Gupta et al, 2012)] X-ray crystallographic structures in complexes with DNA. The best model was selected from a total of 40,000 initial complex models with their distinct consensus contact scores (Fig7B). The consensus contact scores were calculated based on two criteria: (i) interaction of the FEN1 helical region with MutSα protein: FEN1's helical region, with amino acid residues from 245 to 252, which was previously identified to interact with the downstream DNA duplex (Tsutakawa et al, 2011) (now where the mismatch is) and may interact with MutSα protein, and (ii) the interaction between proteins and DNA substrates: The ends of the DNA molecules bound to the two proteins, respectively, should be as close as possible. In the established model, residues of FEN1 in the regions of K244-H253 and V260-Y268 interact directly with the residues in regions of S498-D506 and E529-F539 of MSH2 with a contact area of about 377 Å2 (Fig7C). It is suggested that the strong interaction between MSH2 and FEN1 may stabilize the FEN1 protein to bind with the DNA molecule after the 5′ flap is removed. Due to the strong interaction with FEN1 and energy release in the process of the stabilization of the FEN1/MutSα/mismatch-containing DNA complex, the MutSα protein will pull the FEN1 protein toward the mismatch and be pushed backward. The protein complex could then cleave the nt diester bonds of the Pol α-synthesized error-prone DNA while it loops out the template strand.
Do MSH2 and FEN1 really act together in vivo, as we suggested here, or does MSH2 act in traditional MMR, independently of FEN1? An additional experiment was designed to address this point. We individually knocked down FEN1, MSH2, EXO1, or MLH1, the latter three of which are involved in traditional MMR (Fig8A). We then examined the effects on AEE using the respective NE. Knockdown of FEN1 or MSH2 impaired AEE, whereas down-regulation of EXO1 or MLH1 expression did not (Fig8B). In addition, we examined traditional MMR and AEE reaction efficiencies in cell lines defective in MSH2 or MLH1. As expected, deficiencies in either MSH2 or MLH1 abrogated MMR. In contrast, only MSH2 deficiency, not MLH1 deficiency, impaired AEE (Supplementary Fig S7). Based on these data and the data presented in Figs5 and 6, we conclude that MSH2 and FEN1 form a unique complex, which together play an important role in AEE that appears to largely be independent of MLH1.
Figure 8.
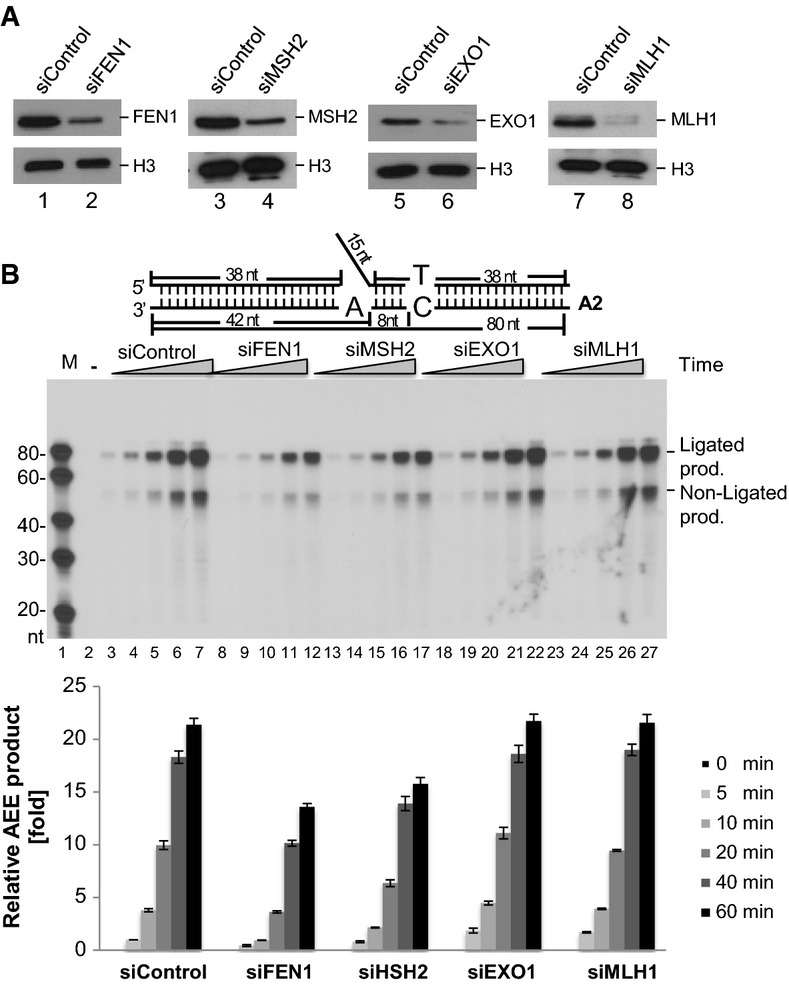
- Western blot analysis of siFEN1, siMSH2, siEXO1, siMLH1, and the control siRNA-infected MEF cells. An antibody against histone H3 was used as a loading control.
- AEE assay with the A2 substrate. The amount of [α-32P] dGTP incorporation is measured to show the AEE efficiency using NEs and NEs with knockdown of FEN1 (lanes 8–12), MSH2 (lanes 13–17), EXO1 (lanes 18–22), or MLH1 (lanes 23–27). The reactions with the control NEs are presented in lanes 3–7. Lane 1 shows the molecular weight markers. Lane 2 shows the reaction without NEs. The bottom histogram presents the quantification of the amount of ligated AEE product (means ± SD) from three independent experiments. Prod., product.
Biological consequences of deficiency in the FEN1/MutSα functional complex
Finally, we asked whether deficiency in AEE has biological consequences. We chose the A159V FEN1 mutation as a tool to answer such a question. We first transformed the human WT and A159V FEN1 mutant genes, which were present in the yeast expression vector pRS, into a RAD27 KO strain (RKY2608) and measured mutation rates. We found that the mutation rate of the mutant strain was approximately 93-fold higher than that of the WT strain (Fig9A). Accumulation of mutations usually causes tumorigenesis. To determine whether the A159V mutation promotes tumorigenesis, we evaluated the cellular transformation frequency using WT/A159V primary MEF cells (Fig9B and Supplementary Fig S8A and B). The number of colonies formed by WT/A159V cells was 4.75-fold higher than the number formed in WT cells. To determine whether A159V mutation increases tumorigenesis, we constructed and characterized a WT/A159V mouse model. Among 77 WT/A159V mice examined between 18 and 22 months of age, 41 mice developed lung tumors and another 15 mice had other types of tumors in various organs. This combined 72% spontaneous cancer incidence is substantially higher than the incidence in WT mice, where only 5 of the 31 WT mice (16%) developed lung tumors (Fig9C and Supplementary Fig S9A–G). Therefore, we conclude that the A159V mutation is associated with increased cancer susceptibility.
Figure 9.
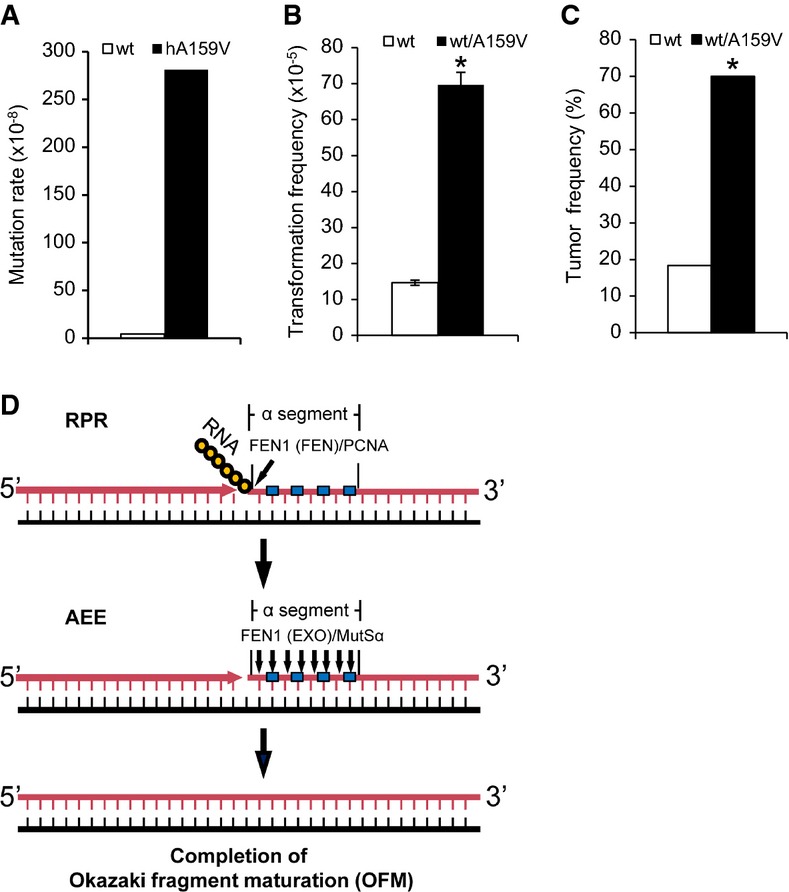
- Mutation rates of a RAD27 deletion yeast strain (RKY2608) containing human WT FEN1 or its mutant A159V.
- Quantification of transformed cells. Values are the mean ± SD of three experiments. *P < 0.01 (Student's t-test).
- Tumor formation percentage in WT mice (n = 31) and WT/A159V mice (N = 77) that were randomly selected for analysis. Mice were evaluated between 18 and 22 months of age. *P < 0.01 (Fisher's exact test).
- A model to demonstrate the separate steps of the Okazaki fragment maturation process: RPR and AEE. In AEE, FEN1 forms a complex with PCNA to remove the RNA primer using the FEN activity mode. If a mismatch error is detected by MutSα, the FEN1/PCNA complex is recruited to form a new functional complex to remove a short stretch (1–12) of nt until the mismatch is removed, using the EXO activity mode. The polymerase then extends the upstream DNA, resulting in a nicked substrate for the ligase to generate high-fidelity newly synthesized DNA. Yellow circles represent ribonucleotides, and cyan squares represent mismatched deoxyribonucleotides. Black lines correspond to DNA templates, and pink lines correspond to newly synthesized DNA. Blue arrows indicate cleavage by the FEN or the EXO activity of FEN1.
To further examine the consequences of the rad27-A159V allele on AEE and genome stability, we introduced the homologous yeast rad27-A157V and rad27-E158D mutations into the yeast genome and analyzed mutagenesis using the CAN1 reporter gene. We also analyzed the distribution of duplication mutations and characteristic of aberrant OFM and of other types of mutations, including base substitutions, frame shifts, and complex mutations (Supplementary Table S2). These data were compared to the corresponding nuclease activity profiles and the RPR and AEE reaction efficiencies that were observed using NEs or after reconstitution (Figs4 and 6 and Supplementary Fig S5). The A159V mutation eliminated both the single-flap endonuclease and nick-specific EXO activities but retained the double-flap endonuclease activity to a significant degree (approximately 60%). NEs from heterozygous A159V MEFs catalyzed the RPR and AEE reactions at approximately 50% efficiency, whereas in the reconstituted reactions, the mutant protein completely failed to catalyze either RPR or AEE. The yeast A157V increased the mutation rate by 61-fold. The mutation spectrum contained 85% duplication mutations, as is characteristically seen in rad27 null mutants (Tishkoff et al, 1997a), with the remainder of the spectrum being single-base mutations, consistent with loss of AEE. In comparison, the E160D mutant enzyme had 100% of the WT double-flap endonuclease activity, but it lost the nick-specific EXO activity completely. In the reconstitution reactions for RPR and AEE, it had nearly WT levels of RPR but failed AEE (Fig6). A corresponding mutation in yeast, E158D, exhibited an approximately 23-fold increase in base substitutions, indels and complex mutations, which accounted for 100% of the mutations. Although the duplications are most likely due to loss of the flap endonuclease activities (RPR), the point mutations are consistent with the reduced FEN-1 EXO activity for AEE (Supplementary Table S2).
Discussion
The current study provides evidence for the existence of a FEN1/MutSα-dependent but EXO1-independent and MLH1-independent mechanism that removes mismatches from the α-segment. The results suggest that the concerted action of endonucleolytic cleavage of displaced primers and exonucleolytic removal of mismatches in the α-segment by FEN1 partly contributes to high-fidelity replication for the portion of the genome that is synthesized by Pol α, because it initiates new DNA chains at origins and on the lagging strand. The actions of FEN1 are coordinated and facilitated by the MutSα-mediated recognition of a mismatch in the DNA duplex downstream from the 5′ nick or gap. MutSα not only recognizes the mismatched base in the substrate but also recruits FEN1 and increases its processivity toward the mismatch via the interaction. Without MSH2, the AEE reaction efficiency is significantly reduced. It is important that neither DNA2 (Bae et al, 2001a) nor EXO1 (Qiu et al, 1999a) was able to replace the function of FEN1 in our AEE reconstitution assays. DNA2, in complex with RPA, is only able to bind and cleave a displaced long flap, leaving a short flap for FEN1 to further process (Bae & Seo, 2000; Bae et al, 2001a,b). By itself, DNA2 would not be able to excise the mismatched base pairs in a DNA duplex. On the other hand, EXO1 is able to remove the nts from the 5′ end but has weak activity for flap cleavage and lacks the protein–protein interaction capacity necessary for a specific functional complex. The other critical difference between the errors incorporated by Pol α and Pol δ/ε is that Pol α-incorporated errors are in the α-segment and mostly within or near the flap during OFM. The 5′ end editing of the α-segment is tightly coordinated with flap cleavage and the joining of Okazaki fragments. These features suggest that FEN1-mediated 5′ end editing proceeds only to mismatches within a limited distance from the flap, in order to avoid a delay in OFM. Repair of the mismatch by the reconstituted purified DNA replication proteins showed that the mismatches at 3–6 nts away from the nick were effectively edited, but those at 12 nt or 18 nt away were not processed via the FEN1-mediated pathway and are more likely to be processed by the traditional MMR pathway. These findings are consistent with a recent report suggesting that lagging-strand replication shapes the mutational landscape of the genome (Reijns et al, 2015).
The AEE mechanism proposed here is advantageous because it effectively removes replication errors while concomitantly maturing Okazaki fragments, which is a rate-limiting step in DNA replication. However, if a mutation occurs in AEE components that deregulates the process and leads to constitutive editing and futile cycles, it could affect DNA ligation and result in spontaneous DNA strand breaks and consequently, various forms of chromosome aberrations. Such a mutation would be similar to the FFAA FEN1 mutation, which specifically disrupts the FEN1/PCNA interaction (Zheng et al, 2007a). The mutation results in DNA strand breaks and aneuploidy, which are hallmarks of human cancer (Zheng et al, 2011, 2012).
We have separated the OFM process into two steps that can be assayed in vitro: RPR and AEE (Fig9D). Even though FEN1 drives both reactions, it has different catalytic modes. In RPR, FEN1 acts in the flap endonuclease mode, whereas in AEE, FEN1 acts in the EXO mode. As previously discussed, the former activity is essential (Zheng & Shen, 2011). A159V is severely defective in double-flap FEN activity and completely deficient in both single-flap FEN and EXO activities. However, the adjacent E160D mutation largely retains FEN activity but is deficient in EXO activity. This explains why the homozygous A159V mutant mouse is embryonically lethal, in sharp contrast to the E160D mutant mouse (Zheng et al, 2007b). This difference also leads us to predict that FEN1 exonuclease-deficient mutants that retain sufficient flap endonuclease activity will be defective in AEE but not in RPR. FEN1-E160D is such a mutation. It eliminates 90% of the EXO activity, which is important for AEE, but retains the flap endonuclease activity critical for RPR (Zheng et al, 2007b). The homologous yeast rad27-E158D mutant was a mutator for base substitutions, frame shift and complex mutations, which may be repaired by FEN1-mediated AEE. The corresponding E160D MEF cells have a proliferation rate similar to WT MEFS, but display high rates of base substitutions. Mutant mice bearing the E160D FEN1 mutation grow and develop as normally as their WT littermates, but develop lung cancer at relatively early ages (Zheng et al, 2007b). The mouse studies thus support the concept that defects in AEE are associated with increased cancer susceptibility.
Materials and Methods
Co-IP and pull-down
In a standard co-IP assay, a non-specific mouse IgG or a mouse monoclonal antibody against hFEN1 (Gene Tex, catalogue# GTX 70185) was conjugated to protein A/G agarose beads. The beads were incubated with NEs of 293T cells (ATCC, catalogue# CRL-11268) and benzonase nuclease (EMD Millipore) overnight at 4°C and washed with PBS buffer containing 0.2% Tween-20. The hMSH2 that was associated with beads was analyzed by Western blotting immunodetection using a rabbit polyclonal antibody against hMSH2 (Abcam, catalogue# ab70270). To pull down the recombinant hFEN1 with recombinant hMSH2, purified hMSH2 or BSA was coated onto CNBR-Sepharose 4B beads and incubated (4°C, overnight) with purified recombinant FEN1 as previously described (Lin et al, 2013). The beads were then washed and boiled at 95°C in SDS–PAGE sample buffer for Western blotting. The experiment was repeated in a reciprocal manner.
Protein purification and nuclease activity assays
We followed previously published protocols to express and purify recombinant hFEN1 and the A159V and E160D mutant proteins (Shen et al, 1996), DNA2 (Ronchi et al, 2013), EXO1 (Zhang et al, 2005), PCNA (Frank et al, 2001), and MutSα (Zhang et al, 2005) and Pol δ complexes (Zhou et al, 2012) (see Supplementary Fig S3). FLAG-tagged MSH2 was purified using an anti-FLAG antibody. The presence of MSH6 was confirmed by Western blotting using an anti-MSH6 antibody (Abcam, catalogue# ab137457). Recombinant Ligase I was purchased from Origene Technologies, Inc. The 32P-labeled flap DNA substrates C1, 2, 3, and 4 were prepared using the oligonucleotides listed in Supplementary Table S1, as described previously (Zheng et al, 2005). The nuclease activity assays were also set up following an established protocol (Zheng et al, 2007a). Briefly, the indicated amount of FEN1 protein and the MutSα complex was incubated with 500 fmol of substrates for varying time periods. Reactions were carried out in a total volume of 10 μl at 37°C and analyzed by denaturing 15% PAGE. The products were visualized by autoradiography and quantified using ImageJ.
RPR assay
NEs from MEF cells were prepared as previously described (Zheng et al, 2005). RPR reactions were reconstituted using mixtures of purified proteins or assayed with NEs using the gapped DNA substrates with DNA flaps and a mismatch in the downstream DNA duplex, as shown in Supplementary Fig S1 (A1 or A2). NEs (1 μg) from MEF cells with various genotypes, or purified recombinant proteins, as indicated in the Figure legends, were incubated with the DNA substrates (500 fmol) in the reaction buffer (50 mM HEPES-KOH (pH 7.5), 45 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.1 mM EDTA, 2 mM ATP, 2 unit creatine phosphokinase, 0.5 mM NAD and 5 mM phosphocreatine) containing 5 μCi [α-32P] dTTP or [α-32P] dATP and 50 μM each of the other three dNTPs. The reactions were carried out at 37°C, and the products were analyzed by 15% denaturing PAGE and autoradiography.
AEE assay
The reaction steps for gap filling, primer cleavage, mismatch excision, and DNA ligation reactions were assayed by the nick translation reaction with the oligonucleotide-assembled substrates that mimic the OFM intermediates, following a modified version of published protocols (Zheng et al, 2007a). Briefly, purified proteins or NEs were incubated with specific gapped substrates for varying time intervals in the same reaction buffer as used in the RPR assay, containing 5 μCi [α-32P] dGTP and 50 μM each of dATP, dCTP, and dTTP or 5 μCi [α-32P] dCTP and 50 μM each of dATP, dGTP, and dTTP.
Mismatch repair assay
In vitro MMR assays were performed essentially as described (Zhang et al, 2005) with some modifications. A circular DNA substrate (25 fmol) containing a G-T mismatch and a nick 5′ to the mismatch (Supplementary Fig S7) was incubated with 75 μg of whole-cell extracts at 37°C for 15 min. DNA samples were recovered by ethanol precipitation and digested with the restriction endonucleases Nsil (repair scoring enzyme), PstI, and BglI. After electrophoresis through a 6% polyacrylamide gel, the DNA products were subjected to Southern blot analysis to identify repaired and unrepaired molecules using [α-32P]-labeled oligonucleotide probes complementary to the nicked DNA strand.
siRNA knockdown
Stealth siRNA oligos against human FEN1 (catalogue# FEN1HSS103627), EXO1 (catalogue# 10620319), MLH1 (catalogue# 119549), or the control siRNA oligos (catalogue# 12935-112) were purchased from Life Technologies. MSH2 (catalogue# SI02663563) was from Qiagen. siRNA oligos were transfected into HeLa cells (ATCC, catalogue# CCL2) using the Lipofectamine 2000 transfection reagent (Life Technologies) according to the instructions from Invitrogen. The knockdown efficiency was verified by Western blotting analysis.
Immunodepletion of human MSH2
To immunodeplete hMSH2, 100 μg of a HeLa NE was incubated with protein A agarose beads (100 μl), which were coated with rabbit polyclonal antibodies to hMSH2 (Abcam, catalogue# ab70270), in the NE buffer at 4°C for 8 h. After a brief centrifugation step, the supernatant was analyzed by Western blot analysis to confirm depletion efficiency.
Generation of an A159V mutant mouse and MEF cell culture
A159V mice were generated according to a protocol that was previously described (Zheng et al, 2007b) and is described in Supplementary Fig S6. WT and WT/159V mutant mice (129S1 strain) of both genders were in-line bred, housed, and cared for in the City of Hope Animal Resource Center. MEFs were isolated from WT/A159V or WT mice (129S1 genetic background) on the 13th day of embryonic development (E13). The embryos were disassociated in trypsin to produce MEF cells. Primary MEF cells were plated onto a 10-cm dish at 37°C in a 5% CO2 incubator. Dulbecco's modified Eagle's medium (Invitrogen), supplemented with 10% fetal bovine serum (Invitrogen) and penicillin–streptomycin (Invitrogen), was used to culture the cells.
Yeast strains, genetic manipulation, mutation rate, and mutation spectrum analysis
The WT (RDKY2672) and isogenic RAD27 deletion strains (MATa, ura3-52, his3Δ200, trp1Δ63, leu2Δ1, ade2Δ1, ade8, lys2-Bgl, hom3-10) were gifts from the Kolodner laboratory (Tishkoff et al, 1997b). To express human FEN1 and the A159V mutant in the RAD27 deletion strain (Δrad27), a DNA fragment encoding the FEN1 protein was subcloned into the pRS314 plasmid. The pRS314 or pRS314-hFEN-1 vector was transformed into Δrad27 yeast. The A157V and E158D RAD27 mutations were knocked into the yeast RDKY2672 genome following a published protocol (Nick McElhinny et al, 2010). Analysis of the Canr mutation rate and mutation spectrum was carried out as previously described (Tishkoff et al, 1997b).
Cell transformation assay
The focus formation assays were conducted according to a previously established protocol (Sweasy et al, 2005). Primary MEF cells were seeded onto a 10-cm dish. After 3 passages, 105 cells were seeded onto a 10-cm dish and incubated for 20–30 days until colonies appeared. The cells then were fixed with methanol and stained with Giemsa solution, and the number of spontaneous colonies was scored under a microscope (ZEISS, Axiovert 135).
Cancer incidence analysis
The nQuery software for statistical analysis was used to estimate the sample size. A minimum of 44 mice for each group was shown to be required to detect a 30% difference in lifespan and cancer phenotypes when compared with the control group with 80% power and 95% confidence based on a previous study (Zheng et al, 2007b). WT and WT/A159V mutant mice (male and female) were randomly selected for determination of the cancer incidence. To determine the lung cancer incidence, lung tissues of mice (18–22 months old) were fixed in 10% formalin and tissue sections were stained with hematoxylin and eosin (H & E). Slides were analyzed in a blinded fashion. All protocols that involved animals were approved by the Institutional Animal Care and Use Committee of City of Hope in compliance with the Public Health Service Policy of the United States and all other federal, state, and local regulations. All aspects of the animal study were adequately reported following the NIH guidelines.
Author contributions
SL has prepared many of the materials used in the assays and performed most of biochemical and cell biological assays. GL designed and prepared RPR and AEE DNA substrates and did preliminary biochemical experiments with NEs. LZ designed many of experiments and supervised both SL and GL for the biochemical and cell biological experiments as well as the transgenic mouse analysis. HD is in charge of generation and maintenance of the transgenic mice as well as of the cell culture work. WL and SA contributed to the biochemical assays and data processing. HL built the docking model of FEN1 and MSH2. HX and YH helped to purify various recombinant proteins used in the biochemical assays. YZ prepared purified and active DNA polymerase δ complex and supervised the reconstitution assays. JO and GML performed the MMR and mutagenesis assays. TAK participated in initial experiment designs and manuscript writing. BS is responsible for overall experiment design and implementation, data interpretation, and manuscript writing.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgments
We thank the City of Hope Pathology, Bioinformatics and Integrated Genomics Core facilities, NCI Designated Cancer Center Support Grant P30 CA033572, Steven Vonderfecht D.V.M., Ph.D., for technical assistance with characterization of the mouse cancer specimens, Liya Gu, Ph.D., for her generous gift of the purified recombinant EXO1 protein, and Nancy Linford, Ph.D., for her critical reading and editing of the manuscript. The work was supported by an NIH Grant RO1 CA076734 to B.H.S., a National Natural Science Foundation of China Grant 31370790 to Y.J.Z., and in part by Project Z01 ES065089 to TAK from the Division of Intramural Research of the National Institutes of Health, National Institute of Environmental Health Sciences, and an international corporation grant from National Natural Sciences Foundation of China (31210103904) to Y.J.H.
Footnotes
- In the 1st affiliation “Colleges of Life Sciences and Agricultural Sciences and Biotechnology” has been corrected to “Colleges of Life Sciences and Agriculture and Biotechnology”.
- Duplicated information in the Acknowledgements section has been removed.
- The Author contributions section has been corrected by listing all authors and providing the correct author abbreviations.
Supporting Information
Supplementary Figures S1–S9, Supplementary Tables S1 and S2
Supplementary Legends
Source Data for Supplementary Figure S2
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S5
Source Data for Supplementary Figure S6
Source Data for Supplementary Figure S7
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
References
- Bae SH, Seo YS. Characterization of the enzymatic properties of the yeast dna2 Helicase/endonuclease suggests a new model for Okazaki fragment processing. J Biol Chem. 2000;275:38022–38031. doi: 10.1074/jbc.M006513200. [DOI] [PubMed] [Google Scholar]
- Bae SH, Bae KH, Kim JA, Seo YS. RPA governs endonuclease switching during processing of Okazaki fragments in eukaryotes. Nature. 2001a;412:456–461. doi: 10.1038/35086609. [DOI] [PubMed] [Google Scholar]
- Bae SH, Kim JA, Choi E, Lee KH, Kang HY, Kim HD, Kim JH, Bae KH, Cho Y, Park C, Seo YS. Tripartite structure of Saccharomyces cerevisiae Dna2 helicase/endonuclease. Nucleic Acids Res. 2001b;29:3069–3079. doi: 10.1093/nar/29.14.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan L, Bambara RA. Flap endonuclease 1. Annu Rev Biochem. 2013;82:119–138. doi: 10.1146/annurev-biochem-072511-122603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–4045. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen AR, Lujan SA, Burkholder AB, Orebaugh CD, Williams JS, Clausen MF, Malc EP, Mieczkowski PA, Fargo DC, Smith DJ, Kunkel TA. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nat Struct Mol Biol. 2015;22:185–191. doi: 10.1038/nsmb.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Keszthelyi A, Muller CA, Miyabe I, Brooks T, Retkute R, Hubank M, Nieduszynski CA, Carr AM. A global profile of replicative polymerase usage. Nat Struct Mol Biol. 2015;22:192–198. doi: 10.1038/nsmb.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger LD, Blanchard MS, Theimer CA, Sengerova B, Singh P, Chavez V, Liu F, Grasby JA, Shen B. The 3′-flap pocket of human flap endonuclease 1 is critical for substrate binding and catalysis. J Biol Chem. 2009;284:22184–22194. doi: 10.1074/jbc.M109.015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank G, Qiu J, Zheng L, Shen B. Stimulation of eukaryotic flap endonuclease-1 activities by proliferating cell nuclear antigen (PCNA) is independent of its in vitro interaction via a consensus PCNA binding region. J Biol Chem. 2001;276:36295–36302. doi: 10.1074/jbc.M103397200. [DOI] [PubMed] [Google Scholar]
- Guo Z, Chavez V, Singh P, Finger LD, Hang H, Hegde ML, Shen B. Comprehensive mapping of the C-terminus of flap endonuclease-1 reveals distinct interaction sites for five proteins that represent different DNA replication and repair pathways. J Mol Biol. 2008;377:679–690. doi: 10.1016/j.jmb.2007.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kanjanapangka J, Liu N, Liu S, Liu C, Wu Z, Wang Y, Loh T, Kowolik C, Jamsen J, Zhou M, Truong K, Chen Y, Zheng L, Shen B. Sequential posttranslational modifications program FEN1 degradation during Cell-cycle progression. Mol Cell. 2012;47:444–456. doi: 10.1016/j.molcel.2012.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Gellert M, Yang W. Mechanism of mismatch recognition revealed by human MutSbeta bound to unpaired DNA loops. Nat Struct Mol Biol. 2012;19:72–78. doi: 10.1038/nsmb.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Kovvali GK, Prakash L, Prakash S. Requirement of the yeast RTH1 5′ to 3′ exonuclease for the stability of simple repetitive DNA. Science. 1995;269:238–240. doi: 10.1126/science.7618086. [DOI] [PubMed] [Google Scholar]
- Kadyrov FA, Genschel J, Fang Y, Penland E, Edelmann W, Modrich P. A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. Proc Natl Acad Sci USA. 2009;106:8495–8500. doi: 10.1073/pnas.0903654106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HI, Henricksen LA, Liu Y, Bambara RA. Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate. J Biol Chem. 2002;277:14379–14389. doi: 10.1074/jbc.M110662200. [DOI] [PubMed] [Google Scholar]
- Koh KD, Balachander S, Hesselberth JR, Storici F. Ribose-seq: global mapping of ribonucleotides embedded in genomic DNA. Nat Methods. 2015;12:251–257. doi: 10.1038/nmeth.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA. DNA replication fidelity. J Biol Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Evolving views of DNA replication (in)fidelity. Cold Spring Harb Symp Quant Biol. 2009;LXXIV:91–101. doi: 10.1101/sqb.2009.74.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- Liberti SE, Larrea AA, Kunkel TA. Exonuclease 1 preferentially repairs mismatches generated by DNA polymerase alpha. DNA Repair (Amst) 2013;12:92–96. doi: 10.1016/j.dnarep.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Sampathi S, Dai H, Liu C, Zhou M, Hu J, Huang Q, Campbell J, Shin-Ya K, Zheng L, Chai W, Shen B. Mammalian DNA2 helicase/nuclease cleaves G-quadruplex DNA and is required for telomere integrity. EMBO J. 2013;32:1425–1439. doi: 10.1038/emboj.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Kissling GE, Kunkel TA. Differential correction of lagging-strand replication errors made by DNA polymerases alpha and delta. Proc Natl Acad Sci USA. 2010;107:21070–21075. doi: 10.1073/pnas.1013048107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi A, Limsirichaikul S, Yoshida S, Iwai S, Masutani C, Hanaoka F, Kool ET, Nishiyama Y, Suzuki M. Palm mutants in DNA polymerases alpha and eta alter DNA replication fidelity and translesion activity. Mol Cell Biol. 2004;24:2734–2746. doi: 10.1128/MCB.24.7.2734-2746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov YI, Frahm C, Nick McElhinny SA, Niimi A, Suzuki M, Kunkel TA. Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr Biol. 2006;16:202–207. doi: 10.1016/j.cub.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Qian Y, Chen V, Guan MX, Shen B. Human exonuclease 1 functionally complements its yeast homologues in DNA recombination, RNA primer removal, and mutation avoidance. J Biol Chem. 1999a;274:17893–17900. doi: 10.1074/jbc.274.25.17893. [DOI] [PubMed] [Google Scholar]
- Qiu J, Qian Y, Frank P, Wintersberger U, Shen B. Saccharomyces cerevisiae RNase H(35) functions in RNA primer removal during lagging-strand DNA synthesis, most efficiently in cooperation with Rad27 nuclease. Mol Cell Biol. 1999b;19:8361–8371. doi: 10.1128/mcb.19.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Kemp H, Ding J, de Proce SM, Jackson AP, Taylor MS. Lagging-strand replication shapes the mutational landscape of the genome. Nature. 2015;518:502–506. doi: 10.1038/nature14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi D, Di Fonzo A, Lin W, Bordoni A, Liu C, Fassone E, Pagliarani S, Rizzuti M, Zheng L, Filosto M, Ferro MT, Ranieri M, Magri F, Peverelli L, Li H, Yuan YC, Corti S, Sciacco M, Moggio M, Bresolin N, et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am J Hum Genet. 2013;92:293–300. doi: 10.1016/j.ajhg.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Nolan JP, Sklar LA, Park MS. Essential amino acids for substrate binding and catalysis of human flap endonuclease 1. J Biol Chem. 1996;271:9173–9176. doi: 10.1074/jbc.271.16.9173. [DOI] [PubMed] [Google Scholar]
- Sweasy JB, Lang T, Starcevic D, Sun KW, Lai CC, Dimaio D, Dalal S. Expression of DNA polymerase beta cancer-associated variants in mouse cells results in cellular transformation. Proc Natl Acad Sci USA. 2005;102:14350–14355. doi: 10.1073/pnas.0505166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff DX, Boerger AL, Bertrand P, Filosi N, Gaida GM, Kane MF, Kolodner RD. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc Natl Acad Sci USA. 1997a;94:7487–7492. doi: 10.1073/pnas.94.14.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff DX, Filosi N, Gaida GM, Kolodner RD. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell. 1997b;88:253–263. doi: 10.1016/s0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, Tomlinson CG, Thompson P, Sarker AH, Shen B, Cooper PK, Grasby JA, Tainer JA. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011;145:198–211. doi: 10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchi JJ, Huang L, Murante RS, Kim Y, Bambara RA. Enzymatic completion of mammalian lagging-strand DNA replication. Proc Natl Acad Sci USA. 1994;91:9803–9807. doi: 10.1073/pnas.91.21.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waga S, Stillman B. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature. 1994;369:207–212. doi: 10.1038/369207a0. [DOI] [PubMed] [Google Scholar]
- Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, Parris T, Kolas NK, Russell R, Hou H, Jr, Kneitz B, Yang G, Kunkel TA, Kolodner RD, Cohen PE, Edelmann W. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003;17:603–614. doi: 10.1101/gad.1060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Zheng L, Zhou M, Chai Q, Parrish J, Xue D, Patrick SM, Turchi JJ, Yannone SM, Chen D, Shen B. Novel function of the flap endonuclease 1 complex in processing stalled DNA replication forks. EMBO Rep. 2005;6:83–89. doi: 10.1038/sj.embor.7400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Dai H, Qiu J, Huang Q, Shen B. Disruption of the FEN-1/PCNA interaction results in DNA replication defects, pulmonary hypoplasia, pancytopenia, and newborn lethality in mice. Mol Cell Biol. 2007a;27:3176–3186. doi: 10.1128/MCB.01652-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Dai H, Zhou M, Li M, Singh P, Qiu J, Tsark W, Huang Q, Kernstine K, Zhang X, Lin D, Shen B. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat Med. 2007b;13:812–819. doi: 10.1038/nm1599. [DOI] [PubMed] [Google Scholar]
- Zheng L, Zhou M, Guo Z, Lu H, Qian L, Dai H, Qiu J, Yakubovskaya E, Bogenhagen DF, Demple B, Shen B. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol Cell. 2008;32:325–336. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Dai H, Hegde ML, Zhou M, Guo Z, Wu X, Wu J, Su L, Zhong X, Mitra S, Huang Q, Kernstine KH, Pfeifer GP, Shen B. Fen1 mutations that specifically disrupt its interaction with PCNA cause aneuploidy-associated cancer. Cell Res. 2011;21:1052–1067. doi: 10.1038/cr.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Shen B. Okazaki fragment maturation: nucleases take centre stage. J Mol Cell Biol. 2011;3:23–30. doi: 10.1093/jmcb/mjq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Dai H, Zhou M, Li X, Liu C, Guo Z, Wu X, Wu J, Wang C, Zhong J, Huang Q, Garcia-Aguilar J, Pfeifer GP, Shen B. Polyploid cells rewire DNA damage response networks to overcome replication stress-induced barriers for tumour progression. Nat Commun. 2012;3:815. doi: 10.1038/ncomms1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Meng X, Zhang S, Lee EY, Lee MY. Characterization of human DNA polymerase delta and its subassemblies reconstituted by expression in the MultiBac system. PLoS ONE. 2012;7:e39156. doi: 10.1371/journal.pone.0039156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1–S9, Supplementary Tables S1 and S2
Supplementary Legends
Source Data for Supplementary Figure S2
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S5
Source Data for Supplementary Figure S6
Source Data for Supplementary Figure S7
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8