

Abstract
A hallmark of the conserved ATM/ATR signalling is its ability to mediate a wide range of functions utilizing only a limited number of adaptors and effector kinases. During meiosis, Tel1 and Mec1, the budding yeast ATM and ATR, respectively, rely on a meiotic adaptor protein Hop1, a 53BP1/Rad9 functional analog, and its associated kinase Mek1, a CHK2/Rad53-paralog, to mediate multiple functions: control of the formation and repair of programmed meiotic DNA double strand breaks, enforcement of inter-homolog bias, regulation of meiotic progression, and implementation of checkpoint responses. Here, we present evidence that the multi-functionality of the Tel1/Mec1-to-Hop1/Mek1 signalling depends on stepwise activation of Mek1 that is mediated by Tel1/Mec1 phosphorylation of two specific residues within Hop1: phosphorylation at the threonine 318 (T318) ensures the transient basal level Mek1 activation required for viable spore formation during unperturbed meiosis. Phosphorylation at the serine 298 (S298) promotes stable Hop1-Mek1 interaction on chromosomes following the initial phospho-T318 mediated Mek1 recruitment. In the absence of Dmc1, the phospho-S298 also promotes Mek1 hyper-activation necessary for implementing meiotic checkpoint arrest. Taking these observations together, we propose that the Hop1 phospho-T318 and phospho-S298 constitute key components of the Tel1/Mec1- based meiotic recombination surveillance (MRS) network and facilitate effective coupling of meiotic recombination and progression during both unperturbed and challenged meiosis.
Introduction
Members of the conserved ATM/ATR family proteins are multi-functional serine/threonine kinases involved in a wide range of processes, including genome duplication, DNA damage repair, cell cycle progression, checkpoint regulation, and meiosis [1–3]. Meiosis is a specialized cell division program, during which a single round of genome duplication is followed by two successive rounds of genome segregation, resulting in halving of the genome. An essential feature of meiosis is that Spo11-catalyzed programmed meiotic DNA double strand breaks (DSBs) are converted to inter-homolog crossovers via meiotic recombination; the crossovers mediate accurate homolog disjunction during the first meiotic division or meiosis I (MI) [4]. During meiotic prophase, the ATM/ATR-based meiotic recombination surveillance (MRS) network ensures that cells do not undergo MI until all Spo11-DSBs are repaired [5, 6].
Central to ATM/ATR signalling is their phosphorylation of a class of proteins referred to as adaptors (or mediators): An adaptor is a scaffold protein that interacts with an effector kinase in an ATM/ATR phosphorylation dependent manner to activate the latter. An activated kinase in turn, phosphorylates relevant downstream targets that are necessary for a developmentally programmed cellular response [2, 7]. Evidence indicates that ATM/ATR utilization of an adaptor and/or effector kinase is regulated by the physiological state of the cell [7]. For example, in response to most forms of DNA damage, Tel1 and Mec1, the budding yeast ATM and ATR, utilize Rad9 (53BP1) and Rad53 (CHK2) as an adaptor and effector kinase, respectively [8, 9]. However, in response to replication stress, a different adaptor, Mrc1 (Claspin) is employed to activate Rad53 [10]. During meiosis, Tel1/Mec1 utilize Hop1, a conserved meiotic chromosome axis protein, and Mek1, a chromosome associated serine/threonine kinase, as a meiosis-specific adaptor and effector kinase, respectively [6, 11–13].
During meiotic prophase in budding yeast, where the molecular basis of ATM/ATR-function is best understood, Tel1 is activated by Spo11-catalysis of programmed DNA double strand breaks (DSBs) [14]; Mec1 activation, on the other hand, is dependent on single-stranded DNA and occurs following DSB resection [5]. Activated Tel1 and Mec1 phosphorylate a number of conserved meiotic proteins, including the above mentioned Hop1, Zip1, a component of the synaptonemal complex, and Rec114, a Spo11-accessory protein required for meiotic DSB formation [6, 15, 16].
An essential meiotic function of Tel1/Mec1 is to promote inter-homolog bias in meiotic recombination [6]. They achieve this via Hop1 phosphorylation, leading to phospho-Hop1-dependent activation of Mek1 [6]. Activated Mek1, in turn, is proposed to phosphorylate relevant target proteins, including Rad54, to ensure the inter-homolog bias in meiotic DSB repair [17, 18]. Another key function of Tel1/Mec1 is to mediate meiotic checkpoint responses. For example, they trigger meiotic arrest in response to accumulation of unrepaired meiotic DSBs in the absence of Dmc1, a conserved meiotic RecA protein [5, 19]. Intriguingly, Tel1 and Mec1 utilize the same adaptor and effector kinase, Hop1 and Mek1, respectively, for promoting the essential inter-homolog bias as well as for implementing meiotic checkpoint arrest [6].
Here we investigated the molecular basis of Tel1/Mec1-dependent signalling cascade mediated by Hop1/Mek1, allowing us to separate essential and checkpoint functions. We present evidence that the dual functionality is facilitated by differential phosphorylation of their meiotic adaptor Hop1 and the phosphorylation-dependent regulation of Mek1 activation.
Results
Hop1-S298 is an in vivo phosphorylation site
Hop1 contains eight ATM/ATR consensus sites (nine in the SK1 strain background), referred to as SQ/TQ motifs, each comprising of a serine (S) or threonine (T) followed by a glutamine (Q) (Fig 1A). Of the eight SQ/TQ motifs, the phospho-T318 is required for the essential recruitment and activation of Mek1, while the threonine at position 181 might play a different role [6]. When replacing any of the remaining SQ/TQ sites to alanine, a residue that cannot be phosphorylated, all of the mutant alleles appear to behave indistinguishably from the wild type during unchallenged meiosis, except for the serine 298 (S298), elimination of which confers a modest reduction in spore viability [6] (below).
Fig 1. Lack of the Hop1-phospho-S298 leads to temperature- and dose- dependent meiotic failure.
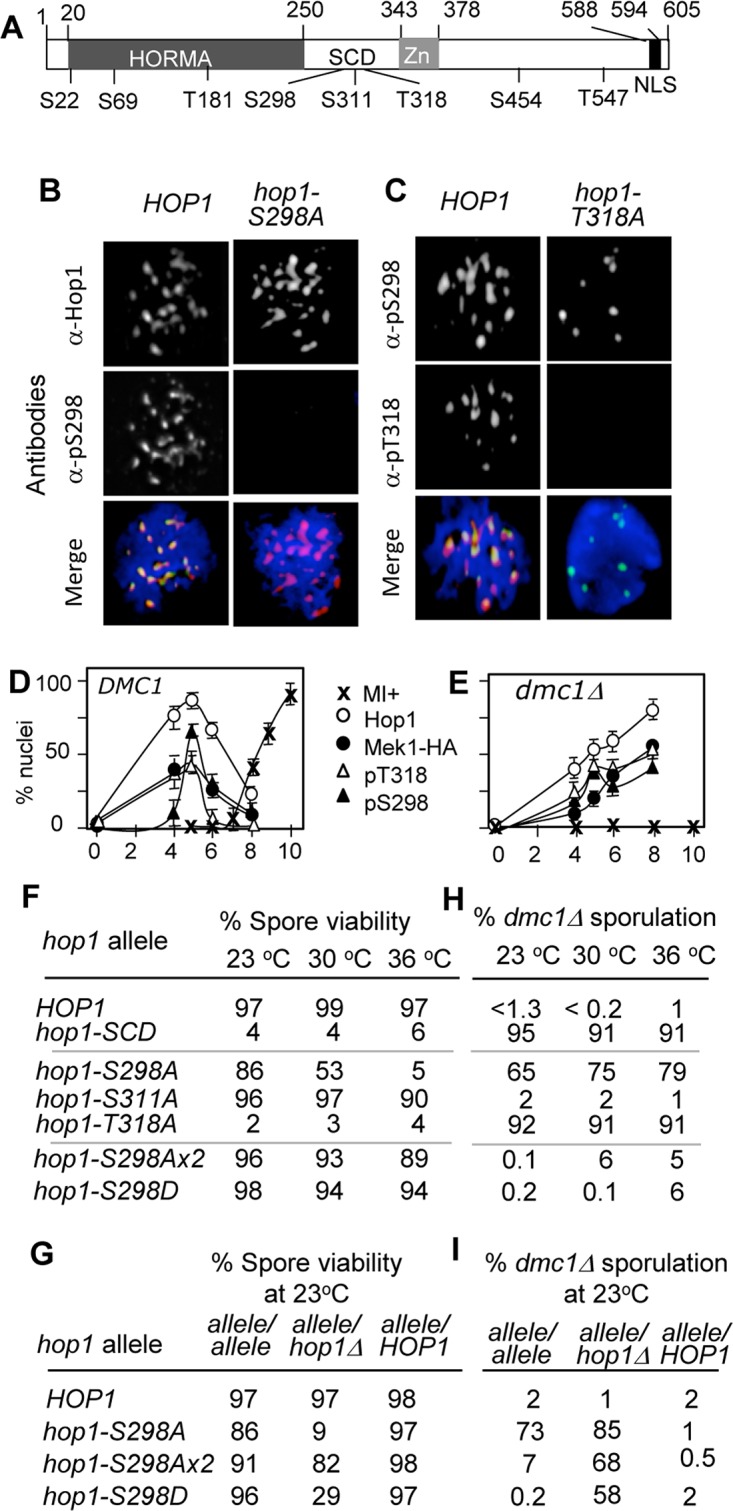
(A) Schematic representation of Hop1 with the locations of eight [S/T]Q motifs. S: serine, T: threonine, SCD: [S/T]Q Cluster Domain. Also shown are the HORMA domain, Zn finger motif, and nuclear localization signal (NLS). (B) and (C) Specificity of the phospho-specific α-pS298 and α-pT318 antibodies. Nuclear spreads of HOP1 and hop1-S298A panel (B) or HOP1 and hop1-T318A panel (C) were prepared from samples taken at 5hours after induction of synchronous meiosis at 23°C. The spreads were stained with DAPI and the antibodies against either the phospho-S298 panel (B) or the phospho-T318 panel (C). (D) and (E). In vivo phosphorylation of Hop1-S298 and T318 during DMC1 (D) or dmc1Δ (E) meiosis at 23°C. Nuclear spreads of a DMC1 or dmc1Δ strain were prepared from samples collected at the indicated time points. The spreads were stained with the antibodies against Hop1, HA (for detection of Mek1-HA), and the two phospho-specific antibodies, α-pS298 and α-pT318. A nucleus exhibiting 10 or more foci of each epitope was scored as a positive. Also shown are the fractions of cells having undergone first meiotic division or meiosis I (MI) at each time point. Errors were calculated from the 95% confidence interval of a binomial distribution. (F) Spore viability of homozygous diploid strains of indicated genotype at specified temperature. For each genotype, at least 80 spores were analysed. A: Alanine; D: aspartic acid, hop1-S298Ax2: an allele containing two tandem copies of hop1-S298A. hop1-SCD: an allele where the S298, S311, and T318 in the SCD are mutated to A [6]. hop1-S311A: an allele where the S311 is mutated to A. (G) Spore viability of indicated HOP1 alleles at 23°C as a either homozygous diploid (allele/allele), hemizygous diploid (allele/ hop1Δ) or heterozygous diploid (allele/HOP1). (H) Sporulation efficiency of dmc1Δ strains in the indicated hop1 mutation background. (I). Sporulation efficiency of dmc1Δ strains in the indicated hop1 mutation background at 23°C as a either homozygous diploid (allele/allele), hemizygous diploid (allele/ hop1Δ) or heterozygous diploid (allele/HOP1).
To confirm that the Hop1-pS298 was an in vivo phosphorylation site, we generated antibodies against the corresponding phospho-peptide, referred to as α-pS298 (Materials and Methods). As a control, we also raised antibodies against a confirmed in vivo phospho-residue, the Hop1 phospho-T318, referred to as α-pT318 [6, 20]. Cytological analysis showed that both the α-pS298 and α-pT318 antibodies generated signals in nuclear spread samples prepared from a WT control and that these signals co-localized with α-Hop1 foci (Fig 1B and 1C). Importantly, the α-pS298 antibodies did not generate any signals in a strain expressing a mutant allele, hop1-S298A, where the corresponding S298 was replaced with a non-phosphorylatable alanine (A) (Fig 1B; S1A and S1B Fig). Similarly, the α-pT318 antibodies did not generate a signal in a hop1-T318A background, where the T318 was replaced with an alanine residue (Fig 1C; S1A and S1B Fig).
The Hop1 phospho-S298 or phospho-T318 signals were observed transiently during meiotic prophase (Fig 1D), the period during which Hop1 is known to undergo transient Tel1/Mec1-dependent phosphorylation [6, 21]. In a dmc1Δ background, Hop1 phosphorylation does not turn over but is maintained in a Tel1/Mec1-dependent manner [6, 22]. We observed that the α-pT318 and α-pS298 signals in a dmc1Δ background did not turn over either, but continued to accumulate (Fig 1E). These observations taken together, we conclude that the Hop1-S298 is an in vivo Tel1/Mec1 phosphorylation site, which becomes phosphorylated during both normal and challenged meiosis.
Prevention of Hop1 phosphorylation at Ser298 confers a dose- and temperature-dependent meiotic failure
Having confirmed in vivo phosphorylation of the Hop1-S298, we proceeded to investigate its function(s). To this end, we characterized the above mentioned non-phosphorylatable allele, hop1-S298A. Spore viability of a hop1-S298A strain was temperature-sensitive in that it dropped from 86% at 23°C to 5% at 36°C (Fig 1F; S1C Fig). In contrast, spore viability of the other hop1 alleles tested (i.e. hop1-SCD, hop1-S311A, and hop1-T318A) was unaffected by changes in temperature (Fig 1F). A strain expressing a phospho-mimetic allele, hop1-S298D, where the S298 was replaced with a negatively charged aspartic acid residue (D) was viable at all temperatures (Fig 1F). Doubling copy number of the hop1-S298A also improved spore viability at 36°C from 5% to 89% (Fig 1F, hop1-S298Ax2), while halving it reduced the viability at 23°C from 86% to 9% (Fig 1G, compare allele/allele and allele/hop1Δ for hop1-S298A).
The temperature- and dose-dependent spore viability of a hop1-S298A strain suggested that the phospho-S298 might be required for Hop1 stability at high temperature. However, analysis showed that neither the mutation nor temperature caused substantial reductions in Hop1 levels, relative to wild type (S1D Fig). We also found that Hop1 chromosome association was normal in a hop1-S298A background at high temperature (data not shown).
Phosphorylation of Hop1 at S298 is required for preventing DMC1-independent DSB repair
Inactivation of Dmc1 triggers Mec1-mediated meiotic arrest, which is dependent on the Hop1 phospho-T318 [5, 6]. To test whether the phospho-S298 was similarly required, we assessed sporulation efficiency of a hop1-S298A dmc1Δ strain. Results showed that the double mutant sporulated efficiently, with its sporulation efficiency ranging from 65% at 23°C to 79% at 36°C (Fig 1H). On the other hand, expression of the phospho-mimetic allele hop1-S298D or multi-copy hop1-S298Ax2 restored the arrest (Fig 1H and 1I). We conclude that the phospho-S298, like the phospho-T318, is required for implementing dmc1Δ arrest.
dmc1Δ-mediated meiotic arrest is triggered by accumulation of unrepaired meiotic DSBs, which activates the checkpoint function of Tel1/Mec1 [19]. The arrest can be bypassed by either the mutations that permit meiotic progression in the presence of unrepaired breaks (e.g. mec1-1, rad24Δ, or rad17Δ) or allowing Rad51 mediated DSB repair (e.g. hed1Δ, hop1-T318A or mek1Δ) [5, 6, 22–24]. Rad51 is the other budding yeast RecA homolog, whose recombinase function becomes inhibited during meiosis by its meiosis-specific inhibitor Hed1 [24, 25]. To test which of the two mechanisms was responsible for the hop1-S298A alleviation of dmc1Δ meiotic arrest, we examined the status of meiotic DSBs in a hop1-S298A dmc1Δ strain. Results showed that breaks did not accumulate in the double mutant (Fig 2A). Since Spo11 catalysis initiates normally in the absence of the Hop1 phospho-S298 [6], the latter suggests that the hop1-S298A alleviation of dmc1Δ arrest is attributable to Rad51-mediated recombination, circumventing accumulation of unrepaired DSBs.
Fig 2. Genetic interaction among hop1-S298A, dmc1Δ, and hed1Δ.

A. PFGE/Southern analysis of ChrIII was performed on samples prepared from a hop1-S298A dmc1Δ or HOP1 dmc1Δ strain. Positions of the full length (FL) and DSBs are indicated on the right side of the gel. Positions of the CHA1 probe (P) and centromere (filled circle) are indicated on the left side of the gel. B. Spore viability of homozygous diploid strains of the indicated genotypes at 23°C. For each genotype, at least 80 spores were analysed.
Hop1-S298 phosphorylation supports high levels of spore viability in the absence of HED1
High spore viability of a hop1-S298A strain at 23°C (Fig 1F, 86%) implies that the phospho-S298 can be dispensable for essential crossover formation under certain conditions. The latter, in turn, raises the possibility that the DMC1-independent break repair in a hop1-S298A dmc1Δ strain at 23°C might proceed with inter-homolog bias and restore spore viability of a dmc1Δ strain. We tested this possibility and found that spore viability of a hop1-S298A dmc1Δ strain was very low (0.8%; Fig 2B). We conclude that DSB repair in a hop1-S298A dmc1Δ background does not proceed with inter-homolog bias.
Deletion of HED1, the gene encoding for a meiosis-specific inhibitor of Rad51, restores spore viability of dmc1Δ cells, indicating that Rad51-mediated DSB repair in a hed1Δ dmc1Δ background can proceed with reduced inter-homolog bias [24, 26]. We observed that hop1-S298A led to a significant reduction in spore viability of a hed1Δ dmc1Δ strain, from 29.6% to 0.6% (Fig 2B). Therefore, the residual inter-homolog bias in Rad51-mediated recombination in a hed1Δ dmc1Δ background is dependent on the Hop1 phospho-S298. Furthermore, we observed synthetic interaction between hop1-S298A and hed1Δeven in the presence of Dmc1, with the spore viability of a hed1Δ hop1-S298A DMC1 strain (47.5%) being notably reduced at 23°C compared to either hed1Δ DMC1 (97.5%) or hop1-S298A DMC1 (86%) (Fig 2B).
Hop1-S298 phosphorylation is dispensable for essential Mek1 activation during normal meiosis
Genetic evidence above suggests that the Hop1 phospho-S298 plays an auxiliary role, along with the essential phosho-T318, to promote spore viability and mediate dmc1Δ meiotic arrest. We wished to address the molecular basis of its function. Since an essential function of the Tel1/Mec1 phosphorylation of Hop1 is to activate Mek1 [6], we proceeded to assess the effects of hop1-S298A on Mek1 phosphorylation. In a HOP1 strain during normal meiosis, Mek1 phosphorylation was modest and transient, observed at 4 and 6 hours (Fig 3C). Comparable levels of Mek1 phosphorylation, reaching ~24% of total Mek1-HA signal at t = 6 hours, were observed in hop1-S298A cells (Fig 3C). As shown previously [6, 20], no Mek1 activation was observed in a hop1-T318A background. We conclude that the Hop1 phospho-S298, unlike the phospho-T318, is dispensable for the essential Mek1 activation during unchallenged meiosis.
Fig 3. Hop1-S298 phosphorylation is required for dmc1Δ-dependent meiotic checkpoint response.

(A) and (B). Fraction of cells undergone meiosis I (MI) in synchronous meiotic cultures of HOP1, hop1-S298A, and hop1-T318A at 23°C in a DMC1 or dmc1Δ background. T50: Time at which 50% of the active culture has completed MI. At least three separate timecourses were considered, for each set of strains for each background. T50 kinetics were calculated from the most representative timecourse for each set of strain and DMC1/dmc1 background. Errors were calculated from the 95% confidence interval of a binomial distribution. (C) and (D). Effects of hop1-S298A and hop1-T318A on Mek1-HA phosphorylation during DMC1 and dmc1Δ meiosis. Samples from the cultures described in panels (A) and (B) were subjected to Western blot analysis using anti-HA antibody. Positions of the unphosphorylated and phosphorylated Mek1-HA species are indicated to the right of the blot. ‘% pMek’ corresponds to the proportion of phosphorylated Mek1-HA species in each lane calculated by dividing the signal found in the ‘pMek1-HA’ area of the gel by the total signal (‘pMek1-HA’ + ‘Mek1-HA’). (E) and (F). Effects of hop1-S298A and hop1-T318A on Hop1 phosphorylation during DMC1 and dmc1Δ meiosis. Samples from the cultures described in panels (A) and (B) were subjected to Western blot analysis using anti-Hop1 antibody. Positions of the unphosphorylated and phosphorylated Hop1-species are indicated to the right of the blot. Shown on the right panels is quantification analysis of the Western images, where the signal in the ‘pHop1’ region in each lane is divided by the total signal (‘pHop1’+ ‘Hop1’) in the corresponding lane.
Hop1-S298 phosphorylation promotes hyper-phosphorylation of Mek1 necessary for mediating dmc1Δ meiotic arrest
Deletion of DMC1 leads to constitutive Mek1 phosphorylation and changes in its mobility shift-pattern that are indicative of an additional phosphorylation event(s) [6, 20] (Fig 3D). Constitutive Mek1 phosphorylation was also observed in a hop1-S298A dmc1Δbackground; however, the pattern of Mek1 mobility shift in the latter remained comparable to HOP1 DMC1 or hop1-S298A DMC1, revealing that the Mek1 hyper-phosphorylation in a dmc1Δ background is dependent on the phospho-S298.
HOP1 dmc1Δ cells arrest before the onset of first meiotic division or meiosis I (MI) (Fig 3B) [19]. In contrast, hop1-S298A dmc1Δ or hop1-T318A dmc1Δ cells underwent MI (Fig 3B). In a hop1-S298A dmc1Δ background, where the basal level Mek1 phosphorylation was maintained (Fig 3D), the onset of MI was delayed by approximately 1 hour (Fig 3A and 3B). In a hop1-T318A background, where Mek1 phosphorylation does not occur, no dmc1Δ-dependent delay was observed (Fig 3A and 3B). Similar correlation between the extent of Mek1-phosphorylation and robustness of dmc1Δ-induced meiotic arrest was observed among different hop1-S298 alleles (i.e. hop1-S298A, hop1-S298D, and hop1-S298Ax2, Fig 1H and 1I) (S2 Fig). Taken together, we conclude that the Hop1 phospho-T318-dependent recruitment and activation of Mek1 is necessary but not sufficient to implement dmc1Δ meiotic arrest; the arrest requires further activation of Mek1 mediated by the Hop1 phospho-S298.
Hop1 phospho-S298 ensures constitutive Hop1/Mek1-signalling in the absence of Dmc1
Following Spo11-catalysis, Hop1 is phosphorylated at multiple residues, including most, if not all, of the eight Tel1/Mec1-consensus sites [6, 20, 21]. We observed comparable levels of Hop1 phosphorylation among HOP1, hop1-S298A, and hop1-T318A strains in a DMC1 background, indicating that neither the phospho-T318 nor the phospho-S298 have a significant effect on the transient Hop1 phosphorylation during unchallenged meiosis (Fig 3E). In contrast, the dmc1Δ-dependent constitutive Hop1 phosphorylation was impaired in both mutants (Fig 3F).
Next, we assessed the effects of hop1-S298A on Hop1- and Mek1- chromosome association. In a DMC1 background, hop1-T318A cells exhibited a modest reduction in transient Hop1-chromosome association and no detectable signal in Mek1 association (Fig 4B and 4D). In a hop1-S298A DMC1 background, both Hop1- and Mek1-chromosome association occurred normally (Fig 4B and 4D), supporting the observation above that the phospho-S298 is dispensable for the essential Mek1 activation during normal meiosis. In the absence of DMC1, the dmc1Δ-dependent maintenance of Hop1/Mek1 chromosome association was impaired in a hop1-S298A as well as a hop1-T319A background (Fig 4A, 4C and 4D).
Fig 4. Hop1-S298 phosphorylation promotes stable Mek1-Hop1 interaction on chromosomes.

(A) Hop1 and Mek1-HA chromosome association during dmc1Δ meiosis at 23°C in a HOP1, hop1-S298A, or hop1-T318A background. (B) and (C) Effects of hop1-S298A or hop1-T318A on Hop1 chromosome association during DMC1 or dmc1Δ meiosis. Fraction of cells exhibiting 10 or more Hop1 foci was scored. (D) and (E) Effects of hop1-S298A or hop1-T318A on Mek1-HA chromosome association during DMC1 or dmc1Δ meiosis. Fraction of cells exhibiting 10 or more Mek1-HA foci was scored. (F) and (G) Effects of hop1-S298A and hop1-T318A on Hop1-Mek1 co-localization during DMC1 and dmc1Δ meiosis. Fraction of nuclei where more than 80% of Mek1-HA foci co-localized with Hop1 foci was scored. (B-G) Errors were calculated from the 95% confidence interval of a binomial distribution. (H) and (I) Effects of hop1-S298A on Hop1-Mek1 interaction on chromosomes. Nuclear spreads of HOP1 dmc1Δ and hop1-S298A dmc1Δ were prepared from samples taken at 6 hours after induction of synchronous meiosis at 23°C. The spreads were stained with DAPI and the antibodies against Hop1 and HA (for detection of Mek1-HA).
Hop1-phospho S298 promotes stable Mek1-Hop1 interaction on chromosomes
Potential effects of the Hop1 phospho-S298 on co-localization between Hop1 and Mek1 were assessed. In a HOP1 background, nearly all Mek1 foci were found nearby a Hop1 signal (Fig 4H). In contrast, the majority of the Mek1 foci in a hop1-S298A background was found without a nearby Hop1 signal (Fig 4I). Quantitative analysis revealed that the fraction of nuclei exhibiting significant co-localization between Hop1 and Mek1 was significantly reduced in a hop1-S298A background, irrespective of the status of DMC1 (Fig 4F and 4G). We conclude that the Hop1 phospho-S298 promotes the continued Hop1-Mek1 interaction on chromosomes following the initial phospho-T318 mediated recruitment of Mek1.
Discussion
Hop1 phospho-T318 and phospho-S298 mediate gradual activation of Hop1 and Mek1
Taken together, evidence thus far suggests that the Tel1/Mec1 activation of Hop1/Mek1 during normal or challenged meiosis occurs in a gradual manner dependent on the Hop1 phospho-T318 and the phospho-S298: (i) For the transient Hop1 phosphorylation during normal meiotic prophase I, neither the phospho-T318 nor the phospho-S298 is required. (ii) For the essential Mek1 recruitment and activation during normal meiosis, only the phospho-T318 is required. And (iii) For the Mek1 hyper-phosphorylation and constitutive Hop1/Mek1 chromosome-association necessary for implementing dmc1Δ meiotic arrest, both the phospho-T318 and the phospho-S298 are required.
We first showed the requirement for the phosphorylation of Hop1 at the SCD on Mek1 recruitment to the axial elements and for its activity [6]. Furthermore, we showed that among the three S/T[Q]s present at the SCD in Hop1, only the phospho-T318 was strictly essential for Hop1 activity over Mek1 initial recruitment and function [6, 20]. We and others proposed that the phospho-dependent activation of Mek1 through phospho-Thr318 relied on its possible interaction with the forkhead-associated (FHA) domain present in Mek1. FHA domains are modular phosphopeptide recognition domains with a striking specificity for phosphorylated threonine residues in target proteins [27–29]. Replacing p-Thr by p-Ser severely compromises binding and the associated function triggered by the FHA-phosphopeptide complex formation [6, 30]. Whereas the presence of a phospho-threonine is still a requirement, some studies have shown that the situation might be more complicated than originally thought, for example, combinations of mono- bi- or tri- phospho-peptide can display differential binding affinity to certain FHA modules in vitro [20, 31, 32].
Interestingly, multi-phospho-peptide binding to FHA modules has been proposed as the mechanism that could induce hierarchical co-operative association of two FHA modules in a sequential manner. This is manifested in proteins that contain segments with multiple phosphorylated threonines as well as serines [31, 32]. Here we present evidence of the existence of synergistic function for one of the two T318 neighbouring phospho-serines, part of the SCD. p-Ser298 is a confirmed phosphorylation site that could enhance or mediate, signal amplification under conditions where persistent and robust, Mek1 association with Hop1, like it occurs in dmc1 mutants, is required. Interestingly, p-Ser298 is surrounded by three other threonines in position -1, +2 and +3, which could also be subjected to phosphorylation, perhaps by a kinase activity promoted by the initial binding of Hop1-pThr318 with FHA-Mek1, and triggered by the presence of phospho-serine298. It is tempting to speculate that phosphorylation within the SCD of Hop1 of additional threonines near p-Ser298 could serve as secondary binding platforms for another FHA-module for a different Mek1 molecule, thus favouring oligomerization and trans-activation of Mek1 [21]. Although several other scenarios might be possible [21, 22, 33–36], the need for more genetic, biochemical and structural data, together with the identification of additional posttranslational modifications in Hop1, Red1 and Mek1 should be the subject of future studies to help identifying the mechanism underlying Mek1 activation.
Another important clue emerging from this study is the confirmation for the need of multiple phosphorylation sites in the context of two interacting molecules during the response to meiotic DSBs. Most ATR/ATM targets, with many of them usually involved in multi-complex formation triggered by DNA damage, contain clusters of S/T[Q]s (SCDs) as opposed to a single reactive phospho-residue [37]. Specific subsets of phosphorylations in Hop1 might select for specific activities in this multi-functional adaptor protein.
Currently, the basis of the phospho-T318-independent Mek1 chromosome-association remains unknown. It is possible that Mek1 is recruited to chromosomes via Red1, another meiotic chromosome axis protein known to form a complex with both Hop1 and Mek1 [13, 38, 39].
Model: Hop1 phospho-T318- and -S298-dependent stepwise activation of Mek1 facilitates Tel1/Mec1-dependent coupling of meiotic recombination and progression
The evidence shown above indicates that the Tel1/Mec1 activation of Hop1/Mek1 proceeds in a stepwise manner dependent on the Hop1 phospho-T318 and phospho-S298: The phospho-T318 mediates essential Mek1 recruitment and phosphorylation (Fig 5ii) and the phospho-S298 promotes stable interaction between Hop1 and Mek1 on chromosomes, following the phospho-T318-dependent Mek1 recruitment (Fig 5iii). While both phospho-T318 and -S298 contribute to an essential function(s) of Hop1, our findings suggest that contribution of the phospho-S298 is minor compared to the essential Hop1 phospho-T318.
Fig 5. Model: Tel1/Mec1 phosphorylation of Hop1 at the T318 and S298 ensures effective coupling of meiotic recombination and progression.

(i) Spo11-catalysis of meiotic DSBs triggers Tel1/Mec1 phosphorylation of chromosome bound Hop1 at multiple residues, including the T318 and S298. (ii) The phospho-T318 mediates the initial Mek1-recruitment and phosphorylation, independently of the phospho-S298. (iii) The phospho-S298 promotes stable Hop1-Mek1 interaction on chromosomes. (iv) The phospho-T318 and phospho-S298 promote spore viability by ensuring inter-homolog repair of meiotic breaks; available genetic evidence suggests that the phospho-T318 and phospho-S298 might be involved in regulating the Dmc1- and Rad51-dependent repair process, respectively (see text). (v) Once the essential crossover requirement is met, Ndt80 is activated, leading to exit from meiotic prophase (vi) and irreversible inactivation of Spo11-complex (vi). (viii) Hop1/Mek1 de-phosphorylation and removal from chromosomes ensue, accounting for the transient activation of the Hop1/Mek1-signalling during unchallenged meiosis. (ix, x) During challenged meiosis (e.g. dmc1Δ), Mek1 undergoes the Hop1 phospho-S298-dependent hyper-phosphorylation (ix), necessary for implementing a meiotic checkpoint response (x).
What could be the role of the phospho-S298? The observed synthetic interaction between hed1Δ and hop1-S298A suggests that the phospho-S298 might have a role in regulating Rad51 activity. For instance, in the absence of Hed1, the phospho-S298 might assume the role of Hed1 and inhibit Rad51-mediated DSB repair. However, the fact that the phospho-S298 is required for viability of hed1Δ dmc1Δ spores (above) would argue against the notion that the phosphorylation prevents Rad51-mediated recombination. Instead, the Hop1 phospho-S298 might be involved in ensuring inter-homolog bias of Rad51-mediated DSB repair in hed1Δ. An implication of the latter would be that Rad51-mediated meiotic recombination, similar to the Dmc1-mediated process, is subjected to regulatory process that promotes inter-homolog bias. It is tempting to speculate that the Hop1 phospho-T318 and phospho-S298 might mediate essential crossover formation by regulating the Dmc1- and Rad51-mediated repair pathways, respectively (Fig 5iv).
Earlier works have shown that Mek1 can phosphorylate other targets which might impact in the outcome of Rad51 strand invasion activity. Rad54, a dsDNA-dependent ATPase, facilitates homologous recombination in concert with Rad51. Phosphorylation of Rad54 by Mek1 attenuates its interaction with Rad51 as well as reducing Rad51 activity [17]. The possibility that Hop1-pS298 could be required to promote this activity might seem obvious, nevertheless, we cannot exclude other more complex scenarios where Rad54 inhibition would not be essential to reinforce IH-bias, for example by Mec1/Hop1-pS298-dependent regulation of the other dsDNA-dependent ATPase, Tid1/Rdh54 [40].
Evidence suggests that the Tel1/Mec1-control of meiotic progression is via Ndt80 activation [15, 41]. Ndt80 is a meiotic transcription factor required for exit from meiotic prophase (Fig 5vi) and irreversible inactivation of the Spo11-complex (Fig 5vii) [15, 42, 43]. Interestingly, we observed that the Hop1 phopho-S298 was required for spore viability of a mutant with reduced Spo11-catalysis (rec114-8D) [15], which suggests that the phospho-S298 might also contribute to viable spore formation by preventing premature inactivation of the Spo11-complex until the requirement for essential crossover formation is satisfied.
During normal meiosis, cells would eventually acquire a sufficient level of crossovers and exit meiotic prophase (Fig 5v and 5vi). Hop1/Mek1 dephosphorylation and removal from chromosomes would ensue, accounting for the transient nature of Hop1/Mek1 activation (Fig 5viii). In the absence of Dmc1, meiotic DSBs accumulate and trigger a Tel1/Mec1- and Hop1/Mek1-dependent meiotic arrest. Here, we demonstrate that the arrest is dependent on the Hop1 phospho-S298-mediated Mek1 hyper-phosphorylation (Fig 5ix and 5x). Currently, the nature of the phospho-S298 and dmc1Δ-dependent Mek1 phosphorylation remains unknown. Notably however, we observed a synthetic interaction between hop1-S298A and mek1-S320A, a mek1 allele lacking a phosphorylation site required for mediating dmc1Δ arrest, suggesting an involvement of the Mek1 phospho-S320 [21, 22] (S3 Fig).
In summary, evidence presented above indicates that the Tel1/Mec1 activation of Hop1/Mek1 during meiotic prophase proceeds in a stepwise manner dependent on Hop1 phospho-T318, phospho-S298, and the status of meiotic recombination. We propose that the phospho-T318 and phospho-S298 constitute key components of the Tel1/Mec1-based meiotic recombination surveillance (MRS) network [15, 44, 45] and that they ensure a successful meiotic outcome during both normal and challenged meiosis by facilitating effective coupling of meiotic recombination and progression.
Materials and Methods
Yeast manipulation
All strains were diploids of the SK1 background; relevant genotypes of the strains are listed in S1 Table. Mutagenesis of HOP1 containing plasmid and integration in hop1Δ strains was performed as in [6]. Integration and copy number were confirmed by digesting DNA from transformed colonies with the restriction enzyme BamHI. Southern blots were then performed where membranes were hybridized using a probe that mapped within the URA3 ORF. Correct integration of a single copy appeared as two bands of approximately14kbp and 6kpb. Multiple integrations appeared as a third band of ~8.4kbp. Additional number of copies of Hop1 plasmids (8.4kbp) were estimated by quantifying the intensity of the third band and was then compared it with the intensities of the 14kbp and the 6kbp bands. hop1-S298Ax2 was considered when the intensity of the 8.4kbp band was approximately equivalent in intensity to each of the other two individual bands (14kbp and 6kbp). Induction of synchronous meiosis was carried out according to a described protocol [16]. All pre-growth was carried out at 30°C; meiotic time courses were carried out at 23°C, 30°C, or 33°C as indicated.
Generation of phospho-specific Hop1 antibodies
Polyclonal antibodies against the Hop1 phospho-T318 and phospho-S298 were obtained as following: The α-pT318 polyclonal antibody [Cambridge Research Biochemicals] was obtained by immunising two rabbits with the antigenic[C]-Ahx-ASIQP-[pT]-QFVSN where C represents the C-terminus of the peptide, Ahx is aminohexanoicacid and pT is a phosphorylated threonine residue. Upon bleeding, antibodies were purified through two affinity columns (each followed by a purification pass), the first adsorbing antibodies that bind to non-phosphorylated peptides and the second adsorbing the phospho-specific antibodies to pT318. The specificity of the antibody was tested using ELISA (enzyme-linked immunosorbent assay) analysis. The polyclonal phospho-specific antibody against phosphorylated serine residue 298 [Eurogentec] was obtained by immunising four guinea pigs with the antigenic peptide [C]-PQNFVT-[pS]-QTTNV, where C represents the C-terminus of the peptide and pS is a phosphorylated serine residue. The α-pS298 antibody was purified in a similar manner to the α-pT318 antibody.
Western blot analysis
Protein extraction and Western blot analysis of Hop1 were carried as previously described [15]. Western blot analysis of Mek1-3HA was carried out using 7.5% acrylamide gels containing 200μM of MnCl2 and 4μM of PhosTag (AAL-107; NARD Institute, Amagasaki, Japan). A mouse monoclonal anti-HA antibody was used for detection of Mek1-HA as previously described [6].
Cytology
The preparation of meiotic nuclear spreads and immunofluorescence analysis were carried out as previously described [6]. The secondary antibodies used to detect the α-pT318 and α-pS298 phospho-specific antibodies were chicken anti-rabbit Alexa-594 [Invitrogen] and goat anti-guinea pig Alexa-594 [Invitrogen], respectively.
Supporting Information
A, B. Effects of hop1-S298A and hop1-T318A on Hop1-S298 or Hop1-T318 phosphorylation during DMC1 or dmc1Δ meiosis at 23°C meiosis. Representation of the relative signals obtained from the quantification of the entire signal detected by western blot in A B using the anti-Hop1, anti-pT318, and anti-pS298 antibodies. C. Homozygous diploids of HOP1 and hop1-S298A were incubated on SPM plate at the indicated temperature for either one (30°C, 33°C, 36°C) or two days (18°C, 23°C). Tetrads were dissected on YPD plates and incubated at 30°C. The images were taken following 2 day incubation. D. Temperature sensitivity of hop1-S298 is not associated with a notable effect on protein stability or phosphorylation. Strains of indicated genotypes were taken through synchronous meiosis at 23°C or 33°C. Samples were collected at the indicated time points and subjected to Western Blot analysis using polyclonal antibodies to Hop1. Positions of unphosphorylated or phosphorylated Hop1 species are as indicated. Shown below each Western blot image is the corresponding ponceau staining gel as a loading control. hop1-S298A: non phosphorylatable allele, hop1-S298D: phospho-mimetic allele, hop1-S298x2: an allele containing two tandem copies of hop1-S298A.
(TIF)
Homozygous diploids of indicated genotypes were taken through synchronous meiosis at 23°C. Samples were collected at the indicated time points and subjected to Western Blot analysis using anti-HA antibody for detection of Mek1-HA. Positions of unphosphorylated or phosphorylated Mek1-HA species are as indicated. Shown below are sporulation efficiency in each culture and quantification analysis of the Western images, where the signal in the ‘pMek1-HA’ region in each lane is divided by the total signal (‘pMek1-HA’+ ‘Mek1-HA’) in the corresponding lane.
(TIF)
Homozygous diploids of indicated genotypes were incubated on SPM plates at 18°C, 30°C, and 36°C for one (30°C, 36°C) or two days (18°C). Tetrads were dissected on YPD plates and incubated at 30°C for two days. Spore viability was calculated as the number of visible spore colonies over the total number of spores dissected. For each strain, at least 160 spores were analysed.
(TIF)
All strains are homozygous diploids of the SK1 background of S. cerevisiae. All strains except HTY2091-2092 and APY67-68/70 were derived from JCY448 (ho::LYS2/ho::LYS2, ura3/ura3, leu2::hisG/leu2::hisG, hop1Δ::LEU2/hop1Δ::LEU2) (Carballo et al. 2008); from JCY511 (ho::LYS2/ho::LYS2, ura3/ura3, leu2::hisG/leu2::hisG, hop1Δ::LEU2/ hop1Δ::LEU2 dmc1Δ::KanMX4/ dmc1Δ::KanMX4) (Carballo et al. 2008); and JCY614 (ho::LYS2/ho::LYS2, ura3/ura3, leu2::hisG/leu2::hisG, hop1Δ::LEU2/hop1Δ::LEU2, MEK1-3HA::URA3/ MEK1-3HA::URA3) (Carballo et al. 2008). HTY2091-HTY2092 are derived from TBR2091 and TBR2092 respectively from (Tsubouchi and Roeder, 2006). APY68-69 were derived from HTY2091 and APY67 was derived from HTY2092.
(PDF)
Supplementary data tables showing the values plotted in the graphs from Figs 1D and 1E, 3A, 3B and 3F, 4B–4G, and S1A and S1B, and S2 Figs.
(XLSX)
Acknowledgments
We would like to thank D. Bishop, V. Börner, E. Hoffmann, N. Hollingsworth, F. Klein, M. Neale, and H. Tsubouchi for strains, reagents and antibodies.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Research was partly funded by the MRC program grant U1175.01.005.00005.01 from RSC and by the MRC centre grant G0801130 from JAC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297(5581):602–6. 10.1126/science.1071398 [DOI] [PubMed] [Google Scholar]
- 2. Harper J, Elledge S. The DNA Damage Response: Ten Years After. Mol Cell. 2007;28:739–45. [DOI] [PubMed] [Google Scholar]
- 3. Carballo JA, Cha RS. Meiotic roles of Mec1, a budding yeast homolog of mammalian ATR/ATM. Chromosome Res. 2007;15(5):539–50. Epub 2007/08/04. 10.1007/s10577-007-1145-y . [DOI] [PubMed] [Google Scholar]
- 4. Bishop D, Zickler D. Early decision: meiotic crossover interference prior to stable strand exchange and synapsis. Cell. 2004;117:9–15. [DOI] [PubMed] [Google Scholar]
- 5. Lydall D, Nikolsky Y, Bishop DK, Weinert T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature. 1996;383(6603):840–3. Epub 1996/10/31. 10.1038/383840a0 . [DOI] [PubMed] [Google Scholar]
- 6. Carballo JA, Johnson AL, Sedgwick SG, Cha RS. Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell. 2008;132(5):758–70. Epub 2008/03/11. S0092-8674(08)00135-9 [pii] 10.1016/j.cell.2008.01.035 . [DOI] [PubMed] [Google Scholar]
- 7. Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol. 2002;14(2):237–45. [DOI] [PubMed] [Google Scholar]
- 8. Emili A. MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol Cell. 1998;2(2):183–9. [DOI] [PubMed] [Google Scholar]
- 9. Sanchez Y, Desany B, Jones W, Liu Q, Wang B, Elledge S. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–60. [DOI] [PubMed] [Google Scholar]
- 10. Alcasabas A, Osborn A, Bachant J, Hu F, Werler P, Bousset K, et al. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol. 2001;3(11):958–65. [DOI] [PubMed] [Google Scholar]
- 11. Hollingsworth NM, Goetsch L, Byers B. The HOP1 gene encodes a meiosis-specific component of yeast chromosomes. Cell. 1990;61(1):73–84. Epub 1990/04/06. 0092-8674(90)90216-2 [pii]. . [DOI] [PubMed] [Google Scholar]
- 12. Hollingsworth NM, Ponte L. Genetic interactions between HOP1, RED1 and MEK1 suggest that MEK1 regulates assembly of axial element components during meiosis in the yeast Saccharomyces cerevisiae . Genetics. 1997;147(1):33–42. Epub 1997/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bailis JM, Roeder GS. Synaptonemal complex morphogenesis and sister-chromatid cohesion require Mek1-dependent phosphorylation of a meiotic chromosomal protein. Genes Dev. 1998;12(22):3551–63. Epub 1998/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell. 2001;7(6):1255–66. . [DOI] [PubMed] [Google Scholar]
- 15. Carballo JA, Panizza S, Serrentino ME, Johnson AL, Geymonat M, Borde V, et al. Budding yeast ATM/ATR control meiotic double-strand break (DSB) levels by down-regulating Rec114, an essential component of the DSB-machinery. PLoS Genet. 2013;9(6):e1003545 10.1371/journal.pgen.1003545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Falk J, Chan A, Hoffmann E, Hochwagen A. A Mec1- and PP4-dependent checkpoint couples centromere pairing to meiotic recombination. Dev Cell. 2010;19:599–611. 10.1016/j.devcel.2010.09.006 [DOI] [PubMed] [Google Scholar]
- 17. Niu H, Wan L, Busygina V, Kwon Y, Allen JA, Li X, et al. Regulation of meiotic recombination via Mek1-mediated Rad54 phosphorylation. Mol Cell. 2009;36(3):393–404. 10.1016/j.molcel.2009.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suhandynata R, Liang J, Albuquerque C, Zhou H, Hollingsworth N. A method for sporulating budding yeast cells that allows for unbiased identification of kinase substrates using stable isotope labeling by amino acids in cell culture. G3. 2014;4(11):2125–35. 10.1534/g3.114.013888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bishop DK, Park D, Xu L, Kleckner N. DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell. 1992;69(3):439–56. Epub 1992/05/01. 0092-8674(92)90446-J [pii]. . [DOI] [PubMed] [Google Scholar]
- 20. Chuang C, Cheng Y, Wang T. Mek1 stabilizes Hop1-Thr318 phosphorylation to promote interhomolog recombination and checkpoint responses during yeast meiosis. Nucleic Acids Res. 2012;40(22):11416–27. 10.1093/nar/gks920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Niu H, Wan L, Baumgartner B, Schaefer D, Loidl J, Hollingsworth NM. Partner choice during meiosis is regulated by Hop1-promoted dimerization of Mek1. Mol Biol Cell. 2005;16(12):5804–18. Epub 2005/10/14. E05-05-0465 [pii] 10.1091/mbc.E05-05-0465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wan L, de los Santos T, Zhang C, Shokat K, Hollingsworth NM. Mek1 kinase activity functions downstream of RED1 in the regulation of meiotic double strand break repair in budding yeast. Mol Biol Cell. 2004;15(1):11–23. Epub 2003/11/05. 10.1091/mbc.E03-07-0499 E03-07-0499 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cartagena-Lirola H, Guerini I, Manfrini N, Lucchini G, Longhese MP. Role of the Saccharomyces cerevisiae Rad53 checkpoint kinase in signaling double-strand breaks during the meiotic cell cycle. Mol Cell Biol. 2008;28(14):4480–93. Epub 2008/05/29. MCB.00375-08 [pii] 10.1128/MCB.00375-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsubouchi H, Roeder GS. Budding yeast Hed1 down-regulates the mitotic recombination machinery when meiotic recombination is impaired. Genes Dev. 2006;20(13):1766–75. 10.1101/gad.1422506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Busygina V, Sehorn MG, Shi IY, Tsubouchi H, Roeder GS, Sung P. Hed1 regulates Rad51-mediated recombination via a novel mechanism. Genes Dev. 2008;22(6):786–95. 10.1101/gad.1638708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lao JP, Cloud V, Huang C-C, Grubb J, Thacker D, Lee C-Y, et al. Meiotic crossover control by concerted action of Rad51-Dmc1 in homolog template bias and robust homeostatic regulation. PLoS Genet. 2013;9(12):e1003978 10.1371/journal.pgen.1003978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hofmann K, Bucher P. The FHA domain: a putative nuclear signalling domain found in protein kinases and transcription factors. Trends in biochemical sciences. 1995;20(9):347–9. Epub 1995/09/01. . [DOI] [PubMed] [Google Scholar]
- 28. Durocher D, Henckel J, Fersht AR, Jackson SP. The FHA domain is a modular phosphopeptide recognition motif. Mol Cell. 1999;4(3):387–94. . [DOI] [PubMed] [Google Scholar]
- 29. Durocher D, Taylor IA, Sarbassova D, Haire LF, Westcott SL, Jackson SP, et al. The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol Cell. 2000;6(5):1169–82. . [DOI] [PubMed] [Google Scholar]
- 30. Yaffe MB, Smerdon SJ. PhosphoSerine/threonine binding domains: you can't pSERious? Structure (London, England: 1993). 2001;9(3):R33–8. Epub 2001/04/05. . [DOI] [PubMed] [Google Scholar]
- 31. Ali AA, Jukes RM, Pearl LH, Oliver AW. Specific recognition of a multiply phosphorylated motif in the DNA repair scaffold XRCC1 by the FHA domain of human PNK. Nucleic Acids Res. 2009;37(5):1701–12. Epub 2009/01/22. 10.1093/nar/gkn1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee H, Yuan C, Hammet A, Mahajan A, Chen ESW, Wu M-R, et al. Diphosphothreonine-Specific Interaction between an SQ/TQ Cluster and an FHA Domain in the Rad53-Dun1 Kinase Cascade. Mol Cell. 2008;30(6):767–78. 10.1016/j.molcel.2008.05.013. 10.1016/j.molcel.2008.05.013 [DOI] [PubMed] [Google Scholar]
- 33. Wu HY, Ho HC, Burgess SM. Mek1 kinase governs outcomes of meiotic recombination and the checkpoint response. Curr Biol. 2010;20(19):1707–16. Epub 2010/10/05. S0960-9822(10)01097-3 [pii] 10.1016/j.cub.2010.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ontoso D, Acosta I, van Leeuwen F, Freire R, San-Segundo PA. Dot1-dependent histone H3K79 methylation promotes activation of the Mek1 meiotic checkpoint effector kinase by regulating the Hop1 adaptor. PLoS Genet. 2013;9(1):e1003262 Epub 2013/02/06. 10.1371/journal.pgen.1003262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tougan T, Kasama T, Ohtaka A, Okuzaki D, Saito TT, Russell P, et al. The Mek1 phosphorylation cascade plays a role in meiotic recombination of Schizosaccharomyces pombe . Cell Cycle. 2010;9(23):4688–702. Epub 2010/11/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lo YH, Chuang CN, Wang TF. Pch2 prevents Mec1/Tel1-mediated Hop1 phosphorylation occurring independently of Red1 in budding yeast meiosis. PLoS One. 2014;9(1):e85687 Epub 2014/01/28. 10.1371/journal.pone.0085687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Traven A, Heierhorst J. SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays. 2005;27(4):397–407. 10.1002/bies.20204 . [DOI] [PubMed] [Google Scholar]
- 38. de los Santos T, Hollingsworth NM. Red1p, a MEK1-dependent phosphoprotein that physically interacts with Hop1p during meiosis in yeast. J Biol Chem. 1999;274(3):1783–90. Epub 1999/01/09. . [DOI] [PubMed] [Google Scholar]
- 39. Woltering D, Baumgartner B, Bagchi S, Larkin B, Loidl J, de los Santos T, et al. Meiotic segregation, synapsis, and recombination checkpoint functions require physical interaction between the chromosomal proteins Red1p and Hop1p. Mol Cell Biol. 2000;20(18):6646–58. Epub 2000/08/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shinohara M, Shita-Yamaguchi E, Buerstedde JM, Shinagawa H, Ogawa H, Shinohara A. Characterization of the roles of the Saccharomyces cerevisiae RAD54 gene and a homologue of RAD54, RDH54/TID1, in mitosis and meiosis. Genetics. 1997;147(4):1545–56. Epub 1997/12/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Acosta I, Ontoso D, San-Segundo P. The budding yeast polo-like kinase Cdc5 regulates the Ndt80 branch of the meiotic recombination checkpoint pathway. Mol Biol Cell. 2011;22(18):3478–90. 10.1091/mbc.E11-06-0482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu L, Ajimura M, Padmore R, Klein C, Kleckner N. NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae . Mol Cell Biol. 1995;15(12):6572–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Allers T, Lichten M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell. 2001;106(1):47–57. . [DOI] [PubMed] [Google Scholar]
- 44. Gray S, Allison R, Garcia V, Goldman A, Neale M. Positive regulation of meiotic DNA double-strand break formation by activation of the DNA damage checkpoint kinase Mec1(ATR). Open Biol. 2013;3(7):130019 10.1098/rsob.130019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Argunhan B, Farmer S, Leung W-K, Terentyev Y, Humphryes N, Tsubouchi T, et al. Direct and indirect control of the initiation of meiotic recombination by DNA damage checkpoint mechanisms in budding yeast. PLoS One. 2013;8(6):e65875 10.1371/journal.pone.0065875 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, B. Effects of hop1-S298A and hop1-T318A on Hop1-S298 or Hop1-T318 phosphorylation during DMC1 or dmc1Δ meiosis at 23°C meiosis. Representation of the relative signals obtained from the quantification of the entire signal detected by western blot in A B using the anti-Hop1, anti-pT318, and anti-pS298 antibodies. C. Homozygous diploids of HOP1 and hop1-S298A were incubated on SPM plate at the indicated temperature for either one (30°C, 33°C, 36°C) or two days (18°C, 23°C). Tetrads were dissected on YPD plates and incubated at 30°C. The images were taken following 2 day incubation. D. Temperature sensitivity of hop1-S298 is not associated with a notable effect on protein stability or phosphorylation. Strains of indicated genotypes were taken through synchronous meiosis at 23°C or 33°C. Samples were collected at the indicated time points and subjected to Western Blot analysis using polyclonal antibodies to Hop1. Positions of unphosphorylated or phosphorylated Hop1 species are as indicated. Shown below each Western blot image is the corresponding ponceau staining gel as a loading control. hop1-S298A: non phosphorylatable allele, hop1-S298D: phospho-mimetic allele, hop1-S298x2: an allele containing two tandem copies of hop1-S298A.
(TIF)
Homozygous diploids of indicated genotypes were taken through synchronous meiosis at 23°C. Samples were collected at the indicated time points and subjected to Western Blot analysis using anti-HA antibody for detection of Mek1-HA. Positions of unphosphorylated or phosphorylated Mek1-HA species are as indicated. Shown below are sporulation efficiency in each culture and quantification analysis of the Western images, where the signal in the ‘pMek1-HA’ region in each lane is divided by the total signal (‘pMek1-HA’+ ‘Mek1-HA’) in the corresponding lane.
(TIF)
Homozygous diploids of indicated genotypes were incubated on SPM plates at 18°C, 30°C, and 36°C for one (30°C, 36°C) or two days (18°C). Tetrads were dissected on YPD plates and incubated at 30°C for two days. Spore viability was calculated as the number of visible spore colonies over the total number of spores dissected. For each strain, at least 160 spores were analysed.
(TIF)
All strains are homozygous diploids of the SK1 background of S. cerevisiae. All strains except HTY2091-2092 and APY67-68/70 were derived from JCY448 (ho::LYS2/ho::LYS2, ura3/ura3, leu2::hisG/leu2::hisG, hop1Δ::LEU2/hop1Δ::LEU2) (Carballo et al. 2008); from JCY511 (ho::LYS2/ho::LYS2, ura3/ura3, leu2::hisG/leu2::hisG, hop1Δ::LEU2/ hop1Δ::LEU2 dmc1Δ::KanMX4/ dmc1Δ::KanMX4) (Carballo et al. 2008); and JCY614 (ho::LYS2/ho::LYS2, ura3/ura3, leu2::hisG/leu2::hisG, hop1Δ::LEU2/hop1Δ::LEU2, MEK1-3HA::URA3/ MEK1-3HA::URA3) (Carballo et al. 2008). HTY2091-HTY2092 are derived from TBR2091 and TBR2092 respectively from (Tsubouchi and Roeder, 2006). APY68-69 were derived from HTY2091 and APY67 was derived from HTY2092.
(PDF)
Supplementary data tables showing the values plotted in the graphs from Figs 1D and 1E, 3A, 3B and 3F, 4B–4G, and S1A and S1B, and S2 Figs.
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.