

Abstract
Vasculogenic mimicry (VM) refers to the process by which highly aggressive tumor cells mimic endothelial cells to form vessel-like structures that aid in supplying enough nutrients to rapidly growing tumors. Histone deacetylases (HDACs) regulate the expression and activity of numerous molecules involved in cancer initiation and progression. Notably, HDAC3 is overexpressed in the majority of carcinomas. However, thus far, no data are available to support the role of HDAC3 in VM. In this study, we subjected glioma specimens to immunohistochemical and histochemical double-staining methods and found that VM and HDAC3 expression were related to the pathological grade of gliomas. The presence of VM correlated with HDAC3 expression in glioma tissues. The formation of tubular structures, as determined by the tube formation assay to evaluate VM, was impaired in U87MG cells when transfected by siRNA or treated with an HDAC3 inhibitor. Importantly, the expression of VM-related molecules such as MMP-2/14 and laminin5γ2 was also affected when HDAC3 expression was altered. Furthermore, U87MG cells were treated with a phosphoinositide 3-kinase (PI3K) inhibitor or/and ERK inhibitor and found that the PI3K and ERK signaling pathways play key roles in VM; whereas, in VM, the two signaling pathways did not act upstream or downstream from each other. Taken together, our findings showed that HDAC3 contributed to VM in gliomas, possibly through the PI3K/ERK–MMPs–laminin5γ2 signaling pathway, which could potentially be a novel therapeutic target for gliomas.
Keywords: ERK, glioma, HDAC3, PI3K (AKT), vasculogenic mimicry
Vasculogenic mimicry was first reported by Maniotis et al.1 in 1999. It refers to the process by which highly aggressive tumor cells independently mimic endothelial cells to form vessel-like structures that aid in supplying enough nutrients to rapidly growing tumors. These VM structures have previously been observed in a number of cancers, such as breast, ovarian, lung, and renal cancers and Ewing sarcoma.2 Furthermore, our previous study indicated that VM exists in gliomas and is a significant prognostic factor for patient survival.3
Before VM was reported, researchers attempted to use anti-angiogenesis as an alternative to frequently ineffective surgical resections and chemoradiotherapies, owing to the fact that glioma growth depends on abundant blood perfusion.4,5 Research that involves targeting endothelial cells has been the basis for drug discovery and development. However, recent studies have found that anti-angiogenic therapy alone may not be effective; or worse, it may elicit greater malignancy.6,7 As a supplementary theory of angiogenesis, VM may account for the failure of antivascular therapy.8,9 Mounting evidence has focused on VM and its close correlation with poor prognosis.2,3,10,11 Many recent studies have shown that some genes and corresponding proteins are involved in VM, including VE-cadherin,12,13 EphA2,14–17 MMPs,17–20 and Ln5γ2 (LAMC2).8–11. The AKT and ERK signaling pathways also play key roles in VM.2,18 Following the identification of the above-described molecules, a classical model of the signaling cascade implicated in VM was suggested.2
Histone acetylation is regulated dynamically by two groups of molecules, histone acetyltransferases and HDACs.21 Histone deacetylases constitute a class of deacetylating enzymes that remove acetyl groups from lysine residues in histone and non-histone proteins.22,23 Many studies have shown that HDACs regulate cell cycle progression, proliferation, and differentiation and are involved in the development of cancer.24–27 In particular, HDAC3 was reported to be overexpressed in the majority of carcinomas, including gliomas, and may be one of the most frequently upregulated genes in cancer.28,29 More importantly, the depletion of HDAC3 by RNAi significantly blocked the activation of ERK and PI3K30; HDAC3 inhibitors also inhibited AKT and ERK signaling pathways.30,31 However, the relationship between HDAC3 and VM in glioma is currently unknown. To expand our knowledge regarding VM and the biological function of HDAC3, the current study was designed in an attempt to identify the contribution of HDAC3 to VM, thereby providing novel therapeutic strategies for gliomas.
Materials and Methods
Tissue specimens
Tissue collection and analysis in this study were approved by the Research Ethics Committee of Southern Medical University (Guangzhou, China). Glioma tissues were obtained from the Department of Pathology, Zhujiang Hospital at Southern Medical University between 2010 and 2013. All tissues were randomly collected from patients who did not undergo any therapy before undergoing surgery. Tumor sections were reviewed by two neuropathologists to verify the diagnosis of glioma according to the 2007 WHO classification of tumors of the central nervous system.
Cell culture
The human U87MG (Laboratory Animal Center, Sun Yat-sen University, GuangZhou, China) glioma cell lines were cultivated in high glucose DMEM (HyClone, Logan, UT, USA) supplemented with 10% FBS (HyClone) in 5% CO2 at 37°C.
Immunohistochemical and CD34-PAS histochemical double staining
For immunohistochemical staining, tumor tissue sections (5 mm) were prepared and deparaffinized in xylene, hydrated by standard procedures described in our previous study.32 To determine the expression of HDAC3, slides were incubated with a rabbit mAb against HDAC3. Five visual fields of each tissue section were selected randomly under a microscope (Leica, Newcastle, UK) at 400× magnification. The number of stained cells and the total number of cells were counted in the five visual fields, and the ratio between the stained and total cells was calculated. The following definitions were used for the stained cell ratio: −/+, <10% negative or weakly positive for expression of HDAC3; ++, 20–50% strongly positive for expression of HDAC3; and +++, >50% very strongly positive for expression of HDAC3. The −/+ rating was considered as low expression of HDAC3; ++ and +++ were both considered as high expression of HDAC3.
To identify the VM structures, CD34/PAS histochemical double staining was carried out. After immunohistochemical staining for CD34, slides were stained following the PAS staining procedures before lightly counterstaining with hematoxylin. The sections were lightly stained with eosin after these procedures. Detailed information of antibodies used in this study is listed in Table1.
Table 1.
Antibodies used in this study
| Antibody | Company | Purpose | Dilution | Product no. |
|---|---|---|---|---|
| HDAC3 | Abcam | IHC | 1:200 | ab32369 |
| CD34 | Abcam | IHC | 1:250 | ab81289 |
| HDAC3 | CST | Western blot | 1:1000 | 3949P |
| Akt antibody | CST | Western blot | 1:1000 | 9272S |
| Phospho-Akt (Ser473) | CST | Western blot | 1:1500 | 4060P |
| p44/42 MAPK (Erk1/2) | CST | Western blot | 1:1000 | 4695P |
| Phospho-p44/42 (Erk1/2) | CST | Western blot | 1:2000 | 4370P |
| Anti-MMP2 antibody | Abcam | Western blot | 1:1000 | ab86607 |
| Anti-MMP9 antibody | Abcam | Western blot | 1:1000 | ab38898 |
| Anti-MMP14 antibody | Abcam | Western blot | 1:1000 | ab53712 |
| Anti-LAMC2 antibody | Abcam | Western blot | 1:2000 | ab96327 |
| EphA2 (D4A2) | CST | Western blot | 1:1000 | 6997P |
| VE-cadherin (BV9) | Santa Cruz | Western blot | 1:200 | Sc-52751 |
| GAPDH loading control | Abcam | Western blot | 1:3000 | ab9485 |
| Goat anti-rabbit | Abcam | Western blot | 1:8000 | ab6721 |
| Goat anti-mouse | Abcam | Western blot | 1:10000 | ab6789 |
AKT, protein kinase B; CST, Cell Signaling Technology (Boston, MA, USA); EphA2, ephrin type-A receptor 2; HDAC, histone deacetylase; IHC, immunohistochemistry; LAMC2, laminin, γ2; Santa Cruz, Santa Cruz Biotechnology (Santa Cruz, CA, USA); VE, vascular endothelial.
For diagnosis of VM, sections were scanning under microscope carefully. CD34−/PAS+ vascular-like structures containing red blood cells formed by glioma cells were identified as positive for VM: VM channels were lined with tumor cells with nuclei stained dark blue by hematoxylin. The channels were rich in ECM that can be highlighted pink or pink-purple by PAS, whereas the luminal surfaces of channels could be highlighted brown (negative for CD34 reaction). In hollows, red blood cells stained by eosin red or grey-red can be observed.
Vasculogenic mimicry channel density
The median number of VM channels was counted under a microscope. Tumor sections were observed under 200× magnification to first identify the accumulation of VM channels. Next, we chose the areas that contained the most VM channels to determine the median number of VM channels observed per field at 400× magnification.
Tube formation assay
Tube formation was observed by 3-D culture, as described in our previous study.33 Briefly, 24-well culture plates were coated with Matrigel Basement Membrane Matrix (0.3 mL/well) (BD Biosciences, Franklin Lakes, NJ, USA), and then allowed to polymerize at 37°C for 60 min. Cells (2.5 × 105 cells/mL) were seeded onto the surface of Matrigel (1 mL/well) and then incubated without serum for 6 h. To investigate the effect of SAHA (Sigma, St. Louis, MO, US), cells were treated with SAHA at indicated concentrations (2 and 4 μM) for 12 h before being seeded onto the Matrigel.34,35 Cultures were photographed by microscope after 6 h. Formation of VM was quantified by the total length of tubes (complete structures) and the number of intersections (complete structures) per field in five randomly chosen fields using image analysis software (Image-Pro Plus, Washington, DC, USA).
Western blot analyses
Cell lysates were harvested with the Total Protein Extraction Kit (KeyGEN, Nanjing, China). Protein concentration was determined by the BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Equal quantities (20 μg) of protein were separated electrophoretically on 10% SDS–polyacrylamide gel and transferred onto PVDF membranes (Millipore, Billerica, MA, USA). The blots were incubated with appropriate primary antibodies, followed by incubation with secondary antibodies. The blots were detected using Pierce ECL plus Western Blotting Substrate (Waltham, MA, USA). A monoclonal GADPH antibody was used for protein loading analyses. Detailed information of the antibodies used in this study is listed in Table1.
Quantitative real-time PCR
For the glioma tissue samples, total RNA was extracted by Qiagen RNeasy FFPE Kit (Qiagen, Hilden, Germany). For cells, total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA). RNA integrity was checked by gel electrophoresis. Reverse transcription was carried out with random primers and Reverse Transcriptase M-MLV (RNase H-; Takara, Dalian, China). The mRNA expression level was determined by SYBR Premix Ex Taq (Tli RNaseH Plus; Takara) and ABI ViiA7 Detection System (Applied Biosystems, Foster, CA, USA); the sequences of target gene-specific primers are listed in Table2. All reactions were carried out with the following program: 30 s at 95°C, followed by 44 cycles of 95°C for 3 s and 60°C for 34 s. For the internal control for normalization, 18S mRNA was used. The relative expression of transcripts was analyzed using the 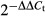 method.
method.
Table 2.
Primer sequences used for quantitative RT-PCR
| Gene | Primer sequence (5′ to 3′) | Size, bp |
|---|---|---|
| HDAC3 | F: CCCTGCGGGATGGCATTGATGA | 123 |
| R: AGCCCAGAGAGTCAGCTCCACA | ||
| MMP-2 | F: GGCGGTCACAGCTACTTCTTC | 105 |
| R: GCAGCCTAGCCAGTCGGATT | ||
| MMP-9 | F: GTGACACCGCTCACCTTCAC | 122 |
| R: GCGTGTGCCAGTAGACCATC | ||
| MMP-14 | F: CTGCGTCCATCAACACTGCCTA | 128 |
| R: GCCCAGCTCCTTAATGTGCTTG | ||
| EphA2 | F: TTAGGGAGAAGGATGGTGAGTT | 140 |
| R: GGTCGAGGGCATGGTGTA | ||
| VE-cadherin | F: TTTCCAGCAGCCTTTCTACCA | 145 |
| R: GGAAGAACTGGCCCTTGTCA | ||
| LAMC2 | F: TCGGGAGCCATGTCATGTGAGTG | 148 |
| R: CCCAGCATCAGGAAGCAAGGAGT | ||
| hu-18S | F: GACTCAACACGGGAAACCTCAC | 122 |
| R: CCAGACAAATCGCTCCACCAAC |
EphA2, ephrin type-A receptor 2; F, forward; LAMC2, laminin, γ2; R, reverse; VE, vascular endothelial.
RNA-mediated interference
The siRNAs were purchased from GenePharma (Shanghai, China). Target sequences of siRNAs were 5′-CCGCCAGACAAUCUUUGAATT-3′ (HDAC3-1), 5′-CGGUGUCCUUCCACAAAUATT-3′ (HDAC3-2), 5′-GCAGGUGUUUGAAGUGUAUTT-3′ (LAMC2), 5′-CCGACAUCAUGAUCUUCUUTT-3′ (MMP-14), and 5′-CGGUGUCCUUCCACAAAUATT-3′ (negative control). For siRNA, U87MG cells were plated onto 6-well dishes at a concentration of 2.5–5 × 105 cells/well and cultured for 24 h. Then, 50 pmol siRNA was transfected into 70% confluent U87MG cells for 24–48 h using Lipofectamine RNAiMAX Reagent (Life Technologies, Carlsbad, CA, USA). Cells were then lysed for Western blot, and the isolated RNA was subjected to reverse transcription. Meanwhile, we used a positive control (GAPDH siRNA) and fluorescein-labeled (FAM-) negative control to ensure the reliability of the method and transfection efficiency.
Statistics
All experiments were carried out at least in triplicate. The data analysis was carried out with spss version 13.0 (SPSS Inc., Chicago, IL, USA). All P-values were two-sided, and P < 0.05 was considered statistically significant.
Results
Relationship between VM and clinicopathological data in glioma tissues
Twenty-six specimens (25.49%) with VM structures were identified out of 102 glioma specimens by CD34-PAS dual staining (Fig.1). Here, clinicopathological data of glioma specimens with VM (n = 26) were compared to those without VM (n = 76). The results are summarized in Table3.
Fig 1.
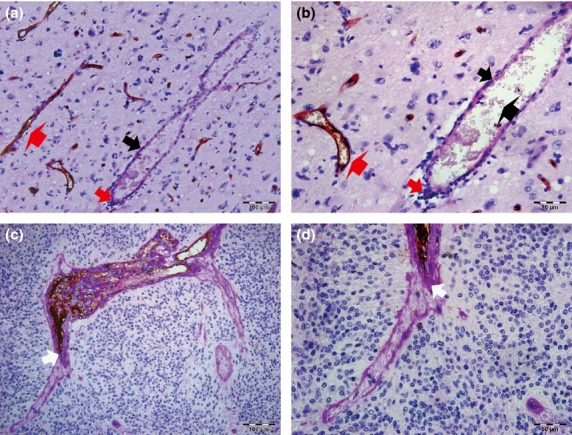
Relationship between vasculogenic mimicry (VM) and clinicopathological data in glioma tissues. (a, b) Endothelial cells were detected by anti-CD34 immunohistochemistry staining, resulting in a brown (or some black-brown) product. Periodic acid–Schiff (PAS)-positive substances formed a basement membrane-like structure that is highlighted pink or pink-purple (thick black arrows). Representative VM channels are lined with tumor cells (thick red arrows) and rich in PAS+ substances, whereas the luminal surfaces of PAS+ channels (thick black arrows) were negative for CD34 reaction. In the hollows, red blood cells (bold black arrow) can be observed. Typical blood vessels (bold red arrows) showed a positive reaction for CD34 on their luminal surface and PAS+ reaction in their wall. (b) is a magnified image of (a). (a) Scale bar = 100 μm (×200). (b) Scale bar = 50 μm (×400). (c, d) Some VM structures were interlinked with CD34+ endothelial cell-lined blood vessels (white arrows). (d) is a magnified figure of (c). (c) Scale bar = 100 μm (×200). (d) Scale bar = 50 μm (×400). All sections were stained with CD34 and PAS.
Table 3.
Relationship between vasculogenic mimicry (VM), histone deacetylase 3 (HDAC3), and clinicopathological data of patients with glioma
| Parameters | Cases | VM | χ2 | P-value | HDAC3 | χ2 | P-value | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Positive | Negative | High | Low | |||||||
| +++ | ++ | –/+ | ||||||||
| Gender | ||||||||||
| Male | 61 | 16 | 45 | 0.044† | 0.834 | 17 | 30 | 14 | 1.677† | 0.432 |
| Female | 41 | 10 | 31 | 7 | 23 | 11 | ||||
| Age, years | ||||||||||
| <40 | 45 | 11 | 34 | 0.982† | 0.610 | 10 | 23 | 12 | 0.425‡ | 0.990 |
| ≥40 to <60 | 32 | 10 | 22 | 10 | 16 | 6 | ||||
| ≥60 | 25 | 5 | 20 | 4 | 14 | 7 | ||||
| Tumor size, cm | ||||||||||
| <5 | 53 | 14 | 39 | 0.824† | 0.943 | 15 | 24 | 14 | 2.549† | 0.280 |
| ≥5 | 49 | 12 | 37 | 9 | 29 | 11 | ||||
| Grade, WHO | ||||||||||
| I | 6 | 0 | 6 | 7.502‡ | 0.048$ | 0 | 2 | 4 | 33.390‡ | 0.000$ |
| II | 40 | 6 | 34 | 7 | 19 | 14 | ||||
| III | 23 | 7 | 16 | 6 | 13 | 4 | ||||
| IV | 33 | 13 | 20 | 11 | 19 | 3 | ||||
| KPS | ||||||||||
| <65 | 45 | 12 | 33 | 0.059† | 0.810 | 6 | 16 | 23 | 3.018† | 0.221 |
| ≥65 | 57 | 14 | 43 | 13 | 24 | 20 | ||||
| HDAC3 | ||||||||||
| Low −/+ | 43 | 6 | 37 | 6.315† | 0.043$ | – | – | – | – | |
| High ++ | 40 | 12 | 28 | – | – | – | ||||
| High +++ | 19 | 8 | 11 | – | – | – | ||||
| VM | ||||||||||
| Positive | 23 | – | – | – | 8 | 12 | 6 | 6.203† | 0.045$ | |
| Negative | 76 | – | – | 11 | 28 | 37 | ||||
†Statistical analyses were carried out using the χ2-test (asymptotic significance, two-sided). ‡Fisher’s exact test (two-sided); $P < 0.05 was considered significant. −/+, <10% negative or weakly positive HDAC3 expression; ++, 20–50% strongly positive HDAC3 expression; +++, >50% very strongly positive HDAC3 expression; KPS, preoperative Karnofsky performance scores.
The results showed that the pathological grade (based on WHO standards) of glioma differed significantly between groups with VM and without VM (P = 0.048). Vasculogenesis mimicry was detected preferentially in high-grade gliomas: 13 of 33 WHO grade IV (39.39%), seven of 23 WHO grade III (30.43%), and six of 40 WHO grade II (15%). Vasculogenesis mimicry was not detected in any WHO grade I gliomas. No significant differences were found in other clinicopathological characteristics such as gender, age, tumor size, or preoperative Karnofsky performance scores.
Upregulation of HDAC3 in VM-positive glioma tissues
HDAC3 protein expression in glioma tissues was evaluated by immunohistochemistry (Fig.2a–c). As presented in Table3, HDAC3 expression was rated −/+ in 43 (42.16%) specimens, ++ in 40 (39.21%) specimens, and +++ in 19 (18.63%) specimens. The expression of HDAC3 significantly correlated with the WHO pathological grade of gliomas (P < 0.001). High HDAC3 expression (HDAC3++ and HDAC3+++) was found in 16.67% (1 of 6) of WHO grade I gliomas, 27.50% (11 of 40) of WHO II, 73.91% (17 of 23) of WHO III, and 87.88% (29 of 33) of WHO IV. More importantly, it was observed that the percent of high expression of HDAC3 in gliomas with VM was 76.92% (20 of 26) compared with 51.31% (39 of 76) of those without VM. This difference was statistically significant (P = 0.045). There was no significant association between the expression of HDAC3 and gender, age, tumor size, or Karnofsky performance score (Table3).
Fig 2.
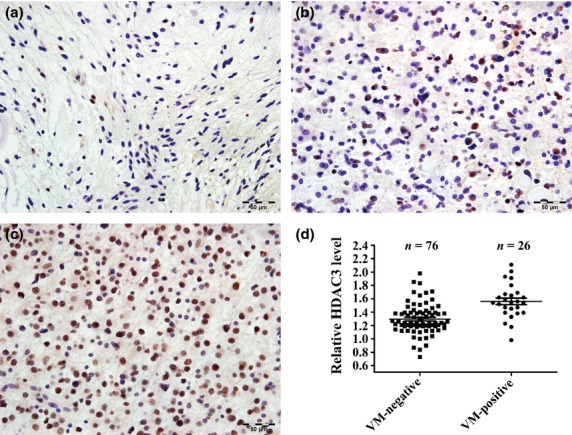
Histone deacetylase 3 (HDAC3) is upregulated in vasculogenic mimicry (VM)-positive glioma tissues. Expression of HDAC3 protein in human glioma tissues was investigated by immunohistochemistry. (a–c) Representative figures for <10% negative or weakly positive HDAC3 expression (−/+) (a), 20–50% strongly positive HDAC3 expression (++) (b), and >50% very strongly positive HDAC3 expression (+++) (c). Scale bar = 50 μm (×400). (d) HDAC3 expression in glioma tissue samples with or without VM. One sample was randomly chosen as a control sample. Gene expression was normalized to that of the housekeeping gene 18S rRNA. Data are presented as fold induction relative to the control sample expression and are represented as mean ± SD.
The mRNA levels of HDAC3 in 102 glioma tissues were also analyzed by qPCR. Prior to qPCR, samples were divided into two groups based on the presence or absence of VM. The results show that the mRNA levels of HDAC3 in VM-positive gliomas were significantly higher than those in VM-negative gliomas (P < 0.001; Fig.2d).
More VM detected in glioma tissues with increased HDAC3 expression
The analysis presented in Table3 shows that VM was detected in six cases (13.95%) with HDAC3 −/+, 12 cases (30%) with HDAC3++, and eight cases (42.11%) with HDAC3+++ (Fig.3a). The positive rate of VM showed a significantly sharp increase with increased HDAC3 expression (P = 0.043). Higher expression of HDAC3 (HDAC3++ and HDAC3+++) showed a greater VM-positive rate than did HDAC3 −/+ (P = 0.022).
Fig 3.
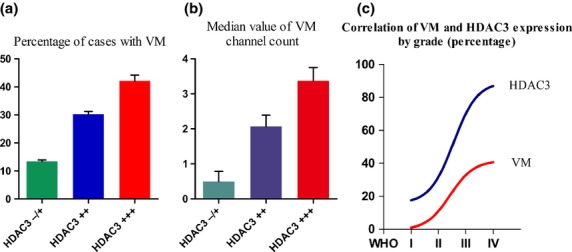
Higher number of vasculogenic mimicry (VM) structures was detected in glioma tissues with increased histone deacetylase 3 (HDAC3) expression. (a) Positive rate of VM showed a sharp increase in tissues with 20–50% strongly positive HDAC3 expression (++) or >50% very strongly positive HDAC3 expression (+++) compared with those with <10% negative or weakly positive HDAC3 expression (−/+). (b) VM channel counts were made in cases with VM. The median value of the VM channels in HDAC3+++ tissues was significantly higher than that in HDAC3 −/+ and HDAC3++ tissues. (c) Correlation between VM and HDAC3 in relation to tumor grade in glioma tissues.
Vasculogenesis mimicry channel density showed that the median value of the VM channels was 0.5 ± 0.29 in HDAC3 −/+ samples, 2.07 ± 0.32 in HDAC3++ samples, and 3.38 ± 0.37 in HDAC3+++ samples (Fig.3b). The highest median value of VM channels was detected in HDAC3+++ cases. Moreover, a significant difference in VM numbers was observed when HDAC3+++ was compared to HDAC3++ or HDAC3++ combined with HDAC3 −/+ groups (P = 0.024 and P = 0.007, respectively). Furthermore, Figure3(c) shows a positive correlation between VM and HDAC3 in relation to tumor grade in glioma tissues.
Downregulation of HDAC3 inhibited VM in U87MG cells
The U87MG cell line was transfected with HDAC3-siRNAs. The tube formation assay was then carried out after the HDAC3-depleted cells had been evaluated by qPCR and Western blot. Quantitative PCR (Fig.4a) showed that HDAC3 mRNA transcripts apparently declined in the HDAC3-1 (P < 0.001) group and HDAC3-2 group (P < 0.001) compared with that in the U87MG group. Additionally, for HDAC3-1 and HDAC3-2 groups, HDAC3 mRNA transcript in the HDAC3-1 group declined the most (P < 0.001). Consistent with the qPCR results, the Western blot analysis (Fig.4b) showed a lower HDAC3 expression in the HDAC3-2 group and, in particular, in the HDAC3-1 group, compared to that in the U87MG group. The tube formation assay results showed that relatively more well-formed tubular structures were found in the HDAC3 group (for number of intersections, 45.71 ± 0.57; for total tube length, 7382 ± 116 μm) (Fig.4c,d). By contrast, the HDAC3-1 group (for number of intersections, 6.57 ± 0.57; for total tube length, 905 ± 29 μm) and HDAC3-2 group (for number of intersections, 29.71 ± 0.61; for total tube length, 4490 ± 59 μm) did not form tubular networks efficiently (Fig.4c). Similar results were also observed when we reduced HDAC3 expression by SAHA, an HDAC3 inhibitor (Doc. S1, Fig. S1).
Fig 4.

Histone deacetylase 3 (HDAC3) influences vasculogenic mimicry (VM) in U87MG cells. (a) U87MG cells were transfected with siRNAs and HDAC3 mRNA levels were detected by quantitative RT-PCR. Gene expression was normalized to that of the housekeeping gene 18S rRNA. Data are presented as fold induction relative to the expression of the U87MG group and are represented as mean ± SD ($P < 0.05). (b) Western blot and densitometry analysis of HDAC3 protein between groups transfected with siRNA ($P < 0.05). (c) U87MG cells subjected to different treatments were seeded into wells of a 24-well plate coated with Matrigel for 6 h then photographed. Scale bar = 100 μm. (d) Total length of VM tubes and the number of intersections per field were compared between groups in (c) ($P < 0.05 compared with U87MG).
Influence of HDAC3 on VM-related molecules
U87MG cells were treated with HDAC3-siRNAs or SAHA, and the expression of HDAC3 and VM-related molecules was determined by qRT-PCR or Western blot analysis. We found that, when HDAC3 was depleted in U87MG cells, the mRNA transcripts (Fig.5a) of MMP-2 (P = 0.001), MMP-14 (P < 0.001), and LAMC2 (P < 0.001) in the HDAC3-1 group were significantly lower than that in U87MG group; the Western blot results were similar (Fig.5b). No other molecules showed differences in expression when compared to the U87MG group. Similarly, the cells subjected to SAHA treatments had lower expression of MMP-2, MMP-14, and LAMC2 compared to that of the U87MG group (Fig.5c).
Fig 5.
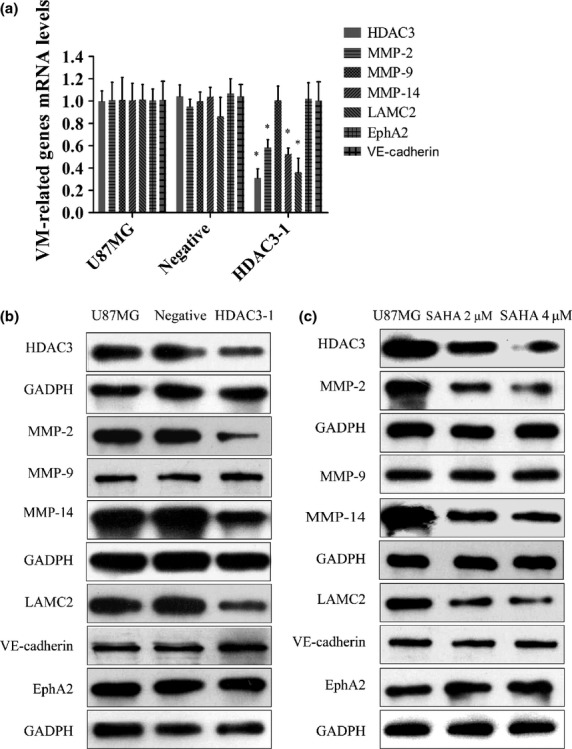
Histone deacetylase 3 (HDAC3) influences vasculogenic mimicry (VM) in U87MG cells. (a) Quantitative RT-PCR analysis of ephrin type-A receptor 2 (EphA2), vascular endothelial (VE)-cadherin, MMP-2, MMP-9, MMP-14, and laminin, γ2 (LAMC2). Gene expression was normalized to that of the housekeeping gene 18S rRNA. Data are presented as fold induction relative to the expression in the U87MG group and are represented as mean ± SD (all $P < 0.05). (b, c) Western blot analysis of EphA2, VE-cadherin, MMP-2, MMP-9, MMP-14, and LAMC2 in different treatment groups. SAHA, suberoylanilide hydroxamic acid.
Histone deacetylase 3 correlated with VM by way of PI3K/ERK–MMPs–Ln5γ2 signaling pathway
To further investigate the mechanism by which HDAC3 regulated VM, we used Western blotting to evaluate the expression of HDAC3, ERK, ρ-ERK, PI3K (AKT), and ρ-PI3K (ρ-AKT) in HDAC3-depleted U87MG cells. Our previous study had shown that MMPs–Ln5γ2 was the final stage of the VM signaling pathway2,19,36 and we had also verified that LMAC2 and MMP-14 were indeed involved in VM (Doc. S1, Fig. S2), hence the expression of the VM-related molecules MMP-14 and LAMC2 was assessed to evaluate VM. Figure6(a) shows that the expression of AKT, ERK, MMP-14, and LAMC2 in the siRNA (HDAC3-1) group decreased significantly, indicating that AKT and ERK are involved in the molecular events that allow HDAC3 to regulate VM. However, to confirm and fully understand how they worked together, another set of experiments were carried out. First, U87MG cells were treated with U0126 (ERK inhibitor; 10 μM, 30 min) (Cell Signaling Technology, Boston, MA, USA) or LY294002 (PI3K inhibitor; 2 μM, 60 min; Cell Signaling Technology), the levels of ρ-ERK or ρ-AKT showed a significant reduction, respectively (Fig.6b). We also found that the two inhibitors caused a similar mild reduction in the expression of both MMP-14 and LAMC2 compared to that in U87MG group. Then U87MG cells were treated with LY294002 plus U0126. Results showed that all molecules, except AKT and ERK, detected by Western blot, showed a significant reduction. Interestingly, the expression of MMP-14 and LAMC2 was more significantly reduced in cells treated with LY294002 plus U0126 than those treated with only one inhibitor (LY294002 or U0126) (Fig.6b).
Fig 6.
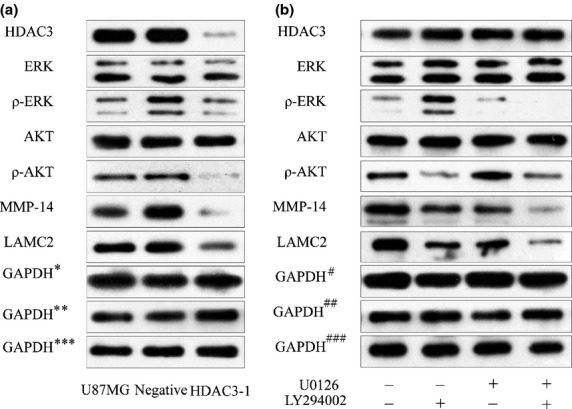
Histone deacetylase 3 (HDAC3) correlates with vasculogenic mimicry (VM): involvement of the phosphoinositide 3-kinase/ERK-MMPs–laminin5γ2 signaling pathway. (a) Western blot analysis of HDAC3, ERK, ρ-ERK, protein kinase B (AKT), ρ-AKT, MMP-14, and laminin, γ2 (LAMC2) in different groups transfected with siRNA. $GAPDH, control for HDAC3, AKT, ERK; $$GAPDH, control for MMP-14, LAMC2; $$$GAPDH, control for ρ-ERK, ρ-AKT.) (b) Western blot analysis of HDAC3, ERK, ρ-ERK, AKT, ρ-AKT, MMP-14, and LAMC2 in different treatment groups. #GAPDH, control for HDAC3, AKT, ERK; ##GAPDH, control for MMP-14, LAMC2; ###GAPDH, control for ρ-ERK, ρ-AKT.
Discussion
The HDAC3 gene, which has been extensively researched in epigenetics, has been reported to be overexpressed in the majority of carcinomas, including gliomas, and may be one of the most frequently up\regulated genes in cancer.27,28 However, there were no data supporting the correlation between HDAC3 expression and VM. In this report, we present evidence that HDAC3 has an important facilitative role on VM in gliomas.
We first found that both VM structures and HDAC3 expression have a positive correlation with tumor grades: the higher the tumor grade, the higher the number of VM structures present or HDAC3 expression. These results are consistent with the findings of a previous study.3,28 Further analysis showed that HDAC3 was upregulated in VM-positive glioma tissues (Table3, Fig.2d); furthermore, VM could be frequently detected in glioma tissues with increased HDAC3 expression (Fig.3a,b). Clearly, all of these results indicate that HDAC3 was closely correlated with VM in glioma tissues. Then we found a significant decrease in VM when HDAC3 expression was altered in U87MG cells (Figs4,S1), which was consistent with the observations in glioma tissues, indicating that the mechanism underlying the role of HDAC3 in promoting the development of VM in gliomas can be elucidated by cellular level experiments.
Certain molecules, such as VE-cadherin,12,13 EphA2,14–17 MMPs,17–20 and LAMC28–11 have been confirmed as VM-related molecules. Of these molecules, we found that MMP-2/14 and LAMC2, but not MMP-9, EphA2, or VE-cadherin, were downregulated in both transfected and inhibitor-treated U87MG cells. These results (Fig.5) indicated that VM was regulated by HDAC3 probably by way of the MMPs and LAMC2 signaling pathways without the players EphA2, MMP-9, or VE-cadherin.
Previous studies reported that SAHA could inhibit cell proliferation through inhibition of the AKT and ERK signaling pathways, also involved in VM.31,37–40 In this study, a significant decrease in levels of both ρ-AKT and ρ-ERK were found when we altered HDAC3 levels by siRNA (Fig.6a), which indicated that AKT and/or ERK signaling pathways may indeed be involved in VM formation regulated by HDAC3. We then used U87MG cells with AKT and ERK inhibitors to further verify AKT and/or ERK involvement and investigate how they worked together, as previous studies had reported that ERK and AKT could interact in various ways.41,42 Vasculogenic mimicry was evaluated by expression of MMP-14 and LAMC2, as we had confirmed that MMP-14 and LAMC2 were indeed involved in VM (Doc. S1, Fig. S2). Results (Fig.6b) showed that ρ-AKT decreased, with no change observed in ρ-ERK, when AKT expression was inhibited by LY294002; similarly, ρ-AKT did not decrease with the inhibition of ERK by U0126, indicating that the ERK or AKT signaling pathways did not act upstream or downstream from each other. However, all molecules except ERK and AKT showed a significant reduction in expression when U0126 and LY294002 were both used and expression of MMP-14 and LAMC2 showed a relatively higher reduction than when cells were treated with only one inhibitor (LY294002 or U0126). These interesting results (Fig.6b) clearly indicated that the PI3K and ERK signaling pathways play key roles in VM. More importantly, ERK or AKT signaling pathways did not act upstream or downstream from each other.
Our study provides a novel insight into the mechanisms underlying VM, and may contribute to the development of a novel therapeutic target for gliomas. However, the methods used in our study are mainly in vitro experiments, and all the experimental data were only verified in U87MG cells. Animal experiments are needed to confirm the data obtained from these cellular level experiments.
In addition, our laboratory previously reported that TGF-β was required for in vitro VM in U251MG cells, and MMP-14 was correlated with TGF-β-induced VM. However, TGF-β can also regulate the activation of the ERK and PI3K signaling pathways, which need the participation of HDACs, in particular, HDAC3.31 Thus, we may further investigate whether the HDAC3 pathways stated in this study are involved in the process by which TGF-β regulates VM (Fig.7). Other studies reported that hypoxia-inducible factor-1α has been shown to induce VM in hepatocellular carcinoma43 and melanoma,44 and, interestingly, HDAC3 has been described as a hypoxia-inducible factor-1α-regulated gene.45 Furthermore, hypoxia enhances HDAC function, for example, HDACs are closely involved in angiogenesis.46 Hence, it would be of interest to investigate whether the HDAC3 pathway is also involved in processes where hypoxia regulates VM (Fig.7). All in all, further delineation of the mechanisms of VM regulated by HDAC3 may potentially provide a novel antiglioma therapeutic target.
Fig 7.
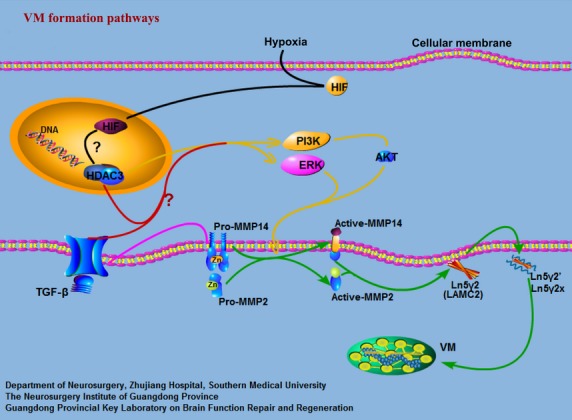
Signaling pathways implicated in the formation of vasculogenic mimicry (VM) networks. Previous observations have suggested a classical model of the signaling cascade implicated in VM. Green arrows indicate the classical signaling pathways implicated in VM in gliomas. In this study, we showed that histone deacetylase 3 (HDAC3) can regulate VM through the phosphoinositide 3-kinase (PI3K)/ERK–MMPs–laminin5γ2 (Ln5γ2) signaling pathways in gliomas. HDAC3 can first regulate the activation of ERK1/2 and PI3K directly or indirectly (yellow arrows). Active PI3K then regulates directly or indirectly the transition of pro-MMP14 into active MMP-14, which subsequently activates pro-MMP2 (green arrows). Both MMP-14 and MMP-2 can promote the cleavage of Ln5γ2 into the pro-migratory fragments 5γ2′ and 5γ2x (green arrows). These fragments result in the formation of VM networks. We have previously shown that transforming growth factor-β (TGF-β) can regulate VM by MMP-14 (pink arrows). However, HDAC3 is need for TGF-β to regulate the activation of the ERK and PI3K signaling pathways. Hence, we may further investigate whether HDAC3 pathways are involved in the process by which TGF-β (red arrows) regulates VM. Other studies reported that hypoxia-inducible factor-1α (HIF-1α) has been shown to induce VM and, interestingly, HDAC3 has been described as a HIF-1α regulated gene. Also, hypoxia enhances HDAC function, such that HDACs are closely involved in angiogenesis. Hence, it would be of interest to investigate whether the HDAC3 pathway is also involved in processes where hypoxia regulates VM (black arrows). AKT, protein kinase B; LAMC2, laminin, γ2.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81272806 to Yi-quan Ke and Grant No. 81302199 to Xin-lin Sun), the Natural Science Foundation of Guangdong Province, China (Grant No. S2012010009088 to Yi-quan Ke), and the Medical Scientific Research Foundation of Guangdong Province, China (Grant No. B2013246 to Xin-lin Sun).
Glossary
- AKT
protein kinase B
- EphA2
ephrin type-A receptor 2
- HDAC
histone deacetylase
- LAMC2
laminin, γ2
- Ln5γ2
laminin5γ2
- PAS
periodic acid–Schiff
- PI3K
phosphoinositide 3-kinase
- qPCR
quantitative PCR
- SAHA
suberoylanilide hydroxamic acid
- TGF-β
transforming growth factor-β
- VE
vascular endothelial
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Doc. S1. Histone deacetylase 3 (HDAC3) inhibitor influenced vasculogenic mimicry (VM) in U87MG cells and MMP-14 and Ln5γ2 were involved in VM.
Fig. S1. Histone deacetylase 3 (HDAC3) inhibitor influenced vasculogenic mimicry in U87MG cells.
Fig. S2. Matrix metalloproteinase-14 and laminin5γ2 are involved in vasculogenic mimicry.
References
- Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–52. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. 2012;18:2726–32. doi: 10.1158/1078-0432.CCR-11-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Ke YQ, Lu GH, et al. Vasculogenic mimicry is a prognostic factor for postoperative survival in patients with glioblastoma. J Neurooncol. 2013;112:339–45. doi: 10.1007/s11060-013-1077-7. [DOI] [PubMed] [Google Scholar]
- Zhang LQ, Zhang XD, Xu J, et al. Potential therapeutic targets for the primary gallbladder carcinoma: estrogen receptors. Asian Pac J Cancer Prev. 2013;14:2185–90. doi: 10.7314/apjcp.2013.14.4.2185. [DOI] [PubMed] [Google Scholar]
- Tian F, Zhang X, Tong Y, et al. PE, a new sulfated saponin from sea cucumber, exhibits anti-angiogenic and anti-tumor activities in vitro and in vivo. Cancer Biol Ther. 2005;4:874–82. doi: 10.4161/cbt.4.8.1917. [DOI] [PubMed] [Google Scholar]
- Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaft DW, Hillen F, Pauwels P, et al. Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res. 2005;65:11520–8. doi: 10.1158/0008-5472.CAN-05-2468. [DOI] [PubMed] [Google Scholar]
- van der Schaft DW, Seftor RE, Seftor EA, et al. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J Natl Cancer Inst. 2004;96:1473–7. doi: 10.1093/jnci/djh267. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhang D, Sun B. Vasculogenic mimicry: current status and future prospects. Cancer Lett. 2007;254:157–64. doi: 10.1016/j.canlet.2006.12.036. [DOI] [PubMed] [Google Scholar]
- El Hallani S, Boisselier B, Peglion F, et al. A new alternative mechanism in glioblastoma vascularization: tubular vasculogenic mimicry. Brain. 2010;4:973–82. doi: 10.1093/brain/awq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix MJ, Seftor EA, Meltzer PS, et al. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci USA. 2001;98:8018–23. doi: 10.1073/pnas.131209798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess AR, Seftor EA, Gruman LM, Kinch MS, Seftor RE, Hendrix MJ. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway: implications for vasculogenic mimicry. Cancer Biol Ther. 2006;5:228–33. doi: 10.4161/cbt.5.2.2510. [DOI] [PubMed] [Google Scholar]
- Kim CH, Yoon JS, Sohn HJ, et al. Direct vaccination with pseudotype baculovirus expressing murine telomerase induces anti-tumor immunity comparable with RNA-electroporated dendritic cells in a murine glioma model. Cancer Lett. 2007;250:276–83. doi: 10.1016/j.canlet.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Sun Q, Zou X, Zhang T, Shen J, Yin Y, Xiang J. The role of miR-200a in vasculogenic mimicry and its clinical significance in ovarian cancer. Gynecol Oncol. 2014;132:730–8. doi: 10.1016/j.ygyno.2014.01.047. [DOI] [PubMed] [Google Scholar]
- Hess AR, Margaryan NV, Seftor EA, Hendrix MJ. Deciphering the signaling events that promote melanoma tumor cell vasculogenic mimicry and their link to embryonic vasculogenesis: role of the Eph receptors. Dev Dyn. 2007;236:3283–96. doi: 10.1002/dvdy.21190. [DOI] [PubMed] [Google Scholar]
- Wang JY, Sun T, Zhao XL, et al. Functional significance of VEGF-a in human ovarian carcinoma: role in vasculogenic mimicry. Cancer Biol Ther. 2008;7(5):758–66. doi: 10.4161/cbt.7.5.5765. [DOI] [PubMed] [Google Scholar]
- Zhang JT, Sun W, Zhang WZ, et al. Norcantharidin inhibits tumor growth and vasculogenic mimicry of human gallbladder carcinomas by suppression of the PI3-K/MMPs/Ln-5gamma2 signaling pathway. BMC Cancer. 2014;14:193. doi: 10.1186/1471-2407-14-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seftor RE, Seftor EA, Koshikawa N, et al. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001;61:6322–7. [PubMed] [Google Scholar]
- Lu XS, Sun W, Ge CY, Zhang WZ, Fan YZ. Contribution of the PI3K/MMPs/Ln-5gamma2 and EphA2/FAK/Paxillin signaling pathways to tumor growth and vasculogenic mimicry of gallbladder carcinomas. Int J Oncol. 2013;42:2103–15. doi: 10.3892/ijo.2013.1897. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–9. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- Tang J, Yan H, Zhuang S. Histone deacetylases as targets for treatment of multiple diseases. Clin Sci. 2013;124:651–62. doi: 10.1042/CS20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz M, Esteller M. New therapeutic targets in cancer: the epigenetic connection. Clin Transl Oncol. 2006;8:242–9. doi: 10.1007/BF02664934. [DOI] [PubMed] [Google Scholar]
- Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- Zhu J, Wan H, Xue C, Jiang T, Qian C, Zhang Y. Histone deacetylase 3 implicated in the pathogenesis of children glioma by promoting glioma cell proliferation and migration. Brain Res. 2013;1520:15–22. doi: 10.1016/j.brainres.2013.04.061. [DOI] [PubMed] [Google Scholar]
- Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004;6:744–50. doi: 10.1593/neo.04277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter MJ, Pybus L, Litherland GJ, et al. HDAC-mediated control of ERK- and PI3K-dependent TGF-beta-induced extracellular matrix-regulating genes. Matrix Biol. 2010;29:602–12. doi: 10.1016/j.matbio.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Kurundkar D, Srivastava RK, Chaudhary SC, et al. Vorinostat, an HDAC inhibitor attenuates epidermoid squamous cell carcinoma growth by dampening mTOR signaling pathway in a human xenograft murine model. Toxicol Appl Pharmacol. 2013;266:233–44. doi: 10.1016/j.taap.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Yu L, Ling GQ, et al. Vasculogenic mimicry and its clinical significance in medulloblastoma. Cancer Biol Ther. 2012;13(5):341–8. doi: 10.4161/cbt.19108. [DOI] [PubMed] [Google Scholar]
- Ling G, Wang S, Song Z, et al. Transforming growth factor-beta is required for vasculogenic mimicry formation in glioma cell line U251MG. Cancer Biol Ther. 2011;12:978–88. doi: 10.4161/cbt.12.11.18155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D, Ong JM, Hu J, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo. Clin Cancer Res. 2007;13:1045–52. doi: 10.1158/1078-0432.CCR-06-1261. [DOI] [PubMed] [Google Scholar]
- Xu J, Sampath D, Lang FF, et al. Vorinostat modulates cell cycle regulatory proteins in glioma cells and human glioma slice cultures. J Neurooncol. 2011;105:241–51. doi: 10.1007/s11060-011-0604-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seftor RE, Seftor EA, Kirschmann DA, Hendrix MJ. Targeting the tumor microenvironment with chemically modified tetracyclines: inhibition of laminin 5 gamma2 chain promigratory fragments and vasculogenic mimicry. Mol Cancer Ther. 2002;1:1173–9. [PubMed] [Google Scholar]
- Pecuchet N, Cluzeau T, Thibault C, Mounier N. Vignot S [Histone deacetylase inhibitors: highlight on epigenetic regulation] Bull Cancer. 2010;97:917–35. doi: 10.1684/bdc.2010.1121. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Oneyama C, Kimura H, Tajima S, Okada M. Down-regulation of the tumor suppressor C-terminal Src kinase (Csk)-binding protein (Cbp)/PAG1 is mediated by epigenetic histone modifications via the mitogen-activated protein kinase (MAPK)/phosphatidylinositol 3-kinase (PI3K) pathway. J Biol Chem. 2011;286:15698–706. doi: 10.1074/jbc.M110.195362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banik D, Khan AN, Walseng E, Segal BH, Abrams SI. Interferon regulatory factor-8 is important for histone deacetylase inhibitor-mediated antitumor activity. PLoS ONE. 2012;7(9):e45422. doi: 10.1371/journal.pone.0045422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni F, Zaliani A, Bertolini G, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci USA. 2002;99:2995–3000. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411–21. doi: 10.1038/nrc1092. [DOI] [PubMed] [Google Scholar]
- Paulis YW, Soetekouw PM, Verheul HM, Tjan-Heijnen VC, Griffioen AW. Signalling pathways in vasculogenic mimicry. Biochim Biophys Acta. 2010;1806(1):18–28. doi: 10.1016/j.bbcan.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Ma JL, Han SX, Zhu Q, et al. Role of Twist in vasculogenic mimicry formation in hypoxic hepatocellular carcinoma cells in vitro. Biochem Biophys Res Commun. 2011;408:686–91. doi: 10.1016/j.bbrc.2011.04.089. [DOI] [PubMed] [Google Scholar]
- Zhang S, Li M, Zhang D, et al. Hypoxia influences linearly patterned programmed cell necrosis and tumor blood supply patterns formation in melanoma. Lab Invest. 2009;89:575–86. doi: 10.1038/labinvest.2009.20. [DOI] [PubMed] [Google Scholar]
- Wu MZ, Tsai YP, Yang MH, et al. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 2011;43(5):811–22. doi: 10.1016/j.molcel.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Kim MS, Kwon HJ, Lee YM, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7:437–43. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Doc. S1. Histone deacetylase 3 (HDAC3) inhibitor influenced vasculogenic mimicry (VM) in U87MG cells and MMP-14 and Ln5γ2 were involved in VM.
Fig. S1. Histone deacetylase 3 (HDAC3) inhibitor influenced vasculogenic mimicry in U87MG cells.
Fig. S2. Matrix metalloproteinase-14 and laminin5γ2 are involved in vasculogenic mimicry.