

Abstract
Background
Tenofovir disoproxil fumarate (TDF) causes kidney toxicity in some patients. We performed genome-wide analyses to identify associations with plasma tenofovir clearance and change in creatinine clearance (CrCl) during the first 6 months after initiating therapy among subjects randomized to TDF/emtricitabine-containing regimens in AIDS Clinical Trials Group protocol A5202.
Methods
Pharmacokinetic analyses involved 501 subjects randomized to the tenofovir arm. The CrCl analyses involved 1096 subjects, including 548 control subjects randomized to abacavir-containing regimens. All had been randomized to also receive atazanavir/ritonavir or efavirenz. Multivariable linear regression and generalized least squares models tested for associations between polymorphisms and tenofovir clearance and CrCl change, with Bonferroni correction. Planned sub-analyses considered candidate genes and polymorphisms.
Results
Median CrCl at baseline was 116 ml/min (interquartile range [IQR] 99.8 to 135.5). Median change in CrCl after 6 months was −0.5 ml/min (−10.7 to +10.8) and 2.2 (IQR −9.9 to +13.2) in tenofovir and abacavir arms, respectively. In genome-wide analyses SLC17A1 rs12662869 was associated with an increase in tenofovir clearance (P = 7.1x10−9). In candidate gene analysis for tenofovir clearance, most polymorphisms evaluated were in ABCC4. In the ABCC4 region, the lowest p-value was for CLDN10 rs12866697 (P=1.4x10−3). Among African Americans, SLC22A2 rs3127573 was associated with a greater 6-month CrCl increase in the tenofovir arm after correcting for multiple comparisons (P = 3.3x10−5).
Conclusions
Among subjects randomized to receive TDF/emtricitabine in A5202, there were no genome-wide significant associations with change in CrCl. This study did not replicate polymorphisms previously implicated in tenofovir-associated renal injury.
Keywords: HIV, tenofovir, pharmacokinetics, creatinine clearance
Introduction
Tenofovir disoproxil fumarate (TDF) is included among recommended first-line regimens for human immunodeficiency virus type 1 (HIV-1) infection [1]. It is converted in vivo to tenofovir, which undergoes intracellular diphosphorylation to its active moiety, tenofovir diphosphate [2]. Although generally safe, effective and well tolerated [3, 4], some HIV-infected patients prescribed TDF experience declines in creatinine clearance (CrCl) [4, 5-7], particularly in regimens that include an HIV-1 protease inhibitor plus low-dose ritonavir [4, 6]. In AIDS Clinical Trials Group (ACTG) protocol A5202, median change in CrCl from baseline to week 96 decreased by 3 mL/min in subjects who received TDF/emtricitabine with atazanavir/ritonavir, but increased by 5 mL/min in subjects who received TDF/emtricitabine with efavirenz [4]. Discontinuation or dose reduction of TDF/emtricitabine for changes in renal function in A5202 was infrequent [4].
Renal elimination of tenofovir involves glomerular filtration and tubular secretion [8]. Tenofovir entry into proximal tubule cells appears to be mediated by two transporters, organic anion transporter 1 (OAT1, also called solute carrier family 22 member 6 (SLC22A6) and OAT2 (also called SLC22A7) [9]. Renal tubular secretion is mediated by efflux transporters including multidrug resistance protein 4 (MRP4, also called ATP-binding cassette, sub-family C, member 4 (ABCC4)) [9], and possibly MRP2 (also called ABCC2), although the importance of MRP2 is uncertain [9]. Declines in CrCl associated with concomitant HIV-1 protease inhibitors may reflect ABCC4 inhibition, with resultant tenofovir accumulation in proximal tubule cells.
Candidate gene studies have suggested associations between single nucleotide polymorphisms (SNPs) and adverse renal effects with tenofovir-containing regimens. A study of 30 patients in France suggested increased risk for proximal renal tubulopathy with an ABCC2 polymorphism (1249G→A, rs2273697, P = 0.02) [10]. A study of 190 Japanese patients suggested increased risk for renal tubular dysfunction with two polymorphisms in ABCC2 (−24T→C, rs717620, and 1249 G→A, each P = 0.02) [11]. An analysis of 115 patients in Spain suggested increased risk of tubular dysfunction with ABCC2 −24T→C (P = 0.03) [29], and two polymorphisms in ABCC10 (rs9349256, P = 0.02; and rs2125739, P = 0.05) [12]. Another study of 30 patients suggested an association between higher peripheral blood mononuclear cell tenofovir diphosphate concentrations and an ABCC4 polymorphism (3463A→G, rs1751034, P = 0.04) [13]. Kidney tubular dysfunction included serum creatinine and/or creatinine clearance differences in some reports [10][11] but not others [12][14]. Previous reports showed limited replication other than perhaps ABCC2 polymorphisms [10][11].
We present two genome-wide association studies (GWAS) based on HIV-infected subjects from a prospective randomized clinical trial. The first GWAS considers plasma tenofovir clearance among subjects randomized to tenofovir (treatment-only study); the second considers change in estimated CrCl among patients randomized to tenofovir versus abacavir (treatment-and-control study). Both tenofovir clearance and change in CrCl are treated as continuous variables. This is the first GWAS of tenofovir pharmacokinetics, and of tenofovir-related change in renal function. We identified SNPs of potential interest, including SNPs that were genome-wide significant after Bonferroni correction. Polymorphisms previously implicated in tenofovir renal toxicity did not replicate.
Methods
Study subjects
AIDS Clinical Trials Group (ACTG) Protocol A5202 (ClinTrials.gov NCT00118898) was a phase IIIb study of once-daily regimens for initial treatment of HIV-1 infection [3, 4]. Briefly, 1,858 HIV-infected subjects were randomized to receive either TDF/emtricitabine (300 mg/200 mg) or abacavir/lamivudine (600 mg/300 mg), with either open-label atazanavir (300 mg) plus ritonavir (100 mg), or efavirenz (600 mg). Serum creatinine determinations were performed before entry, at entry, at weeks 4, 8, 16 and 24, and every 12 weeks thereafter until week 96 after the last subject enrolled. Creatinine clearance was calculated using the Cockcroft-Gault formula based on ideal body weight [15].
Tenofovir assays and plasma sampling
Pharmacokinetic samples include those collected between weeks 4 and 24 of A5202. A sparse sampling strategy was designed to measure antiretroviral concentrations in 3 plasma samples per subject. Plasma collection times included a 24-hour post-dose sample followed by an observed-dose sample 3-4 hours post-dose, and another sample 5-15 hours post-dose. The TDF dose of 300 mg is equivalent to 245 mg of tenofovir disoproxil and to 136 mg of tenofovir. Steady-state plasma concentrations of tenofovir were measured using tandem mass spectrometry detection [16].
Pharmacokinetic model development
A total of 2,172 plasma tenofovir determinations from 818 participants were analyzed using a nonlinear mixed-effects modeling approach (NONMEM version VII; ICON, Ellicott City, MD). One- and two-compartment disposition models with first-order absorption were tested to determine the pharmacokinetic structural model. First-order conditional estimation method with interaction (FOCEI) was used throughout. The final model was a two-compartment model that estimated apparent oral and intercompartmental clearances, and volumes of distribution of central and peripheral compartments. Because concentration data were lacking in the absorption phase, the absorption rate constant was fixed to 1 h−1 based on previous data [17]. Exponential errors with log-normal distribution were used for intersubject variability of pharmacokinetic parameters. A proportional error model was assigned to residual variability. The final structural pharmacokinetic model was assessed by successful convergence and goodness-of-fit plots. Individual Bayesian estimates of oral clearance values of tenofovir were estimated from the final model.
Genetic polymorphisms
Genetic consent was obtained under protocol A5128 [18]. Of 1,858 subjects with clinical data, 1,356 consented to A5128, including 677 randomized to tenofovir-containing regimens (350 with efavirenz, 327 with atazanavir/ritonavir) and 679 randomized to abacavir-containing regimens (349 with efavirenz, 330 with atazanavir/ritonavir). Genome-wide genotype data on 1,221 subjects from Illumina Human-1M-Duo were from a separate immunogenomics project [19]. The Vanderbilt Institutional Review Board and the ACTG approved this use of genotype data.
For pharmacokinetic association analyses, we considered candidate SNPs of potential relevance to tenofovir [10-12, 14, 21-27]. Initially, 25 candidate SNPs in 11 genes were identified using PharmGKB (ABCB1, ABCC10, ABCC2, ABCC4, AK2, AK3, NME1, SLC22A6, SLC22A8 and SLC22A11) [28], of which 15 were in our genotype data. Proxies were identified for 3 of the other 10 (a total of 15 proxies) through SNP annotation and proxy (SNAP) search [29], using an r2 threshold of 0.8, providing 30 SNPs (representing 18 candidate SNPs) for pharmacokinetic association analysis. For separate analyses we considered all available SNP data between transcription start and end positions of these 11 genes, based on the human reference genome hg18/NCBI36 [30]. We identified 594 such SNPs: 110 in ABCB1, 12 in ABCC10, 38 in ABCC2, 378 in ABCC4, 8 in AK2, 18 in AK3, 6 in NME1, 6 in SLC22A6, 14 in SLC22A8, 4 SLC22A11 and 0 in NME1.
For CrCl association analyses, we identified 91 SNPs previously associated with any renal trait at P < 1.0 x 10−5 in any cohort, as posted to the NHGRI GWAS Catalog [31], of which 43 were available in our genotype data. These GWAS Catalog traits were chronic kidney disease, chronic kidney disease and serum creatinine levels, creatinine levels, end-stage renal disease, renal function and chronic kidney disease, renal function-related traits (BUN), renal function-related traits (eGRF creatinine), renal function-related traits (sCR), renal function-related traits (urea), glomerulosclerosis, IgA nephropathy, nephropathy, nephropathy (idiopathic membranous), and nephrotic syndrome (acquired). Proxies were identified for 34 of the other 48 SNPs (a total of 169 proxies) as described above, providing 212 SNPs (representing 77 candidate SNPs) for CrCl association analysis.
Quality control of genetic data
Quality control (QC) was done using PLINK version 1.07 [20]. Before QC, 546 subjects met inclusion criteria for pharmacokinetic (1,212 for CrCl) association analyses as described in the statistical analyses section, and with ~1.2 million SNPs. We excluded subjects with >2% missing genotypes, extreme heterozygosity (|F| > 0.1), for duplicates or relatedness (pihat > 0.125), or sex mismatch. We excluded SNPs that deviated from Hardy-Weinberg equilibrium within principal component (PC)-derived race/ethnicity groups (P < 10−6), with <98% genotyping efficiency, and minor allele frequency (MAF) < 5%. Quality control was performed for the combined group (African Americans, European Americans and Hispanic Americans, hereafter called White, Black, and Hispanic, respectively) and separately for each race/ethnicity group.
We generated PCs to infer and adjust for genetic ancestry. Before generating PCs we removed non-autosomal SNPs, and SNPs from two regions of high linkage disequilibrium (LD), 25.5 MB to 33.5 MB spanning the MHC region on chromosome 6, and from 8.0 MB to 12.0 MB on chromosome 8. Subsets of SNPs in low pairwise LD (r2 < 0.2) were used to generate PCs using smartpca in EIGENSTRAT [32]. Race/ethnicity was derived by analyzing these samples in concert with HapMap 3 samples from 11 populations [19].
Imputing genotypes
Imputation was done by Dr. Marylyn Ritchie's laboratory at Pennsylvania State University. Briefly, post QC data were imputed to 1000 Genomes [33] after converting to genome build 37 using liftOver [34] and stratifying by chromosome to parallelize imputation processing. ShapeIt2 [35] was used to check strand alignment and to phase data. The IMPUTE2 algorithm [36] was used to impute additional genotypes that were available in the 1000 Genomes reference panel, but not directly genotyped. Each chromosome was segmented into 6 MB regions with at least 3500 reference variants in each region. Imputed genotypes were included if posterior probabilities exceeded 0.9. Chromosomes 23 through 26 were not imputed.
Quality of imputed data was assessed following the Electronic Medical Records and Genomics (eMERGE) protocol [37]. Each chromosome from each phase was checked for 100% concordance with genotyped data. No batch effects were found. Imputed SNPs were dropped for imputation scores < 0.3, genotyping call rates < 98% and minor allele frequencies < 0.05.
Tenofovir clearance association analyses
Pharmacokinetic association analyses included only subjects that had been randomized to tenofovir-containing arms and had available clinical, pharmacokinetic, and genotype data. Multivariable linear regression models were fit. Plasma tenofovir clearance values approximated a normal distribution (Supplemental Digital Content Figure 1). Analyses were performed on all subjects, and separately in each group (White, Black, and Hispanic) based on PCs. Meta-analysis of the three stratified models was also performed.
Tenofovir clearance, estimated from the pharmacokinetic model, was regressed using multiple linear regression on genotype, adjusting for sex, age, body mass index (BMI), concomitant efavirenz versus atazanavir/ritonavir use, and baseline CrCl. In the combined analysis, we also adjusted for the first two PCs (adjusting for additional PCs did not substantially change results; see Supplemental Digital Content Table 1) and self-reported race coded as White, Black, Hispanic, and other. For QC we tested for the known genome-wide association between UGT1A1 SNPs and baseline plasma bilirubin concentration [38].
As a sensitivity analysis, we repeated the TDF clearance analyses as described above, but evaluated the combined effect of genotype and a genotype-by-efavirenz/atazanavir interaction using a likelihood ratio test with 2 degrees of freedom.
Creatinine clearance association analyses
Creatinine clearance association analyses included subjects randomized to TDF or abacavir arms, and with available clinical and genotype data. All determinations within 200 days after randomization were included in analyses; 200 days was chosen to represent 6 months plus a grace period. At least two CrCl determinations after baseline were available from 91% of subjects.
The TDF and abacavir arms were expected to result in different changes in creatinine clearance over time, and we were interested in identifying SNPs that influence this difference. We addressed this using a one-degree-of-freedom test for significance of a three-way interaction among time, treatment, and genotype. We used a generalized least squares regression model with compound symmetric correlation structure, with creatinine clearance (hereafter called time-dependent CrCl change) as the outcome and including all main, two-way and three-way interactions of time, treatment arm (TDF or abacavir), and genotype. Generalized least squares models, instead of linear mixed effects models, were chosen for analyses with multiple measurements per subject because of their computational efficiency and so not to assume a distributional form for the random effects. The model was adjusted for sex, age, BMI, self-reported race, concomitant antiretroviral (efavirenz or atazanavir/ritonavir), baseline CrCl, and the first two PCs. Baseline CrCl was the value at randomization (i.e., day 0); for subjects without data on day 0, we used the first available value before randomization (preferred) or after randomization. Three additional models, stratified by PC-inferred race, were fit in the same manner as above, but without adjusting for self-reported race and PCs. Meta-analysis of regression output from the stratified models was also done. For CrCl we evaluated the 212 candidate GWAS Catalog SNPs.
As sensitivity analyses, we repeated analyses as described above, but using change in CrCl from baseline to 6 months (defined as the value closest to day 183 ± 30 days) as the phenotype. We hereafter call this the 6-month CrCl change. This cut-off is based on reported time to change in creatinine, much of which is apparent within the first 6 months of initiating TDF-containing regimens. Time was excluded in this model, and the p-value for two-way interaction between genotype and treatment arm (TDF or abacavir) was calculated. Because patients prescribed tenofovir in A5202 were more likely to experience a decline in CrCl if also prescribed a ritonavir-boosted protease inhibitor [4], we performed an additional sensitivity analysis by repeating the analyses for 6-month CrCl change with the inclusion of an interaction between treatment arm and concomitant ARV.
Statistical software and technicalities
Genetic data management and statistical analyses were done using PLINK version 1.07 [20] and R version 3.0.1. Meta-analyses were performed in PLINK, and the random effects p-values reported. Analysis scripts are available upon request. Except where indicated otherwise, we used Bonferroni correction to determine significance thresholds, with P < 5.0 x 10−8 for genome-wide analyses, and 0.05 divided by number of SNPs in candidate gene and SNP analyses. QQ-plots of each analysis showed no evidence of genomic inflation (Supplemental Digital Content Figures 2-7). QQ-plots of tenofovir clearance and CrCl residuals suggested linear models were appropriate (Supplemental Digital Content Figures 8-10).
Results
Study subjects and genetic data
Characteristics of subjects in tenofovir and abacavir arms are shown in Table 1. Randomization provided similar distributions of sex (86-87% male), concomitant antiretroviral (49% atazanavir and 51% efavirenz), age (median 38-39 years), BMI (median 24.8 kg/m2), baseline creatinine clearance (median 116 ml/min), and race/ethnicity between study arms. Figure 1 describes data management and QC steps in the combined group analyses. After QC, there were 501 subjects and ~890,000 SNPs for pharmacokinetic analyses, and 1096 subjects (548 randomized to TDF-containing regimens) and ~840,000 SNPs for creatinine clearance analyses. There were ~ 4.3 million imputed SNPs for both analyses.
Table 1.
Baseline characteristics of study subjects
| Variable | TDF/FTC (n = 501) | ABC/3TC (n = 548) |
|---|---|---|
| Male; No. (%) | 434 (87) | 472 (86) |
| Self-reported race/ethnicity; n (%) | ||
| White | 243 (49) | 243 (44) |
| Black | 149 (30) | 189 (36) |
| Hispanic | 100 (20) | 102 (18) |
| Other | 9 (2) | 14 (2) |
| Concomitant antiretroviral; n (%) | ||
| Atazanavir/r | 246 (49) | 269 (49) |
| Efavirenz | 255 (51) | 279 (51) |
| Age in years; median (IQR) | 39.0 (31.0, 45.0) | 38.0 (31.0, 45.0) |
| BMI in kg/m2; median (IQR) | 24.8 (22.3, 27.9) | 24.8 (22.3, 27.8) |
| Baseline CrCl in ml/min; median (IQR) | 116.0 (99.8, 135.5) | 116.6 (99.2, 138.0) |
TDF/FTC = tenofovir disoproxil fumarate with emtricitabine; ABC/3TC = abacavir with lamivudine; n = number; kg/m2 = kilogram per square meter; mL/min = milliliter per minute; BMI = body mass index; CrCl = creatinine clearance; IQR = interquartile range.
Figure 1. Disposition of study subjects and SNPs through the data management and QC process.
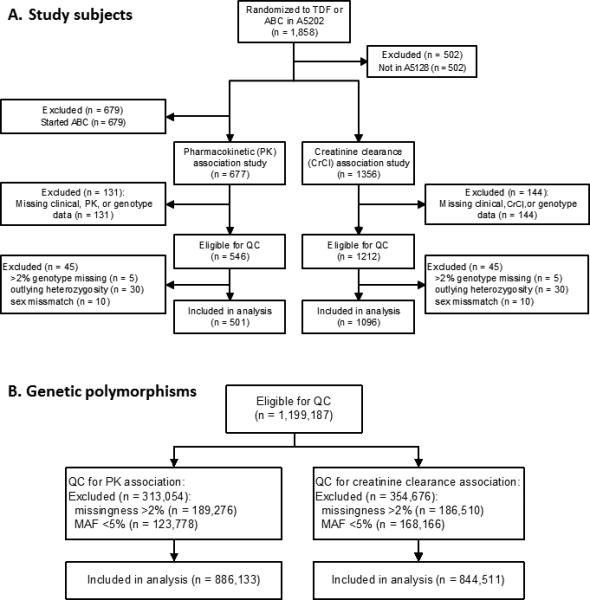
Panel A is the disposition of study subjects. The number of subjects included in PK and CrCl association analyses varied depending on the SNPs included in the analysis, with a median (IQR) of 501 (500 to 501) PK analysis subjects, and 1039 (1038 to 1040) CrCl analysis subjects. Panel B is the disposition of genetic polymorphisms.
Tenofovir clearance pharmacokinetic association analyses
For pharmacokinetic association analyses, median plasma tenofovir clearance was 46.8 L/hr (IQR 39.7 to 53.7). Table 2 shows the 20 SNPs with the smallest p-values in meta-analyses based on GWAS with genotyped SNPs, GWAS with imputed SNPs, association analyses with candidate SNPs, and association analyses with candidate genes. Minor allele frequencies of these 20 SNPs are in Supplemental Digital Content Table 2. In the GWAS analysis with imputed SNPs, rs12662869 in SLC17A1 was associated with increased tenofovir clearance (P = 7.1x10−9); five SLC17A1 polymorphisms in strong LD with rs12662869 (rs6926425, rs1179086, chr6:25792711:D, rs1575535, rs1165216) were also among the top ten SNPs with the lowest p-values. Results of similar analyses, but for all subjects adjusting for PC-derived ancestry, and separately within each race/ethnicity group, are provided in Supplemental Digital Content Table 3. Of 30 candidate SNPs evaluated, 15 (50%) were in ABCC4. In a LocusZoom plot of this region, the lowest p-value was in CLDN10 (rs12866697, P=1.4x10−3), not ABCC4 (Figure 2, Panel A). As a positive control, log-transformed baseline bilirubin concentration was analyzed [38]; multiple UGT1A1 SNPs were associated (P = 2.2 x 10−11 for UGT1A1 rs887829), confirming our ability to detect true associations.
Table 2.
Meta-analysis Results of Pharmacokinetic Associations (top 20 SNPs)
| GWAS (genotyped SNPs) | GWAS (imputed SNPs) | Candidate SNPs (30 genotyped SNPs tested) | Candidate SNPs (594 genotyped SNPs tested) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | chr | gene | Pa | SNP | chr | gene | Pa | SNP | chr | gene | Pb | SNP | chr | gene | Pc |
| rs113162402 | 17 | SEPT4c | 1.40E-07 | rs12662869 | 6 | SLC17A1 | 7.10E-09 | rs3818486 | 13 | ABCC4 | 3.59E-02 | rs3847258 | 9 | AK3 | 2.93E-02 |
| rs359770 | 18 | SMIM21, ZNF516c | 4.00E-06 | rs6926425 | 6 | SLC17A1 | 5.40E-08 | rs2185631 | 6 | ABCC10 | 4.81E-02 | rs16921966 | 9 | AK3 | 3.28E-02 |
| rs359769 | 18 | SMIM21, ZNF516c | 4.10E-06 | rs1179086 | 6 | SLC17A1 | 1.10E-07 | rs6421690 | 11 | SLC22A11 | 7.01E-02 | rs3818486 | 13 | ABCC4 | 3.59E-02 |
| rs438697 | 1 | LPHN2, TTLL7c | 8.20E-06 | chr6:25792711:D | 6 | SLC17A1 | 1.70E-07 | rs4148486 | 13 | ABCC4 | 8.97E-02 | rs10798924 | 1 | AK2 | 3.67E-02 |
| rs1833170 | 16 | C16orf72, GRIN2Ac | 8.40E-06 | rs2586052 | 17 | CUEDC1 | 1.70E-07 | rs2389204 | 13 | ABCC4 | 1.07E-01 | rs12429339 | 13 | ABCC4 | 3.90E-02 |
| re17199679 | 9 | KLF4, IKBKAPc | 9.30E-06 | rs1575535 | 6 | SLC17A1 | 1.70E-07 | rs7924450 | 11 | SLC22A11 | 1.21E-01 | rs11591185 | 1 | AK2 | 4.64E-02 |
| rs6429839 | 1 | LPHN2, TTLL7c | 1.20E-05 | rs1165216 | 6 | SLC17A1 | 3.90E-07 | rs11231803 | 11 | SLC22A11 | 1.24E-01 | rs2185631 | 6 | ABCC10 | 4.81E-02 |
| rs4662167 | 1 | LPHN2, TTLL7c | 1.20E-05 | rs113162402 | 17 | SEPT4 | 4.40E-07 | rs3818493 | 13 | ABCC4 | 1.29E-01 | rs2268691 | 1 | AK2 | 5.29E-02 |
| rs7829911 | 8 | SBSPON, RPL7c | 1.50E-05 | rs1408270 | 6 | SLC17A3 | 5.80E-07 | rs9349256 | 6 | ABCC10 | 1.43E-01 | rs1611822 | 13 | ABCC4 | 5.93E-02 |
| rs1165176 | 6 | SLC17A1 | 2.00E-05 | rs2685506 | 17 | CUEDC1 | 6.20E-07 | rs2274405 | 13 | ABCC4 | 1.71E-01 | rs17268129 | 13 | ABCC4 | 5.98E-02 |
| rs1420040 | 16 | GRIN2A | 2.00E-05 | rs3807044 | 6 | KCNK16 | 6.20E-07 | rs7331142 | 13 | ABCC4 | 1.77E-01 | rs1751033 | 13 | ABCC4 | 7.18E-02 |
| rs765285 | 6 | SLC17A1 | 2.10E-05 | rs390296 | 1 | LPHN2, TTLL7c | 1.90E-06 | rs9524827 | 13 | ABCC4 | 1.82E-01 | rs9561765 | 13 | ABCC4 | 7.94E-02 |
| rs1165177 | 6 | SLC17A1 | 2.10E-05 | rs437749 | 1 | LPHN2, TTLL7c | 1.90E-06 | rs9394952 | 6 | ABCC10 | 2.00E-01 | rs1564351 | 13 | ABCC4 | 8.01E-02 |
| rs1185569 | 6 | SLC17A1 | 2.10E-05 | rs409651 | 1 | LPHN2, TTLL7c | 1.90E-06 | rs1045642 | 7 | ABCB1 | 2.38E-01 | rs7330330 | 13 | ABCC4 | 8.22E-02 |
| rs4319926 | 2 | ST6GAL2, RGPD4-AS1c | 2.20E-05 | rs423768 | 1 | LPHN2, TTLL7c | 1.90E-06 | rs2273697 | 10 | ABCC2 | 2.80E-01 | rs4148487 | 13 | ABCC4 | 8.36E-02 |
| rs4442993 | 2 | ST6GAL2, RGPD4-AS1c | 2.20E-05 | rs6693748 | 1 | LPHN2, TTLL7c | 1.90E-06 | rs717620 | 10 | ABCC2 | 2.87E-01 | rs4148442 | 13 | ABCC4 | 8.68E-02 |
| rs563189 | 1 | LPHN2, TTLL7c | 2.50E-05 | rs7552456 | 1 | LPHN2, TTLL7c | 1.90E-06 | rs2125739 | 6 | ABCC10 | 3.19E-01 | rs4148451 | 13 | ABCC4 | 8.68E-02 |
| rs10503961 | 8 | DUSP26, UNC5Dc | 2.50E-05 | rs10874339 | 1 | LPHN2, TTLL7c | 1.90E-06 | rs4148477 | 13 | ABCC4 | 3.35E-01 | rs1729745 | 13 | ABCC4 | 8.89E-02 |
| rs7144413 | 14 | SCFD1, COCHc | 2.70E-05 | rs12141972 | 1 | LPHN2, TTLL7c | 1.90E-06 | rs4148478 | 13 | ABCC4 | 3.35E-01 | rs4148486 | 13 | ABCC4 | 8.97E-02 |
| rs11844480 | 14 | SCFD1, COCHc | 2.80E-05 | rs10874341 | 1 | LPHN2, TTLL7c | 1.90E-06 | rs17222723 | 10 | ABCC2 | 4.27E-01 | rs9524858 | 13 | ABCC4 | 9.17E-02 |
CHR = Chromosome; SNP = SNP identifier; P = P-value.
Significance threshold was 5×10−8 for the genome-wide analyses.
Significance threshold was 0.002 for the subset of 30 SNPs.
Significance threshold was 8.4×10−5 for the subset of 594 SNPs.
Highlighted: imputed SNP found to be associated with tenofovir clearance
Figure 2. LocusZoom plots of ABCC4 gene region for association with tenofovir pharmacokinetics and change in creatinine clearance by meta-analysis.
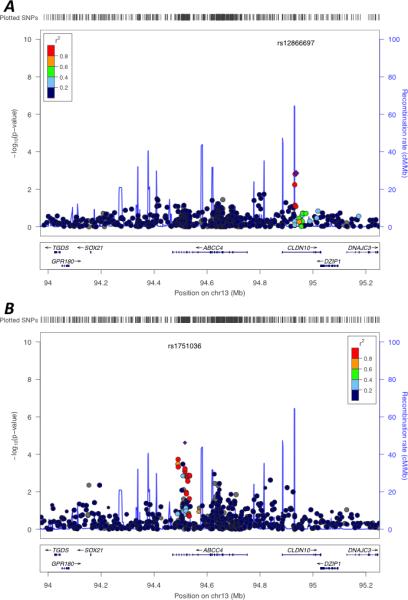
The region of ABCC4 (± 500 KB) is shown. Genes in the region are shown at the bottom. Filled circles represent p-values for SNPs in our data. Markers are color coded to represent their degree of correlation (r2) with the lowest p-value SNP as estimated internally by LocusZoom using the hg18/HapMap Phase II CEU genome build [45]. The blue lines correspond to the recombination rate [45]. Panel A represents tenofovir pharmacokinetic analyses. The lowest p-value SNP for tenofovir pharmacokinetics in this region, rs12866697, is represented by the purple diamond. Panel B represents change in creatinine clearance analyses. The lowest p-value SNP for change in creatinine clearance in this region, rs1751036, is represented by the purple diamond. The 12 SNPs with the lowest p-values for change in creatinine clearance are rs7330330, rs7331488, rs4148540, rs2766475, rs1678387, rs1678409, rs1678365, rs1189466, rs1751043, rs943289, rs1189435, and rs1189434.
Based on results from a prior study [4], we performed an additional GWAS with genotyped SNPs (non-imputed) including a genotype-efavirenz or atazanavir/r interaction on TDF clearance. The results, based on a likelihood ratio test, were similar: No SNPs were genome-wide significant, and SNPs with the smallest p-values were those seen in the initial analyses (results not shown).
Creatinine clearance association analyses
The median change in CrCl from baseline to 6 months was −0.5 ml/min (IQR −10.7 to +10.8) and 2.2 (IQR −9.9 to +13.2) in the tenofovir and abacavir arms, respectively. No SNP was significantly associated with a differential time-dependent CrCl change between the tenofovir and abacavir arms in the first 200 days of therapy, after Bonferroni correction. The 20 SNPs with the lowest meta-analysis p-values in GWAS with genotyped SNPs, GWAS with imputed SNPs, and association analyses with candidate SNP meta-analyses for time-dependent CrCl change are shown in Table 3. Minor allele frequencies of these 20 SNPs are in Supplemental Digital Content Table 2. Results of similar analyses, but for all subjects adjusting for PC-derived ancestry, and separately within each race/ethnicity group, are provided in Supplemental Digital Content Table 4. In the entire population, rs1751036 in ABCC4 was among the top 20 SNPs for time-dependent CrCl change (P = 2.4 x 10−5, Supplemental Digital Content Table 4). In a LocusZoom plot of this region, 12 ABCC4 SNPs in LD with rs1751036 at r2 > 0.8 were associated with time-dependent change in creatinine clearance at unadjusted P < 0.01 (Figure 2, Panel B).
Table 3.
Meta-analysis Results of Creatinine Clearance Associations (top 20 SNPs)
| GWAS (genotyped SNPs) | GWAS (imputed SNPs) | Candidate SNPs (from GWAS Catalog) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | chr | gene | Pa | SNP | chr | gene | Pa | SNP | chr | gene | Pc |
| rs121882 | 5 | ARHGAP26 | 1.2E-05 | rs2682618 | 12 | TMEM132B, TMEM132Cc | 4.8E-06 | rs10941692 | 5 | MRPS30, HCN1c | 2.6E-02 |
| rs4243086 | 15 | CYP11A1, SEMA7Ac | 1.5E-05 | rs34574970 | 9 | KLF9, TRPM3c | 9.1E-06 | rs7805747 | 7 | PRKAG2 | 2.8E-02 |
| rs9602954 | 13 | MOB1AP1, DDX6P2c | 1.5E-05 | rs34536198 | 1 | KIRREL | 9.6E-06 | rs12520150 | 5 | MRPS30, HCN1c | 4.0E-02 |
| rs3914576 | 15 | NDN, PWRN2c | 2.0E-05 | rs121882 | 5 | ARHGAP26 | 1.1E-05 | rs9473932 | 6 | TFAP2B, PKHD1c | 5.1E-02 |
| rs419129 | 6 | GSTA3, GSTA4c | 2.5E-05 | chr1:158038705:D | 1 | KIRREL | 1.1E-05 | rs2082424 | 19 | CEP89 | 5.6E-02 |
| rs17039196 | 4 | RAPGEF2, FSTL5c | 2.9E-05 | rs35676418 | 7 | AC005029.1, AC005022.1c | 1.2E-05 | rs7246178 | 19 | CEP89 | 5.9E-02 |
| rs17622266 | 7 | LINC01450 | 3.0E-05 | rs9602954 | 13 | MOB1AP1, DDX6P2c | 1.3E-05 | rs660895 | 6 | HLA-DQA1, HLA-DRB1c | 7.2E-02 |
| rs6963950 | 7 | AC005029.1, AC005022.1c | 3.0E-05 | rs4243086 | 15 | CYP11A1, SEMA7Ac | 1.4E-05 | rs17272197 | 19 | SLC7A9 | 1.5E-01 |
| rs7002294 | 8 | PVT1 | 4.0E-05 | rs7783070 | 7 | AC005029.1, AC005022.1c | 1.6E-05 | rs1133029 | 20 | SULF2, PREX1c | 1.5E-01 |
| rs7034027 | 9 | TRPM3 | 4.2E-05 | rs113869768 | 4 | RAPGEF2, FSTL5c | 1.8E-05 | rs653178 | 12 | ATXN2 | 1.5E-01 |
| rs2273289 | 1 | PLOD1 | 4.3E-05 | rs10780944 | 9 | TRPM3 | 2.0E-05 | rs2057291 | 20 | GNAS | 1.5E-01 |
| rs10758871 | 9 | KDM4C, TMEM261c | 4.5E-05 | rs4605427 | 20 | DOK5, CBLN4c | 2.0E-05 | rs6677604 | 1 | CFH | 1.6E-01 |
| rs1976756 | 15 | NDN, PWRN2c | 4.5E-05 | rs3914576 | 15 | PWRN4 | 2.3E-05 | rs4664308 | 2 | PLA2R1 | 2.4E-01 |
| rs7033976 | 9 | TRPM3 | 4.6E-05 | rs17039181 | 4 | RAPGEF2, FSTL5c | 2.6E-05 | rs16902083 | 5 | HCN1 | 2.5E-01 |
| rs3750494 | 9 | CDK5RAP2 | 4.7E-05 | rs72959075 | 4 | RAPGEF2, FSTL5c | 2.6E-05 | rs11705804 | 3 | LRIG1, SUCLG2c | 2.5E-01 |
| rs749074 | 14 | GPATCH2L | 5.5E-05 | rs4322895 | 20 | DOK5, CBLN4c | 2.6E-05 | rs1556751 | 9 | PIP5K1B | 2.5E-01 |
| rs4524177 | 20 | DOK5, CBLN4c | 5.6E-05 | rs4322896 | 20 | DOK5, CBLN4c | 2.6E-05 | rs11864909 | 16 | PDILT | 2.5E-01 |
| rs2682621 | 12 | TMEM132B, TMEM132Cc | 5.9E-05 | rs4619678 | 20 | DOK5, CBLN4c | 2.7E-05 | rs3925075 | 16 | ITGAM, ITGAXc | 2.6E-01 |
| rs11067378 | 12 | TBX3, MED13Lc | 6.4E-05 | rs4346453 | 20 | DOK5, CBLN4c | 2.7E-05 | rs16853741 | 3 | MECOM | 2.7E-01 |
| rs12701851 | 7 | LINC01450 | 6.7E-05 | rs4622788 | 20 | DOK5, CBLN4c | 2.7E-05 | rs2151421 | 9 | PIP5K1B | 2.8E-01 |
Significance threshold was 5×10−8 for the genome-wide analyses.
b Significance threshold was 0.0002 for the subset of 212 SNPs.
For intergenic SNPs, indicated are the nearest known gene 3′ and 5′ genes.
Sensitivity analyses were performed using the outcome 6-month CrCl change (i.e., CrCl at 6-months minus CrCl at baseline). The 20 SNPs with the lowest meta-analysis p-values in GWAS with genotyped SNPs, GWAS with imputed SNPs, and association analyses with candidate SNP meta-analyses for 6-month CrCl change are shown in Supplemental Digital Content Table 5; similar to primary analyses, no SNP was significant after adjusting for multiple comparisons.
In candidate SNP analyses stratified by population, rs3127573 in SLC22A2 was significantly associated with a positive 6-month CrCl change among African Americans (Supplemental Digital Content Table 6, P = 3.3×10−5), and was also among the top 20 SNPs in the analysis of combined group (Supplemental Digital Content Table 6, P = 0.036). This SNP was also among the top 20 SNPs in the genome-wide analysis of 6-month CrCl change among African Americans (Supplemental Digital Content Table 5), had the second lowest P-value (P = 0.0018) among 212 candidate SNPs in the time-dependent CrCl change analyses among African Americans (Supplemental Digital Content Table 7), and was among the top 20 SNPs in the 212 candidate SNPs analysis of the combined group with time-dependent CrCl change (Supplemental Digital Content Table 7, P = 0.09).
Analyses of 6-month CrCl change in which the interaction between treatment arm and efavirenz or atazanavir/r was included yielded results similar to those in which this interaction was not included. Except for the above SNP in the 212 candidate SNPs analysis of the African American group (rs3127573, P = 4.1×10−5), no SNPs were significantly associated with the outcome.
Discussion
Tenofovir disoproxil fumarate is extensively prescribed worldwide. The present report describes the first GWAS of associations with tenofovir pharmacokinetics, and the first GWAS of change in CrCl with tenofovir-containing regimens. A polymorphism in SLC17A1 was significantly associated with tenofovir clearance (rs12662869, P = 7.1x10−9). However, no polymorphism achieved genome-wide significance (P < 5.0 x 10−8) for association with change in CrCl. The tenofovir clearance GWAS was complemented by targeted analyses involving 594 SNPs in genes suggested to affect tenofovir disposition, and an even more focused analysis involving 30 candidate SNPs suggested to affect tenofovir disposition. No polymorphism was significant in either of these analyses. Our CrCl GWAS was complemented by a more targeted analyses of 212 SNPs associated with any renal trait in prior GWAS. Again, no polymorphism was significant in these targeted analyses.
Several aspects of the present study favored our likelihood of identifying true genotypephenotype associations if present. Both CrCl and tenofovir clearance analysis involved over 500 subjects, considerably more than in previous candidate gene association studies of tenofovir renal toxicity [10-12, 15]. The extent of genotype data analyzed far exceeded previous candidate gene analyses [10-12, 15]. Clinical data were from a prospective randomized clinical trial, which included rigorous quantification of change in creatinine clearance over time, and which showed TDF/emtricitabine with atazanavir/ritonavir to be less favorable in this regard [4, 38, 39]. For CrCl analyses, availability of well matched randomized TDF and abacavir arms allowed the use of the arm containing abacavir (which is not nephrotoxic) as a control to test for associations between SNPs and CrCl change present in the TDF arm but not the abacavir arm, thus permitting an investigation of SNPs that might interact with tenofovir to change CrCl. Substantial numbers of White, Black and Hispanic subjects afforded the opportunity to examine associations in the combined population, and in each population separately, an approach that has proven valuable in pharmacogenomic analyses of other antiretrovirals [38, 39]. Longitudinal models in our analyses allowed us to capture associations between genotype and change in CrCl over time.
There are possible reasons for the paucity of significant associations in the present analyses. With GWAS the threshold for significance after correcting for multiple comparisons is stringent. However, functional polymorphisms that affect drug disposition and/or pharmacodynamics may be genome-wide significant with modest sample sizes. For example, genetic prediction of abacavir hypersensitivity would be genome-wide significant (P<5.0x10−8) with 15 cases and 200 controls [40], and statin-induced myopathy with 85 cases and 90 controls [41]. In addition, we complemented our GWAS with more focused candidate gene/SNPs analyses with less stringent P-value thresholds, which still did not identify significant associations. It is possible that effects of genetic polymorphisms are context dependent, and may not have been detected with concomitant efavirenz or atazanavir/ritonavir (analyses with efavirenz or atazanavir/r interactions also showed insufficient evidence that the effect of genotype on tenofovir clearance or 6-month CrCl change differed by efavirenz or atazanavir/r), but could have been apparent with other concomitant antiretrovirals. We cannot exclude the possibility that previously reported associations were spurious, as reported P-values were marginally significant and would not have withstood correction for multiple comparisons even for the few SNPs genotyped.
We considered plasma tenofovir clearance in the present analyses, despite intracellular tenofovir diphosphate being the presumed toxic moiety. While plasma tenofovir pharmacokinetics do not equate with drug exposure within renal tubular cells, we hypothesized that functional drug transporter gene polymorphisms that affect drug disposition across cell membranes might also affect plasma drug clearance. Analyses with imputed SNPs detected a genome-wide association of SLC17A1 rs12662869 with plasma tenofovir clearance, and additional SNPs in strong LD with rs12662869 in this gene were among the top SNPs detected. Although not a gene known to be associated with tenofovir clearance, the encoded protein sodium-dependent phosphate transport protein 1 is a urate transporter speculated to be relevant to the kidney [42]. Its potential relevance to tenofovir disposition may warrant further study.
Within each analysis, there was some overlap for a few SNPs, especially in the combined and meta-analyses. Although not significant, most of the top SNPs in pharmacokinetic candidate SNP analyses were in ABCC genes, which have been reported to be involved in tenofovir clearance and creatinine clearance among patients receiving tenofovir [8-12]. A previous study of the association of ABCC10 polymorphisms with kidney tubular dysfunction identified an association between rs9349256 (odds ratio = 2.3, P = 0.02) and rs2125739 (OR = 2.0, P = 0.05) and tubular dysfunction [12]. In our study, rs9349256 was among the top 20 (of 30) candidate SNPs evaluated in the combined, Black, and White group analyses, and was among the top 20 (of 594) candidate SNPs evaluated in the White population (P = 0.10), which also suggests an association of this SNP with tenofovir clearance.
Although none of the SNPs in the ABCC family were significant at the genome-wide or candidate SNP level, a SNP near ABCC4, rs12866697 located in CLDN10, was the top SNP in the ABCC4 region (± 500 KB). A biological study in mice linked loss of CLDN10 to hypermagnesemia and nephrocalcinosis because this gene is involved in paracellular sodium permeability [43], but the relevance of this to tenofovir is not apparent. We cannot exclude the possibility that SNPs in CLDN10 regulate ABCC4 expression.
Only the sensitivity analysis of the 212 candidate SNPs identified SNP rs3127573 as significant in the CrCl associations. However, this SNP appeared among the top 20 SNPs in some of our genome-wide and candidate SNPs analyses as outlined in the results section. Previous studies have suggested a potential role of SLC22A6 and SLC22A7 in renal proximal tubules [9, 25]. Furthermore, a study of the association of SLC22A2 polymorphisms with phenotypes of net tubular creatinine secretion in which rs3127573 was one of two SNPs genotyped in patients with end-stage renal disease found an association between end-stage renal disease and rs3127573: odds ratios [95% CI] 1.39 [1.16-1.67] [44]. Our results are in the same direction as this finding, affirming the association of some SLC22A2 SNPs with renal phenotypes.
There were limitations to the present study. Renal toxicity associated with tenofovir in A5202 was modest, so did not include extreme phenotypes. The present analysis focused on change in CrCl as the primary phenotype, but it is possible that other markers of renal tubular function are more affected by genotype. The sample size within each PC-derived race/ethnicity was small, although studies have reported significance with even fewer individuals [10-12]. It is also possible that analyses with ~4.3 million imputed SNPs were limited in power by small sample sizes. Nevertheless, we detected a genome-wide association in SLC17A. It is conceivable that alternative statistical approaches (e.g. joint multiple-SNP analysis) could identify associations not identified herein. Finally, intracellular tenofovir exposure and secretory clearance of tenofovir, which are likely more directly relevant to renal toxicity than is plasma exposure, was not measured in A5202.
In summary, we identified a genome-wide significant association with plasma tenofovir clearance, but not with change in CrCl among patients randomized to TDF-containing regimens in A5202. Further research is warranted to replicate the SLC17A association with tenofovir disposition, and to assess whether previously suggested genetic associations with tenofovir-associated renal tubular injury depend on context, such as specific concomitant medication.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to the many persons with HIV infection who volunteered for A5202 and A5128. In addition, they acknowledge the contributions of study teams and site staff for protocols A5202 and A5128. We are grateful to Marylyn D. Ritchie, PhD for providing imputed genotype data. The project described was supported by Award Number U01AI068636 from the National Institute of Allergy and Infectious Diseases (NIAID) and supported by National Institute of Mental Health (NIMH), National Institute of Dental and Craniofacial Research (NIDCR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. This work was supported by the AIDS Clinical Trials Group funded by the National Institute of Allergy and Infectious Diseases (AI-068636, AI-038858, AI-068634, AI-038855). Grant support included AI-069439, RR-024975, AI-054999 (DWH), AI-077505 (HJR), BRS-ACURE-06-00140-T001 (GDM), and AI-069434 (ACC).
Clinical Research Sites that participated in ACTG protocol A5202, and collected DNA under protocol A5128, were supported by the following grants from NIAID: AI-069477, AI-027675, AI-073961, AI-069474, AI-069432, AI-069513, AI-069423, AI-050410, AI-069452, AI-69450, AI-054907, AI-069428, AI-069439, AI-069467, AI-045008, AI-069495, AI-069415, AI-069556, AI-069484, AI-069424, AI-069532, AI-069419, AI-069471, AI-025859, AI-069418, AI-050409, AI-069423, AI-069501, AI-069502, AI-069511, AI-069434, AI-069465, AI-069471, AI-069494, AI-069424, AI-069472, AI-069501, AI-069470, AI-046376, AI-069511, AI-072626, AI-069472, AI-038858, AI-069472, AI-069472, AI-069471, AI-027661, AI-027661, AI-034853, AI-069472, AI-069447, AI-032782, AI-027658, AI-27666, AI-027661, AI-058740, AI-046370, AI-069423, AI-069470, AI-069511, AI-069470, AI-069494, and by the following grants from the National Center for Research Resources (NCRR): RR-00051, RR-00046, RR-025747, RR-025777, RR-024160, RR-024996, RR-024156, RR024160, and RR-024160. Study drugs were provided by Bristol-Myers Squibb Co. and GlaxoSmithKline, Inc.
Footnotes
Conflict of Interest/Disclosure:
David W. Haas has been principal investigator on a research grant to Vanderbilt from Merck, and has been a consultant to Merck.
Eric S. Daar has received research grants from Bristol Myers Squibb, Abbott, Merck, Pfizer, ViiV, Gilead and a consultant/advisor to Bristol Myers Squibb, Merck, ViiV and Gilead.
Other authors declare no potential conflicts.
References
- 1.The Panel on Clinical Practices for Treatment of HIV Infection. [February 21, 2014];Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Available at: http://www.aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf.
- 2.Kearney BP, Flaherty JF, Shah J. Tenofovir disoproxil fumarate: clinical pharmacology and pharmacokinetics. Clin Pharmacokinet. 2004;43:595–612. doi: 10.2165/00003088-200443090-00003. [DOI] [PubMed] [Google Scholar]
- 3.Sax PE, Tierney C, Collier AC, Fischl MA, Mollan K, Peeples L, et al. Abacavir-lamivudine versus tenofovir-emtricitabine for initial HIV-1 therapy. N Engl J Med. 2009;361:2230–40. doi: 10.1056/NEJMoa0906768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daar ES, Tierney C, Fischl MA, Sax PE, Mollan K, Budhathoki C, et al. Atazanavir plus ritonavir or efavirenz as part of a 3-drug regimen for initial treatment of HIV-1. Ann Intern Med. 2011;154:445–56. doi: 10.1059/0003-4819-154-7-201104050-00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerard L, Chazallon C, Taburet AM, Girard PM, Aboulker JP, Piketty C. Renal function in antiretroviral-experienced patients treated with tenofovir disoproxil fumarate associated with atazanavir/ritonavir. Antivir Ther. 2007;12:31–9. [PubMed] [Google Scholar]
- 6.Goicoechea M, Liu S, Best B, Sun S, Jain S, Kemper C, et al. Greater tenofovir-associated renal function decline with protease inhibitor-based versus nonnucleoside reverse-transcriptase inhibitor-based therapy. J Infect Dis. 2008;197:102–8. doi: 10.1086/524061. [DOI] [PubMed] [Google Scholar]
- 7.Horberg M, Tang B, Towner W, Silverberg M, Bersoff-Matcha S, Hurley L, et al. Impact of Tenofovir on Renal Function in HIV-Infected Antiretroviral Naive Patients. J Acquir Immune Defic Syndr. 2009;53:62–9. doi: 10.1097/QAI.0b013e3181be6be2. [DOI] [PubMed] [Google Scholar]
- 8.Barditch-Crovo P, Deeks SG, Collier A, Safrin S, Coakley DF, Miller M, et al. Phase i/ii trial of the pharmacokinetics, safety, and antiretroviral activity of tenofovir disoproxil fumarate in human immunodeficiency virus-infected adults. Antimicrob Agents Chemother. 2001;45:2733–9. doi: 10.1128/AAC.45.10.2733-2739.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ray AS, Cihlar T, Robinson KL, Tong L, Vela JE, Fuller MD, et al. Mechanism of active renal tubular efflux of tenofovir. Antimicrob Agents Chemother. 2006;50:3297–304. doi: 10.1128/AAC.00251-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Izzedine H, Hulot JS, Villard E, Goyenvalle C, Dominguez S, Ghosn J, et al. Association between ABCC2 gene haplotypes and tenofovir-induced proximal tubulopathy. J Infect Dis. 2006;194:1481–91. doi: 10.1086/508546. [DOI] [PubMed] [Google Scholar]
- 11.Nishijima T, Komatsu H, Higasa K, Takano M, Tsuchiya K, Hayashida T, et al. Single nucleotide polymorphisms in ABCC2 associate with tenofovir-induced kidney tubular dysfunction in Japanese patients with HIV-1 infection: a pharmacogenetic study. Clin Infect Dis. 2012;55:1558–67. doi: 10.1093/cid/cis772. [DOI] [PubMed] [Google Scholar]
- 12.Pushpakom SP, Liptrott NJ, Rodriguez-Novoa S, Labarga P, Soriano V, Albalater M, et al. Genetic variants of ABCC10, a novel tenofovir transporter, are associated with kidney tubular dysfunction. J Infect Dis. 2011;204:145–53. doi: 10.1093/infdis/jir215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiser JJ, Aquilante CL, Anderson PL, King TM, Carten ML, Fletcher CV. Clinical and genetic determinants of intracellular tenofovir diphosphate concentrations in HIV-infected patients. J Acquir Immune Defic Syndr. 2008;47:298–303. doi: 10.1097/qai.0b013e31815e7478. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez-Novoa S, Labarga P, Soriano V, Egan D, Albalater M, Morello J, et al. Predictors of kidney tubular dysfunction in HIV-infected patients treated with tenofovir: a pharmacogenetic study. Clin Infect Dis. 2009;48:e108–16. doi: 10.1086/598507. [DOI] [PubMed] [Google Scholar]
- 15.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 16.King JR, Yogev R, Jean-Philippe P, Graham B, Wiznia A, Britto P, et al. Steady-state pharmacokinetics of tenofovir-based regimens in HIV-infected pediatric patients. Antimicrob Agents Chemother. 2011;55:4290–4294. doi: 10.1128/AAC.01334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baheti G, Kiser JJ, Havens PL, Fletcher CV. Plasma and intracellular population pharmacokinetic analysis of tenofovir in HIV-1-infected patients. Antimicrob Agents Chemother. 2011;55:5294–9. doi: 10.1128/AAC.05317-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haas DW, Wilkinson GR, Kuritzkes DR, Richman DD, Nicotera J, Mahon LF, et al. A multi-investigator/institutional DNA bank for AIDS-related human genetic studies: AACTG Protocol A5128. HIV Clin Trials. 2003;4:287–300. doi: 10.1310/MUQC-QXBC-8118-BPM5. [DOI] [PubMed] [Google Scholar]
- 19.Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–7. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olagunju A, Owen A, Cressey TR. Potential effect of pharmacogenetics on maternal, fetal and infant antiretroviral drug exposure during pregnancy and breastfeeding. Pharmacogenomics. 2012;13:1501–22. doi: 10.2217/pgs.12.138. [DOI] [PubMed] [Google Scholar]
- 22.Michaud V, Bar-Magen T, Turgeon J, Flockhart D, Desta Z, Wainberg MA. The dual role of pharmacogenetics in HIV treatment: mutations and polymorphisms regulating antiretroviral drug resistance and disposition. Pharmacol Rev. 2012;64:803–33. doi: 10.1124/pr.111.005553. [DOI] [PubMed] [Google Scholar]
- 23.Kiser JJ, Carten ML, Aquilante CL, Anderson PL, Wolfe P, King TM, et al. The effect of lopinavir/ritonavir on the renal clearance of tenofovir in HIV-infected patients. Clin Pharmacol Ther. 2008;83:265–72. doi: 10.1038/sj.clpt.6100269. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez-Novoa S, Labarga P, Soriano V. Pharmacogenetics of tenofovir treatment. Pharmacogenomics. 2009;10:1675–85. doi: 10.2217/pgs.09.115. [DOI] [PubMed] [Google Scholar]
- 25.Bleasby K, Hall LA, Perry JL, Mohrenweiser HW, Pritchard JB. Functional consequences of single nucleotide polymorphisms in the human organic anion transporter hOAT1 (SLC22A6). J Pharmacol Exp Ther. 2005;314:923–31. doi: 10.1124/jpet.105.084301. [DOI] [PubMed] [Google Scholar]
- 26.Yee SW, Chen L, Giacomini KM. Pharmacogenomics of membrane transporters: past, present and future. Pharmacogenomics. 2010;11:475–9. doi: 10.2217/pgs.10.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirt D, Urien S, Ekouevi DK, Rey E, Arrivé E, Blanche S, et al. Population pharmacokinetics of tenofovir in HIV-1-infected pregnant women and their neonates (ANRS 12109). Clin Pharmacol Ther. 2009;85:182–9. doi: 10.1038/clpt.2008.201. [DOI] [PubMed] [Google Scholar]
- 28.McDonagh EM, Whirl-Carrillo M, Garten Y, Altman RB, Klein TE. From pharmacogenomic knowledge acquisition to clinical applications: the PharmGKB as a clinical pharmacogenomic biomarker resource. Biomark Med. 2011;5:795–806. doi: 10.2217/bmm.11.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson AD, Handsaker RE, Pulit S, Nizzari MM, O'Donnell CJ, de Bakker PIW. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patrias K. In: Citing Medicine: The NLM Style Guide for Authors, Editors, and Publishers [Internet] 2nd edition. Wendling D, editor. National Library of Medicine (US); Bethesda (MD): 2007. [2011 Sep 15]. Chapter 24, Databases/Retrieval Systems on the Internet. 2007 Oct 10. Available from: http://www.ncbi.nlm.nih.gov/books/NBK7273/ [Google Scholar]
- 31.Hindorff LA, MacArthur J, Morales J, Junkins HA, Hall PN, Klemm AK, et al. [May 20, 2014];A Catalog of Published Genome-Wide Association Studies. Available at: http://www.genome.gov/gwastudies.
- 32.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 33.1000 Genomes Project Consortium. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.liftOver [Internet] Available from: http://genome.ucsc.edu/cgi-bin/hgLiftOver.
- 35.Delaneau O, Zagury J-F, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 36.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verma SS, de Andrade M, Tromp G, Kuivaniemi H, Pugh E, Namjou B, et al. Imputation and quality control steps for combining multiple genome-wide datasets. Front. Genet. 2014;5:370. doi: 10.3389/fgene.2014.00370. doi: 10.3389/fgene.2014.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson DH, Venuto C, Ritchie MD, Morse GD, Daar ES, McLaren PJ, Haas DW. Genomewide association study of atazanavir pharmacokinetics and hyperbilirubinemia in AIDS Clinical Trials Group protocol A5202. Pharmacogenet Genomics. 2014;24:195–203. doi: 10.1097/FPC.0000000000000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holzinger ER, Grady B, Ritchie MD, Ribaudo HJ, Acosta EP, Morse GD, et al. Genome-Wide Association Study of Plasma Efavirenz Pharmacokinetics in AIDS Clinical Trials Group Protocols. Pharmacogenet Genomics. 2012;22:858–67. doi: 10.1097/FPC.0b013e32835a450b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson MR, Bacanu SA, Mosteller M, Li L, Bowman CE, Roses AD, et al. Genome-wide approaches to identify pharmacogenetic contributions to adverse drug reactions. Pharmacogenomics J. 2009;9:23–33. doi: 10.1038/tpj.2008.4. [DOI] [PubMed] [Google Scholar]
- 41.Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med. 2008;359:789–9. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 42.Moss DM, Neary M, Owen A. The role of drug transporters in the kidney: lessons from tenofovir. Front Pharmacol. 2014;5:248. doi: 10.3389/fphar.2014.00248. doi: 10.3389/fphar.2014.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Breiderhoff T, Himmerkus N, Stuiver M, Mutig K, Will C, Meij IC, et al. Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc Natl Acad Sci U S A. 2012;109:14241–6. doi: 10.1073/pnas.1203834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reznichenko A, Sinkeler SJ, Snieder H, van den Born J, de Borst MH, Damman J, et al. SLC22A2 is associated with tubular creatinine secretion and bias of estimated GFR in renal transplantation. Physiol Genomics. 2013;45:201–9. doi: 10.1152/physiolgenomics.00087.2012. [DOI] [PubMed] [Google Scholar]
- 45.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.