

Abstract
Increased autophagy/mitophagy is thought to contribute to cerebellar dysfunction in Purkinje cell degeneration mice. Intriguingly, cerebellar Purkinje cells are highly vulnerable to hypoxia-ischemia (HI), related at least in part to their high metabolic activity. Whether or not excessive or supraphysiologic autophagy plays a role in Purkinje cell susceptibility to HI is unknown. Accordingly, we evaluated the role of autophagy in the cerebellum after global ischemia produced by asphyxial cardiac arrest in postnatal day (PND) 16–18 rats, using siRNA-targeted inhibition of Atg7, necessary for microtubule-associated protein light chain 3-II (LC3-II) and Atg12-Atg5 complex formation. Two days before a 9 min asphyxial cardiac arrest or sham surgery, Atg7 or control siRNA was injected intracisternally to target the cerebellum. Treatment with Atg7 siRNA: 1) reduced Atg7 protein expression in the cerebellum by 56%; 2) prevented the typical ischemia-induced formation of LC3-II in the cerebellum 24 h after asphyxial cardiac arrest; 3) improved performance on the beam-balance apparatus on days 1–5; and 4) increased calbindin-labeled Purkinje cell survival assessed on day 14. Improved Purkinje cell survival was more consistent in female vs. male rats, and improved beam-balance performance was only seen in female rats. Similar responses to Atg7 siRNA i.e. reduced autophagy and neurodegeneration vs. control siRNA were seen when exposing sex-segregated green fluorescent protein-LC3 tagged mouse primary cortical neurons to oxygen glucose deprivation in vitro. Thus, inhibition of autophagy after global ischemia in PND 16–18 rats leads to increased survival of Purkinje cells and improved motor performance in a sex-dependent manner.
Keywords: Asphyxia, Atg7, cardiac arrest, cerebellum, hypoxia-ischemia, ischemic brain injury, oxygen glucose deprivation, Purkinje neuron, small interfering RNA
Introduction
Accelerated cerebellar Purkinje cell degeneration is seen in mice with mutations in one of now several identified pcd alleles, with the Purkinje cell degeneration mouse the first to be characterized [1]. These pcd mice demonstrate cerebellar ataxia and gait disturbances beginning around postnatal day (PND) 21 and lose >99% of Purkinje cells by PND 42 [1]. This profound Purkinje cell vulnerability has been reported to be at least in part due to excessive autophagy [2]. Autophagy is an intracellular degradation pathway that is involved in the homeostatic turnover of aging proteins and organelles, including mitochondria. Autophagic degradation of mitochondria—termed “mitophagy”, can be triggered by externalization of cardiolipin [3] or severe membrane depolarization [4]. Autophagy proceeds via a complex interplay of autophagy-related genes (Atg), with Atg7 representing a central player in its induction [5], although an Atg5/7-independent pathway has been reported [6]. Atg7 is an ubiquitin E1-like enzyme that controls the critical step of converting Atg8/microtubule-associated protein light chain 3-I (LC3-I) to LC3-II via covalent attachment of phosphatidylethanolamine [7,8] and for the formation of Atg12-Atg5 complexes [9]. Increased autophagy in the injured brain has been reported after multiple insults, including traumatic brain injury and hypoxia-ischemia (HI) [10–15]. However, the role of autophagy after HI has been controversial, as attempts to elucidate its role after HI have been limited by the lack of specific pharmacological inhibitors, the requirement for basal autophagy in normal neurodevelopment complicating studies in transgenic mice, and limited distribution of small interfering RNA (siRNA) in the brain when injected in vivo [16–19].
In addition to being prone to neurodegeneration via dysregulation of autophagy [2], cerebellar Purkinje cells are exquisitely vulnerable to HI [20,21], both perhaps related to the fact that Purkinje cells have one of the highest metabolic rates of any class of neurons. Purkinje cell vulnerability to HI and proclivity toward autophagy-induced neurodegeneration, in combination with the cerebellum's proximity to the intracisternal space, provided us with the opportunity to directly evaluate the role of autophagy after global brain HI in vivo, using a siRNA strategy. In the present study, we were able to effectively reduce both Atg7 protein abundance and HI-induced autophagy in the cerebellum using injection of Atg7 siRNA into the intracisternal space. We then evaluated the role of autophagy after HI by determining the effect of Atg7 knockdown in the cerebellum on Purkinje cell survival and vestibulomotor performance in our model of pediatric asphyxial cardiac arrest [22,23]. Lastly, we verified the capacity for Atg7 knockdown to inhibit ischemia-induced autophagic neurodegeneration using sex-segregated green fluorescent protein (GFP)-LC3 tagged (GFP-LC3+/−) mouse primary cortical neurons exposed to oxygen-glucose deprivation (OGD) in vitro.
1. Materials and methods
1.1. Asphyxial cardiac arrest
Studies were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. A total of 90 rats were used for the in vivo studies reported here, with 72 rats used for siRNA treatment studies.
An established model of pediatric asphyxial cardiac arrest was utilized for this study using PND 16–18 rat pups [22,23]. This developmental age was chosen because in the rat it is similar in terms of cerebral metabolism and blood flow, dendritic pruning, and synaptogenesis, to children 1–4 years of age [24]. PND 16–18 Sprague-Dawley rats (35–40g) were anesthetized with 3% isoflurane/50% N2O/balance oxygen in a Plexiglas chamber until unconscious. Rats were intubated with an 18-gauge angiocatheter and mechanically ventilated with 1% isoflurane/50% N2O/balance oxygen for surgery. Tidal volumes and ventilatory rates were adjusted to maintain PaCO2 35 to 45 mmHg. Femoral arterial and venous catheters (PE10) were inserted via inguinal cut down. Mean arterial blood pressure, heart rate, and temperature were monitored continuously. Vecuronium (1 mg/kg, intravenously) was administered 10 min before asphyxia. Isofluorane/N2O was discontinued 2 min before asphyxia to allow washout of anesthetic. 1 min before asphyxia, the FiO2 was reduced to 0.21 to avoid hyperoxygenation. The endotracheal tube was disconnected from the ventilator for 9 min to induce asphyxial cardiac arrest. Resuscitation consisted of reconnecting the tracheal tube to the ventilator at a FiO2 of 1.0, intravenous administration of epinephrine 0.005 mg/kg and sodium bicarbonate 1 mEq/kg, and rapid manual chest compression until return of spontaneous circulation. Vascular catheters were removed, rats were extubated, observed for 1 h, then returned to their mothers. Shams underwent all procedures except asphyxia and resuscitation.
To characterize neurodegeneration in the cerebellum in this model a separate group of male rats were sacrificed at 24 or 72 h after asphyxial cardiac arrest with naïve rats serving as control, and processed for histological assessment of neurodegeneration (n = 3/group) or electron microscopy (n = 3/group). Hemotoxylin and eosin staining and Fluorojade labeling of degenerating neurons were performed in paraffin sections, and ultrastructural analysis was performed in 65 nm ultrathin sections of cerebellum by transmission electron microscopy as previously described [22,25].
1.2. Intracisternal administration of siRNA
Intracisternal (i.c.) injection was performed using a modification of the technique described by Consiglio et al. [26] for application in rat pups. Naïve postnatal day (PND) 14–16 Sprague-Dawley rats were anesthetized with 3% isoflurane/50% N2O/balance oxygen in a Plexiglas chamber until unconscious. The head was flexed and using sterile technique a 27-gauge butterfly needle was inserted vertically and centrally approximately 2-mm posterior to the intra-aural line. The i.c. space was identified by the appearance of CSF in the tubing, and verified by aspiration of ~25 μl of CSF (to accommodate injection). A total of 800 pmol of Atg7 siRNA (5'-GCAUCAUCUUUGAAGUGAA-3'; Sigma, PDSIRNA) or non-targeting control siRNA (Fischer, D-001206-13-20) were combined with 25 μl of jetSI (Polyplus Transfection, 55–126), a commercial cationic polymer transfection reagent, formulated with dioleoylphosphatidyl ethanolamine (DOPE) (Sigma, P1223) for i.c. injection. Commercially obtained control siRNA consisted of missense sequences which have not been shown to have off-target effects. A syringe containing 25 μl of siRNA containing solution was injected slowly over 1 min to prevent leakage from the puncture site, then the needle was withdrawn. Inhaled anesthetics were discontinued and animals were allowed to recover with their mothers prior to randomization to asphyxial cardiac arrest or sham surgery.
The dose and timing of i.c. siRNA injection was based on pilot experiments in separate naïve rats. A volume of 25 μl of solution—artificial CSF containing Evans blue dye, was the maximum tolerated by rats at this age. Evans blue dye was distributed prominently within and around the cerebellum 24 h after injection. In our hands, the maximum amount of siRNA that can be added to 25 μl is 800 pmol. Subsequent experiments in naïve rats (n = 3/treatment group) using doses of 250–800 pmol of Atg7 siRNA injected 24 – 48 h prior to sacrifice, demonstrated that Atg7 knockdown was optimized with 800 pmol siRNA 48 h after i.c. injection (56%). After i.c. injection Atg7 knockdown was seen in the cerebellum, but not the cortex or hippocampus using western blot (data not shown). Accordingly, for the remainder of experiments 800 pmol siRNA was injected i.c. 48 h before asphyxia or sham surgery and we focused on effects in the cerebellum.
1.3. Western blot analysis
For determination of effective siRNA knockdown, western blot analysis was performed as previously described [25,27] on cerebellar samples from male naïve rats 48 h after siRNA injection, and in male rats 24 h after asphyxial cardiac arrest (72 h after Atg7 or control siRNA injection). Briefly, rats were anesthetized and transcardially perfused with ice-cold saline. Brains were removed and the cerebellum was isolated. Samples were homogenized with lysis buffer (20 mM HEPES-KOH, pH 7.4, 10 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, 1 mM DTT, 1 mM PMSF, 2 mg/ml aprotinin). Lysates were serially centrifuged to separate cellular proteins, with the P2 fraction containing autophagosomes, mitochondria, and small organelles. Samples were stored at −80°C in 10% glycerol. Protein concentration was determined using a Bradford-based protein assay. Proteins were loaded into 15% acrylamide gels, separated electrophoretically, then transferred to a polyvinyl difluoride membrane. The membranes were incubated in 1:1000 dilution of monoclonal antibody against Atg7 (Sigma, A2856) or LC3 (MBL International, M115-3) at room temperature for 1 h, washed in phosphate-buffered saline (PBS) containing 0.1% Tween 20, then incubated in the appropriate secondary antibody for 1 h. The membranes were then incubated in chiemiluminescence reagents, and exposed to X-ray film. After imaging membranes were washed and re-incubated using an antibody against cytochrome oxidase IV (COX IV) to serve as a loading control. Relative optical densities (ROD) of Atg7, LC3-II, and COX IV were calculated for each sample.
1.4. Functional outcome assessment
To assess vestibulomotor function, male and female rats were evaluated using the beam balance and inclined plane tests on days 1–5 after asphyxial cardiac arrest or sham injury. For the beam balance test, rats were placed on a suspended narrow wooden beam (1.5 cm wide) and the latency that the rat remained on the beam was recorded for a maximum of 60 sec. The rat was considered to have passed the test once it remained on the beam for 45 sec. This criterion was based on the duration where 90% of uninjured rats at these ages (PND 17-23) successfully pass the test.
The inclined plane test consisted of a flat board at an initial angle of 45°. The angle was increased by 5° increments to a maximum of 80°. The rat was placed on the board, and the steepest angle that it can maintain its position on the board for 10 sec is recorded. The rat was considered to have passed the test once it could maintain its position on the platform at 75°. This criterion was based on the angle where 90% of uninjured rats at these ages (PND 17-23) successfully pass the test.
1.5. Histological assessment
After completion of functional testing the rats were allowed to survive for a total of 14 days after asphyxia or sham surgery before being sacrificed for assessment of Purkinje neuron survival. Purkinje neurons are vulnerable to global ischemia after asphyxial cardiac arrest in both developing [22] and adult rats [21]. Purkinje neurons were distinguished using calbindin immunohistochemistry. The 14 day survival time was chosen to reduce the possibility that siRNA treatment effects after asphyxia were temporary; and was based on recommendations for experimental models of cerebral ischemia [28]. Briefly, rats were anesthetized with 3% isofluorane and transcardially perfused with heparinized ice-cold saline, followed by 2% paraformaldehyde. Brains were dissected and further post-fixed overnight, then transferred to a 30% sucrose solution until equilibration. Brains were frozen in liquid nitrogen, and mounted on a freezing stage of a sliding microtome. Sagittal sections of the cerebellum at a thickness of 10 μm were obtained at 0, 0.5, and 1 mm from midline. For immunohistochemistry, PBS was used for all rinses. Sections were treated with 3% hydrogen peroxide, washed, then blocked with 3% goat serum. Sections were then incubated in monoclonal anti-Calbindin-D-28K (Sigma-Aldrich, C9848) diluted in 1.5% serum at 1:10,000 overnight at 4°C. Following washes in PBS, the secondary biotinylated mouse anti-IgG (Vector, PK-4002) was applied for 1 h at room temperature, then visualized using an avidin-biotin-HRP complex (ABC, Vector, PK-4002) and diaminobenzidine tetrahydrochloride (DAB, Vector, SK-4100). Slides were rinsed, dehydrated in alcohols, cleared in xylenes, and coverslips were applied using Permount (Fisher).
Calbindin-positive Purkinje neurons were visualized using a Nikon eclipse 90i microscope with the Nikon DS-Ri1 camera. Photos were captured with NIS-Elements AR 3.10 imaging software at a magnification of 10×. Calbindin-positive Purkinje neurons were quantified in each sagittal section using Image J and the ITCN (Image-based Tool for Counting Nuclei) plugin for Image J developed by The Center for Bio-Image Informatics at The University of California, Santa Barbara.
1.6. In vitro oxygen glucose deprivation in primary cortical neurons
Sex-segregated primary cortical neurons were obtained from ED 16#–17 GFP-LC3+/− mice as described [29]. Atg7 siRNA knockdown was achieved using mouse Atg7 (5′-GCAUCAUCUUUGAAGUGAA-3′; Sigma) with pooled non-targeting control siRNA (5′-AUGAACGUGAAUUGCUCAA-3′, 5′UAA GGCUAUGAAGAGUAC-3′, 5′-AUGUAUUGGCCUGUAUUAG-3′, 5′-UAGCGACUAAACACAUCAA-3′; Dharmacon) used as control as described [29]. Primary neurons were transfected on DIV 4 with 20 nmol Atg7 or control siRNA using Lipofectamine 2000 (Invitrogen). Experiments were performed 72 h after transfection.
For OGD, Neurobasal medium and B27 supplements (Gibco) were removed from neurons on day in vitro (DIV) 10 and replaced with custom-made medium lacking sodium pyruvate and L-aspartate, and with a final concentration of 0.5 mM D-glucose and 2 mM L-glutamine. The glucose deprived cultures were placed into a pre-warmed Billups-Rothenberg modular incubator chamber containing 50 ml sterile distilled-deionized water at 37°C. The chamber was flushed with 95% argon and 5% CO2 for 15 min and then sealed. The chamber was then placed in an incubator at 37°C for 2 h. Afterward cultures were returned to the incubator containing 95% air and 5% CO2.
Autophagy and neurodegeneration were assessed as previously described [29]. In brief, autophagy was quantified by measuring formation of GFP-LC3 enriched vesicles in neurons grown on glass-bottomed 25 mm Petri dishes (MatTek Corp.). GFP-LC3 fluorescence was enhanced using an anti-GFP conjugated antibody (Invitrogen) and visualized using an inverted Olympus Fluoview 1000 microscope. Fluorescent intensity above threshold values was quantified in 8 areas (3000 × 3000 pixels) per dish using 6 dishes per group, with threshold values defined as the mean fluorescent intensity of control GFP-LC3 neurons. Cell death was determined by measuring lactate dehydrogenase (LDH) release and propidium iodide (PI) uptake.
1.7. Statistical analysis
Data are expressed as mean ± standard deviation (SD). Physiological variables were compared using repeated measures analysis of variance (ANOVA). Between group comparisons of western blot data were made using analysis of variance (ANOVA) with Tukey's post hoc test. The effect of treatment on calbindin-labeled Purkinje cell survival at multiple levels through the cerebellum was analyzed using two-factor ANOVA. Functional outcome (beam balance and inclined plane testing) data are expressed as time to passing the test with between group comparisons made using the Log rank test. Since there were no within-sex differences in sham-treated Atg7 siRNA and control siRNA groups, motor performance data were combined into one female sham group, and one male sham group, for analysis. Differences between groups in the in vitro experiments were determined by ANOVA with Tukey's post hoc test.
2. Results
2.1. Purkinje cell degeneration after asphyxial cardiac arrest in PND 17 Rats
We first sought to delineate the extent of Purkinje cell degeneration in this model of pediatric cardiac arrest. Fig. 1 shows Purkinje cells that are hyper-eosinophilic, Fluorojade C stained, and electron dense consistent with neurodegeneration. Some cerebellar granule cells were also hyper-eosinophilic and electron dense, exhibiting chromatin clumping. Calbindin immunostaining distinguished Purkinje cells from other cell types, and was therefore later used for quantification. A robust reduction in calbindin immunoreactivity was observed after asphyxial cardiac arrest compared with sham injury.
Figure 1. Purkinje neurodegeneration after asphyxial cardiac arrest in male PND 17 rats.

A. Hematoxylin and eosin (H&E) staining in coronal sections from a naïve rat showing normal cerebellar morphology of the Purkinje and granule cell layers. B. H&E staining in a rat 72 h after asphyxial cardiac arrest showing classic hyper-eosinophilic Purkinje cells indicative of neurodegeneration. C. Fluorojade C staining in coronal sections from a naïve rat without evidence of Purkinje or granule cell degeneration. D. Fluorojade C staining in a rat 72 h after asphyxial cardiac arrest showing Fluorojade-positive neurons indicative of neuronal degeneration in some Purkinje and granule cells. E–I. Ultrastructural evaluation of cerebellum in a rat 72 h after asphyxial cardiac arrest showing hyperchromatic and shrunken Purkinje cells with microvacuoles representing end stage neuronal degeneration (E and F; bar = 2 μm) corresponding to hyper-eosinophilic and Fluorojade-positive neurons at the light microscopic level. In addition, dark and shrunken granule neurons were also occasionally observed (G; bar = 2 μm). Higher magnification also revealed microvacuoles with various degenerating organelles in Purkinje cells (H and I; bar = 500 nm). J and K. Mid-sagittal sections of calbindin-immunolabeled cerebellum. Loss of calbindin-positive Purkinje cells is seen in a rat 14 d after asphyxial cardiac arrest (K) vs. a rat 14 d after sham surgery (J). PrF = Primary fissure.
2.2. Intracisternal injection of Atg7 siRNA inhibited autophagy in the cerebellum
The effect of Atg7 knockdown on autophagy in the cerebellum after asphyxial cardiac arrest was assessed by measuring the relative abundance of LC3-II (Fig. 2). At 24 h after 9 min asphyxial cardiac arrest, increased LC3-II abundance was seen in the cerebellum from male rats treated with control siRNA vs. naïve rats (P < 0.05; n = 3/group). Treatment with Atg7 siRNA prevented formation of LC3-II in cerebellum compared with control siRNA (P < 0.05; n = 3/group). Treatment with Atg7 siRNA also reduced Atg7 protein abundance vs. control siRNA after asphyxial cardiac arrest (P < 0.05). Of note, Atg7 was increased in cerebellum from rats treated with control siRNA after asphyxial cardiac arrest vs. naïve rats (P < 0.05). Accordingly, the effect of HI alone on Atg7 abundance in brain independent of siRNA treatment may warrant further study.
Figure 2. Prevention of ischemia-induced autophagy in cerebellum using Atg7 siRNA.
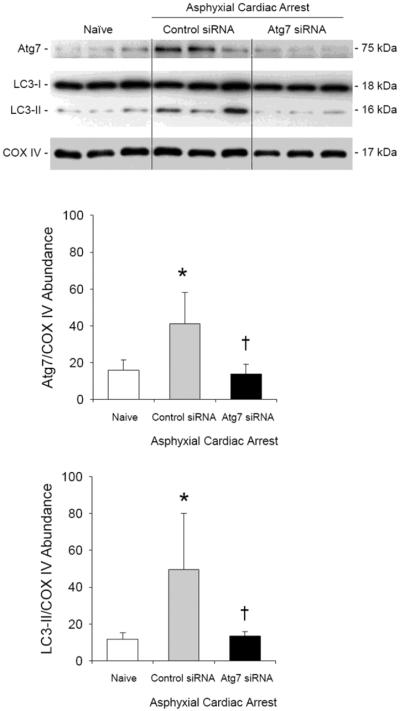
Western blot analysis of LC3-I and LC3-II in cerebellum of naïve male rats and male rats 24 h after asphyxial cardiac arrest. At 48 h before asphyxial arrest, rats were randomized to receive control or Atg7 siRNA via intracisternal injection. In rats treated with control siRNA, relative LC3-II protein abundance adjusted for cytochrome oxidase IV (COX IV) as a loading control increased after asphyxial cardiac arrest vs. naïve rats (*P < 0.05). Treatment with Atg7 siRNA prevented an ischemia-induced increase in LC3-II:COX IV vs. control siRNA. Consistent with Fig. 2, Atg7:COX IV ratios were also reduced in Atg7 vs. control siRNA treated rats after asphyxia. Mean ± SD, n = 3/group, *P < 0.05 vs. naïve, †P < 0.05 vs. control siRNA, ANOVA with Tukey's post hoc test.
2.3. Intracisternal injection of Atg7 siRNA improved beam balance performance after asphyxial cardiac arrest
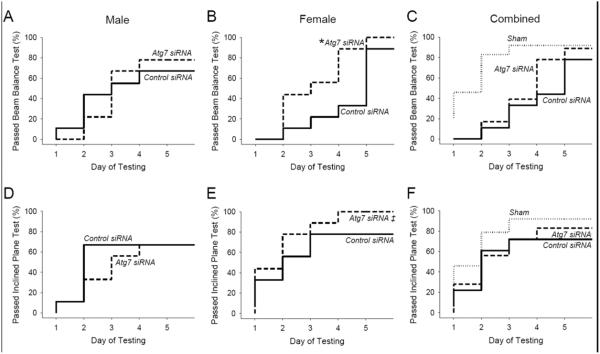
For functional outcome studies a total of 63 rats were randomized to receive i.c. injection of 800 pmol (25 μl) of Atg7 or control siRNA 48 h before asphyxial cardiac arrest or sham surgery. Of these, 3 rats died before completion of functional outcome testing (1 male after control siRNA injection and asphyxia and 2 males after control siRNA injection and sham surgery). These rats were replaced to balance the groups. Thus, vestibulomotor function was assessed in 9 male and 9 female rats per treatment group after asphyxial cardiac arrest and 6 male and 6 female rats per treatment group after sham surgery using the beam balance and incline plane apparatuses on days 1–5. After asphyxial arrest, female rats treated with Atg7 siRNA were able to satisfactorily pass the beam balance test (Fig. 3) more quickly than female rats treated with control siRNA (P = 0.02). However, this treatment effect was not seen in male rats (P = 0.96). There was no significant difference in performance on the inclined plane apparatus in rats treated with Atg7 vs. control siRNA, nor were injury-effects detected. These data suggest that inhibition of autophagy in the cerebellum after asphyxial cardiac arrest improves vestibulomotor function in a sex-dependent manner, favoring female rats.
Figure 3. Reduction in ischemia-induced motor dysfunction using Atg7 siRNA.

A–C. Beam balance performance on days 1–5 after asphyxial cardiac arrest or sham surgery. Data are presented as the time required to successfully pass the test (defined as the duration where 90% of age-matched sham rats pass) in male rats (A), female rats (B), and male and female rats combined (C). D–F. Inclined plane performance on days 1–5 after asphyxial cardiac arrest or sham surgery. Data are presented as the time required to successfully pass the test (defined as the duration where 90% of age-matched sham rats pass) in male (D), female rats (E), and male and female rats combined rats (F). For sham n = 12/group, for asphyxial cardiac arrest n = 9/sex/group, female and male rats combined for panels C and F, *P = 0.02 vs. control siRNA, Log rank test.
2.4. Intracisternal injection of Atg7 siRNA improved Purkinje cell survival after asphyxial cardiac arrest
Fourteen days after asphyxial cardiac arrest Purkinje cell survival was assessed using calbindin staining (1 male rat in each of the treatment groups died between functional outcome testing and 14 d sacrifice). Calbindin is a calcium binding protein with a strong affinity for Purkinje cells [30,31]. Mid-sagittal sections of calbindin immunolabeled cerebellum in rats after asphyxial cardiac arrest or sham surgery are shown in Fig. 1J and K. Total calbindin-positive Purkinje cells were counted in sagittal sections obtained at 0.0, 0.5, and 1.0-mm from midline. Atg7 siRNA treatment significantly improved Purkinje cell survival after asphyxial cardiac arrest compared with control siRNA treatment in female but not male rats (P = 0.002 vs. 0.125, respectively; n = 9 females and 8 males/group; Fig. 4). Interestingly, greater numbers of Purkinje cells were seen in sham male vs. female rats (P < 0.05; n = 6/sex), suggesting a sex-difference in cerebellar development and/or ultimate Purkinje cell number at this age. A highly significant beneficial effect of Atg7 siRNA treatment after asphyxial cardiac arrest was also demonstrated when Purkinje cell survival was expressed as percent Purkinje cells per sex-matched sham (Fig. 4C; P = 0.003; n = 17/group). Overall Purkinje cell survival adjusted for baseline differences between female and male rats was 91.6 ± 15.5% in the Atg7 siRNA group vs. 78.6 ± 21.8% in the control siRNA group (P = 0.05). For consistency of presentation, higher magnification images from calbindin immunolabeled sections encompassing the terminus of the cerebellar primary fissure from representative rats from each group are shown in Fig. 4D. Of note, relative to other regions of the cerebellum, more Purkinje cell loss was observed in the primary fissure (Fig. 1J, K).
Figure 4. Prevention of ischemia-induced Purkinje neurodegeneration using Atg7 siRNA.
Calbindin-immunolabeled Purkinje cell counts per sagittal section measured at 0.0, 0.5 and 1.0 mm from midline in male (A) and female (B) rats 14 d after asphyxial cardiac arrest or sham surgery. Increased Purkinje cell survival after asphyxial cardiac arrest was seen in rats treated with Atg7 siRNA vs. control siRNA, with the treatment effect appearing more robust in female rats vs. their male counterparts. To account for sex-differences in baseline (sham) Purkinje cell numbers, data are also expressed as the percentage of Purkinje cells per sex-matched sham (C). D. Representative calbindin-immunolabeled sections showing Purkinje cell survival in male and female rats treated with control or Atg7 siRNA using the cerebellar primary fissure as a point of reference are shown. Mean ± SD, for sham n = 6/sex/group, for asphyxial cardiac arrest n = 9 females and 8 males/group, female and male rats combined for panel C (n = 17/group), *P < 0.005 vs. control siRNA, two-factor ANOVA.
The differences in Purkinje neuron survival could not be explained by group differences in physiological derangements early after asphyxial cardiac arrest. Physiological variables for each group used to assess outcome provided in Tables 1 and 2. Typical physiological changes, particularly a reduction in blood pressure and transient acidosis, were observed early after asphyxial cardiac arrest vs. baseline values and these changes were similar among all female or male sex and treatment groups.
Table 1.
Physiological Variables: Female Rats.
| Group | Weight (gm) | T (°C) | HR (bpm) | MAP (mmHg) | pH | PaCO2 (mmHg) | PaO2 (mmHg) |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Sham control siRNA | 35.2±1.2 | 37.3±0.1 | 398±10 | 62.5±3.4 | 7.36±0.01 | 36.3±0.5 | 198.4±14.8 |
|
| |||||||
| Sham Atg7 siRNA | 36.0±1.3 | 37.4±0.1 | 420±9 | 60.8±0.8 | 7.36±0.01 | 36.0±0.4 | 232.8±10.0 |
|
| |||||||
| Asphyxia control siRNA | |||||||
| Baseline | 37.3±0.1 | 404±4 | 60.0±2.0 | 7.37±0.01 | 36.0±0.4 | 220.8±4.3 | |
| 10 min | 37.1±0.1 | 373±4a | 62.2±1.9 | 7.30±0.02a | 38.9±2.7 | 326.8±18.1a | |
| 30 min | 37.3±0.1 | 403±12 | 45.0±1.7a | 7.38±0.02 | 36.0±1.5 | 343.2±19.1a | |
| 60 min | 35.6±1.3 | 37.3±0.1 | 404±9 | 46.7±1.2a | 7.39±0.01 | 37.5±1.1 | 371.9±12.0a |
|
| |||||||
| Asphyxia Atg7 siRNA | |||||||
| Baseline | 37.2±0.1 | 418±9 | 65.6±1.9 | 7.36±0.01 | 35.8±0.9 | 216.9±10.8 | |
| 10 min | 37.1±0.0 | 378±9a | 66.1±1.8 | 7.29±0.01a | 39.3±1.4a | 323.4±17.2 | |
| 30 min | 37.3±0.1 | 426±10 | 49.4±2.1a | 7.38±0.02 | 36.4±1.3 | 345.1±15.1a | |
| 60 min | 35.9±1.0 | 37.2±0.1 | 424±9 | 48.9±1.4a | 7.39±0.01 | 35.6±0.9 | 371.4±11.7a |
Abbreviations: T, temperature; HR, heart rate; bpm, beats per minute; MAP, mean arterial pressure.
P < 0.05 vs. baseline.
Table 2.
Physiological Variables: Male Rats.
| Group | Weight (g) | T (°C) | HR (bpm) | MAP (mmHg) | pH | PaCO2 (mmHg) | PaO2 (mmHg) |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Sham control siRNA | 32.2±1.2 | 37.3±0.1 | 417±12 | 60.8±2.0 | 7.35±0.02 | 38.1±1.4 | 217.6±5.7 |
|
| |||||||
| Sham Atg7 siRNA | 32.3±1.2 | 37.2±0.1 | 408±12 | 57.5±1.1 | 7.34±0.01 | 38.4±1.0 | 224.7±6.8 |
|
| |||||||
| Asphyxia control siRNA | |||||||
| Baseline | 37.2±0.1 | 408±9 | 61.7±1.7 | 7.34±0.01 | 37.8±0.7 | 222.5±7.3 | |
| 10 min | 37.1±0.1 | 389±7 | 64.4±2.3 | 7.28±0.01a | 38.3±2.1 | 320.9±22.4a | |
| 30 min | 37.3±1.0 | 37.3±0.0 | 410±9 | 46.1±3.0a | 7.36±0.01 | 36.5±1.4 | 339.1±15.0a |
| 60 min | 37.2±0.1 | 423±12 | 48.9±1.1a | 7.37±0.01a | 38.6±1.3 | 357.0±15.6a | |
|
| |||||||
| Asphyxia Atg7 siRNA | |||||||
| Baseline | 37.3±0.0 | 398±10 | 57.8±1.2 | 7.34±0.01 | 36.1±0.5 | 221.8±5.4 | |
| 10 min | 37.1±0.0 | 389±7 | 62.2±3.2 | 7.30±0.02 | 35.8±0.9 | 357.6±10.0a | |
| 30 min | 37.2±0.1 | 411±7 | 48.3±0.8a | 7.40±0.01a | 34.3±0.9 | 361.6±8.2a | |
| 60 min | 34.2±1.4 | 37.2±0.0 | 396±10 | 48.9±1.6 | 7.41±0.01a | 36.0±0.3 | 385.2±7.5a |
Abbreviations: T, temperature; HR, heart rate; bpm, beats per minute; MAP, mean arterial pressure.
P < 0.05 vs. baseline.
2.5. Atg7 siRNA inhibits autophagy and reduces neuronal death after OGD in vitro
An increase in GFP-LC3 enriched vesicles was observed in neurons from both male (XY) and female (XX) mice at 24 h after OGD, with earlier (2 h) and more prominent increases seen in XY vs. XX neurons (Fig. 5; P < 0.05 vs. control). This increase in GFP-LC3 enriched vesicle formation was inhibited by Atg7 siRNA (Fig. 5A and B; P < 0.05 vs. control siRNA), consistent with GFP values above threshold representing autophagosomes after OGD. Consistent with autophagy-induced neurodegeneration, Atg7 siRNA treatment reduced cell death at 2 and 24 h after OGD vs. control siRNA in both XY and XX neurons as evidenced by LDH release (Fig. 5D; P < 0.05) and PI uptake (Fig. 5E; P < 0.05). Cell death after OGD was more prominent in XY vs. neurons identified using both LDH release and PI uptake.
Figure 5. Oxygen-glucose deprivation (OGD) produces sex-dependent autophagy in primary cortical neurons from GFP-LC3+/− mice.

The GFP signal was enhanced immunohistochemically. A. Identification of GFP-LC3 enriched vesicles consistent with autophagosome formation at 2 and 24 h after OGD. B. Quantification of GFP-LC3 fluorescent intensity peaks over threshold (fluorescence in control cells) showing that Atg7 siRNA reduces OGD-induced autophagosome formation in XY- and XX-neurons (n = 6/group; *P < 0.05 vs. control siRNA, †P < 0.05 vs. control; ANOVA with Tukey's test). C. GFP immunohistochemistry showing neuronal loss and formation of GFP-enriched vesicles after OGD. D and E. Treatment with Atg7 siRNA reduced LDH release and increased neuron survival detected by PI labeling at 2 and 24 h after OGD in both XY- and XX-neurons vs. control siRNA (LDH, n = 6/group; PI, n = 4/group; *P < 0.05 vs. control siRNA; ANOVA with Tukey's test).
3. Discussion
We found that administration of Atg7 siRNA via intracisternal injection prevented the increase in LC3-II formation in cerebellum typically observed after asphyxial cardiac arrest, consistent with inhibition of injury-induced autophagy. This targeted inhibition resulted in improved Purkinje cell survival and vestibulomotor function in a sex-dependent fashion, with effects observed predominantly—or with less variability—in female rats. These data suggest that inhibition of autophagy is beneficial in terms of Purkinje cell survival after HI as a consequence of asphyxial cardiac arrest in the developing brain; and are consistent with a heightened vulnerability of Purkinje cells to increased and/or dysregulation of autophagy [2].
We were able to inhibit Atg7 in the cerebellum by delivering Atg7 siRNA using a cationic polymer transfecting reagent formulated with dioleoylphosphatidylethanolamine, and administering the siRNA via local delivery into the intracisternal space. This siRNA delivery system has been shown by Hassani et al. to effectively down-regulate gene expression in neurons after intra-cerebroventricular injection into mice pups [32]. In our study, a reduction in Atg7 relative protein abundance by 56% (Fig. 2) effectively prevented induction of autophagy typically produced in this model of cerebral HI (Fig. 3). This incomplete reduction in Atg7 protein in tandem with complete prevention of ischemia-induced autophagy may be therapeutically desirable, given the importance of basal Atg7 for neuronal health and homeostasis, including in Purkinje cells [33,34]. Mice that lack Atg7 in the CNS develop growth retardation, motor and behavioral deficits, and die within 28 weeks of birth [33]. Koike et al. subjected these mice to HI injury via left carotid artery occlusion followed by exposure to low oxygen, and found that neonatal mice deficient in CNS Atg7 showed nearly complete protection as assessed by caspase-3 activation and pyramidal neuron death in the hippocampus 7 days after injury compared with wild-type mice [18]. Due to the basal effects of complete Atg7 deficiency, they were unable to address the potential role of ischemia-induced autophagy in older mice. Taken together, the work by Koike at al. and the present study in older postnatal rats suggest that while long-term disruption of autophagy is detrimental in the brain, short-term blunting of the ischemia-induced autophagy response may prove beneficial in the developing brain.
The use of siRNA to inhibit autophagy in the brain in vivo has been reported in another model of cerebral ischemia in adult rats. Zheng et al. [35] administered lentiviral vectors encoding Beclin 1 short hairpin RNA (LV-shBeclin1) into the ipsilateral ventricle 7 days prior to transient middle cerebral artery occlusion. Administration of LV-shBeclin1 resulted in down-regulation of Beclin 1 after cerebral ischemia with decreased infarct volume, reduced histological injury in the ipsilateral cortex and hippocampus, and lower neurological deficit scores. They also showed a reduction in apoptosis in immature neurons, with increased neurogenesis. These data imply that inhibition of autophagy by RNA interference of Beclin 1 improves histological and neurological outcome after cerebral ischemia. Cross talk between autophagy and apoptosis is present due to the interactions of Beclin 1 with Bcl-2 and Bcl-xL, and furthermore caspase mediated cleavage of Beclin 1 forms fragments that inhibit autophagy and activate apoptosis [36–38]; thus complicating analysis of the impact of Beclin 1 as a direct effect on apoptosis cannot excluded. Nonetheless, these data are consistent with the present study and the concept that preventing ischemia-induced increases in autophagy is beneficial following brain injury. That said and given the challenges in using siRNA in brain in vivo, we performed parallel knockdown experiments in primary neurons after OGD in vitro. The findings that Atg7 siRNA reduced autophagosome formation and neurodegeneration (Figure 5) were consistent with the in vivo experiments.
Consistent with a previous study from our laboratory showing sex-differences in autophagy under conditions of starvation in vitro [29], sex-differences in response to Atg7 knockdown were seen in the current study; although in apparent contrast. Female rats had a tighter response to Atg7 siRNA treatment in terms of Purkinje cell survival (91.0 vs. 76.2% in Atg7 vs. control siRNA groups, respectively) compared with male rats (92.3 vs. 81.8% in Atg7 vs. control siRNA groups, respectively) after asphyxial cardiac arrest, although the statistical power for the male groups was 0.2, indicating that with a larger sample size significant differences may have been detected in male rats as well. Female rats subjected to asphyxial cardiac arrest also had improved beam balance performance after treatment with Atg7 vs. control siRNA. A treatment effect on motor testing was not seen in male rats. The observed benefit of Atg7 knockdown primarily in female rats contradicted our previous in vitro study [29], and it was initially anticipated that the effects of Atg7 knockdown would likewise be more robust in male vs. female rats after cerebral ischemia. Interestingly, after OGD Atg7 knockdown reduced autophagic neurodegeneration in neurons from both male and female mice (Figure 5), and like starvation, showed more prominent autophagy in neurons from males vs. females. It is possible that partial Atg7 knockdown is simply less effective in vivo than in vitro, and thus we were only able to detect a treatment effect in female rats after asphyxial arrest given our sample sizes. Alternatively, since mortality in rats receiving siRNA was only seen in males (4 receiving non-targeting siRNA and 1 receiving Atg7 siRNA), death in the more severely impacted rats may have biased our results toward a lack of treatment effects in males.
We found that baseline (sham) numbers of Purkinje neurons were greater in male compared with female rats on PND 30–32. Sex differences in the rat brain have been described by Yanai [39], who reported that male Wistar and Long-Evans rats had larger brains than females at all ages (1 to 60 days). Additionally, males had 7.2% more Purkinje cells than females, with Long-Evans rats having 9.6% more Purkinje cells than Wistar rats (P<0.01) [39]. No study to our knowledge has compared Purkinje cell counts in male and female Sprague-Dawley rats (which were utilized in the present study) at this developmental stage. In the present study, the sex-difference in Purkinje cell number may represent differences in cerebellar development and/or ultimate Purkinje cell number. This absolute difference in Purkinje cell number may affect how male and female rats deal with Purkinje cell death after asphyxial cardiac arrest, consequently impacting their vestibulomotor function.
There are limitations in the present study. First, while inhibition of Atg7 using siRNA allows for more specific inhibition of the autophagic pathway than existing pharmacological agents, Atg7 has also been implicated in roles outside of autophagy. A deficiency of Atg7 during metabolic stress has been shown to increase p53-dependent apoptosis [40], although this mechanism certainly cannot account for the neuroprotection seen in the current context we did not directly examine for apoptosis in this study. Second, the effectiveness of Atg7 siRNA injected into the intracisternal space in our hands was limited to protein knockdown in the cerebellum, thus, the role of autophagy after HI in other brain regions such as the hippocampus could not be determined. Nevertheless, this approach allowed us to specifically target the cerebellum. Third, complete genetic disruption of Atg7 is known to result in profound neurodegeneration, including in Purkinje cells [33,34]. Despite this fact, partial reductions in Atg7 protein appear to be neuroprotective after asphyxial cardiac arrest. The mechanisms underlying this difference may relate to the completeness and/or chronicity of the Atg7 deficiency. Differences in the molecular regulation of basal versus injury-induced autophagy have been reported in cultured neurons [41,42], and neuroprotection is generally associated with inhibition of injury-induced increases, rather than basal levels, of autophagy markers.
4. Conclusions
In conclusion, inhibition of autophagy induced by global ischemia leads to improved beam balance performance and increased survival of cerebellar Purkinje cells in a sex-sensitive manner. These data are consistent with a detrimental role for increased autophagy in brain after asphyxial cardiac arrest in the developing brain.
Highlights.
Asphyxial cardiac arrest in juvenile rats leads to Purkinje cell neurodegeneration
Atg7 knockdown inhibits ischemia-induced autophagy in the cerebellum
Atg7 knockdown improves beam-balance performance post-ischemia in female rats
Atg7 knockdown improves Purkinje cell survival after asphyxial cardiac arrest
Atg7 knockdown reduces hypoxia-induced autophagy and neurodegeneration in vitro
Acknowledgements
We would like to thank Henry Alexander and Christina Hosler for expert technical assistance. Supported by NIH grants R01 NS084604, R01 HD045968, R01 AG026389 and 5T32 HD040686, and the Children's Hospital of Pittsburgh of UPMC Scientific Program. C.T.C. was a Julie Martin Mid-Career Awardee in Aging Research supported by The Ellison Medical Foundation and AFAR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- 1.Mullen RJ, Eicher EM, Sidman RL. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci U S A. 1976;73:208–212. doi: 10.1073/pnas.73.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakrabarti L, Eng J, Ivanov N, Garden GA, La Spada AR. Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain. 2009;2:24. doi: 10.1186/1756-6606-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 6.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 7.Kouno T. Solution Structure of Microtubule-associated Protein Light Chain 3 and Identification of Its Functional Subdomains. Journal of Biological Chemistry. 2005;280:24610–24617. doi: 10.1074/jbc.M413565200. [DOI] [PubMed] [Google Scholar]
- 8.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 9.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang YB, Li SX, Chen XP, Yang L, Zhang YG, et al. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci Bull. 2008;24:143–149. doi: 10.1007/s12264-008-1108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu CL, Chen S, Dietrich D, Hu BR. Changes in autophagy after traumatic brain injury. Journal of Cerebral Blood Flow & Metabolism. 2007;28:674–683. doi: 10.1038/sj.jcbfm.9600587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diskin T, Tal-Or P, Erlich S, Mizrachy L, Alexandrovich A, et al. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J Neurotrauma. 2005;22:750–762. doi: 10.1089/neu.2005.22.750. [DOI] [PubMed] [Google Scholar]
- 13.Smith CM, Chen Y, Sullivan ML, Kochanek PM, Clark RSB. Autophagy in acute brain injury: Feast, famine, or folly? Neurobiology of Disease. 2011;43:52–59. doi: 10.1016/j.nbd.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu C, Wang X, Xu F, Bahr BA, Shibata M, et al. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia–ischemia. Cell Death and Differentiation. 2004;12:162–176. doi: 10.1038/sj.cdd.4401545. [DOI] [PubMed] [Google Scholar]
- 15.Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115:68–78. doi: 10.1111/j.1471-4159.2010.06905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008 doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 17.Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009 doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- 18.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Fougerolles A, Vornlocher HP, Maraganore J, Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov. 2007;6:443–453. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cervos-Navarro J, Diemer NH. Selective vulnerability in brain hypoxia. Crit Rev Neurobiol. 1991;6:149–182. [PubMed] [Google Scholar]
- 21.Paine MG, Che D, Li L, Neumar RW. Cerebellar Purkinje cell neurodegeneration after cardiac arrest: effect of therapeutic hypothermia. Resuscitation. 2012;83:1511–1516. doi: 10.1016/j.resuscitation.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 22.Fink EL, Alexander H, Marco CD, Dixon CE, Kochanek PM, et al. Experimental model of pediatric asphyxial cardiopulmonary arrest in rats. Pediatr Crit Care Med. 2004;5:139–144. doi: 10.1097/01.pcc.0000112376.29903.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manole MD, Foley LM, Hitchens TK, Kochanek PM, Hickey RW, et al. Magnetic resonance imaging assessment of regional cerebral blood flow after asphyxial cardiac arrest in immature rats. J Cereb Blood Flow Metab. 2009;29:197–205. doi: 10.1038/jcbfm.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rice D, Barone S., Jr. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–533. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai Y, Hickey RW, Chen Y, Bayir H, Sullivan ML, et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28:540–550. doi: 10.1038/sj.jcbfm.9600551. [DOI] [PubMed] [Google Scholar]
- 26.Consiglio AR, Lucion AB. Technique for collecting cerebrospinal fluid in the cisterna magna of non-anesthetized rats. Brain Res Brain Res Protoc. 2000;5:109–114. doi: 10.1016/s1385-299x(99)00062-8. [DOI] [PubMed] [Google Scholar]
- 27.Chu CT, Plowey ED, Dagda RK, Hickey RW, Cherra SJ, 3rd, et al. Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models. Methods Enzymol. 2009;453:217–249. doi: 10.1016/S0076-6879(08)04011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gladstone DJ, Black SE, Hakim AM. Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33:2123–2136. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- 29.Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, et al. Starving neurons show sex difference in autophagy. J Biol Chem. 2009;284:2383–2396. doi: 10.1074/jbc.M804396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bitoun E, Finelli MJ, Oliver PL, Lee S, Davies KE. AF4 Is a Critical Regulator of the IGF-1 Signaling Pathway during Purkinje Cell Development. Journal of Neuroscience. 2009;29:15366–15374. doi: 10.1523/JNEUROSCI.5188-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitney ER, Kemper TL, Rosene DL, Bauman ML, Blatt GJ. Calbindin-D28k is a more reliable marker of human Purkinje cells than standard Nissl stains: A stereological experiment. Journal of Neuroscience Methods. 2008;168:42–47. doi: 10.1016/j.jneumeth.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Hassani Z, Lemkine GF, Erbacher P, Palmier K, Alfama G, et al. Lipid-mediated siRNA delivery down-regulates exogenous gene expression in the mouse brain at picomolar levels. The Journal of Gene Medicine. 2005;7:198–207. doi: 10.1002/jgm.659. [DOI] [PubMed] [Google Scholar]
- 33.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 34.Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr., Iwata J, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng YQ, Liu JX, Li XZ, Xu L, Xu YG. RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol Sin. 2009;30:919–927. doi: 10.1038/aps.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death and Differentiation. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS Journal. 2011;278:403–413. doi: 10.1111/j.1742-4658.2010.07965.x. [DOI] [PubMed] [Google Scholar]
- 38.Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717–1719. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- 39.Yanai J. Strain and sex differences in the rat brain. Acta Anat (Basel) 1979;103:150–158. doi: 10.1159/000145006. [DOI] [PubMed] [Google Scholar]
- 40.Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225–228. doi: 10.1126/science.1218395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, et al. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cherra SJ, 3rd, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, et al. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010;190:533–539. doi: 10.1083/jcb.201002108. [DOI] [PMC free article] [PubMed] [Google Scholar]