

ABSTRACT
RNA interference (RNAi) is considered an ancient antiviral defense in diverse organisms, including insects. Virus infections generate double-strand RNAs (dsRNAs) that trigger the RNAi machinery to process dsRNAs into virus-derived short interfering RNAs (vsiRNAs), which target virus genomes, mRNAs, or replication intermediates. Viruses, in turn, have evolved viral suppressors of RNAi (VSRs) to counter host antiviral RNAi. Following recent discoveries that insects mount an RNAi response against DNA viruses, in this study, we found that Autographa californica multiple nucleopolyhedrovirus (AcMNPV) infection similarly induces an RNAi response in Spodoptera frugiperda cells by generating a large number of vsiRNAs postinfection. Interestingly, we found that AcMNPV expresses a potent VSR to counter RNAi. The viral p35 gene, which is well known as an inhibitor of apoptosis, was found to be responsible for the suppression of RNAi in diverse insect and mammalian cells. The VSR activity of p35 was further confirmed by a p35-null AcMNPV that did not suppress the response. In addition, our results showed that the VSR activity is not due to inhibition of dsRNA cleavage by Dicer-2 but acts downstream in the RNAi pathway. Furthermore, we found that the VSR activity is not linked to the antiapoptotic activity of the protein. Overall, our results provide evidence for the existence of VSR activity in a double-stranded DNA virus and identify the responsible gene, which is involved in the inhibition of RNAi as well as apoptosis.
IMPORTANCE Our findings demonstrate the occurrence of an insect RNAi response against a baculovirus (AcMNPV) that is highly utilized in microbial control, biological and biomedical research, and protein expression. Moreover, our investigations led to the identification of a viral suppressor of RNAi activity and the gene responsible for the activity. Notably, this gene is also a potent inhibitor of apoptosis. The outcomes signify the dual role of a virus-encoded protein in nullifying two key antiviral responses, apoptosis and RNAi.
INTRODUCTION
RNA interference (RNAi) is a highly conserved sequence-specific gene silencing mechanism in diverse eukaryotes, involved in the regulation of some critical processes, including virus-host interactions in insects, plants, fungi, and mammals (1, 2). Viral infections in insects induce an RNAi-based antiviral response through processing of virus-specific double-strand RNAs (dsRNAs) into virus-derived short interfering RNAs (vsiRNAs) by the action of Dicer-2 (DCR-2). vsiRNAs are then loaded into Argonaute-2 (Ago-2), a part of the protein complex known as the RNA-induced silencing complex (RISC). This complex cleaves target RNAs based on sequence complementarity of the loaded small RNA (3). The RNAi-based antiviral response can be mounted through another type of functional noncoding small RNA, microRNA (miRNA). After processing by DCR-1, mature miRNAs are loaded into Ago proteins and guide the miRNA-RISC to mRNA target sequences (4). Unlike siRNA, in animals, miRNA sequences are not completely complementary to the target sequences.
Antiviral RNAi has been noted as an immune response against virus infections in several insects. In Aedes and Culex vector mosquitoes, for example, RNAi plays a critical role in interactions with different viruses such as Sindbis virus (SINV), West Nile virus, and dengue virus, among others (5–7). In the model insect Drosophila melanogaster, RNAi was reported as an immune response against acute and latent viral infections, including cricket paralysis virus (CrPV), Flock House virus (FHV), SINV, and Drosophila C virus (DCV) infections (8–10). Considering RNAi as an immune reaction against viral infection, it is not surprising that viruses have also evolved factors to circumvent this host antiviral response. These factors are mostly virus-encoded proteins, designated viral suppressors of RNAi (VSRs) (1). Almost all plant viruses and some insect RNA viruses have been shown to encode VSRs. The B2 protein of FHV and the 1A protein of dicistroviruses (CrPV and DCV) are classic examples of VSRs reported for insect viruses (11, 12). Structurally, VSRs are diverse due to their production by different viruses (13). Likewise, in terms of function, VSRs interfere with host RNAi in different ways, including sequestering dsRNA and siRNA, thereby preventing dsRNA dicing and siRNA assembly into the RISC, respectively; inhibiting components of the host RISC machinery (Ago protein) by direct protein-protein interactions; and interfering with the spread of RNA silencing (reviewed in reference 1). In insects, VSRs have been reported mainly for RNA viruses. However, an instance of VSR activity from a DNA virus was reported for Heliothis virescens ascovirus (HvAV-3e), with its encoded RNase III degrading siRNAs (14). More recently, a potential VSR was identified in invertebrate iridescent virus 6 (IIV6), which has an RNA-binding domain, inhibits Dicer-2-mediated cleavage of dsRNA, and also binds to siRNAs, blocking their loading into the RISC (15). In the shrimp Litopenaeus vannamei, dsRNA-mediated silencing was suppressed by white spot syndrome virus (WSSV) (a dsDNA virus) (16), although the mechanism of suppression or the gene involved is not known.
The family Baculoviridae is a family of large rod-shaped insect viruses with double-stranded DNA circular genomes ranging from 80 to 180 kb, which encode 100 to 200 predicted proteins (17). The most studied baculovirus species is Autographa californica multiple nucleopolyhedrovirus (AcMNPV), the type species of the Alphabaculovirus genus of the Baculoviridae family, with a 134-kbp genome containing 156 open reading frames (ORFs). Baculovirus genomes contain some species-specific genes, or genes specific to certain baculovirus lineages, as well as groups of conserved genes in the family. Moreover, in the course of evolution, baculovirus genomes have been subjected to high levels of gene loss and gene acquisition from their hosts (18). Of these adapted genes, the p35 and iap (inhibitor of apoptosis) genes play important roles in baculovirus-host interactions by inhibition of host cell apoptosis (reviewed in reference 19). Despite host antiviral responses, such as the global shutdown of protein synthesis and apoptosis, expression of baculovirus genes occurs and is even temporally regulated throughout the infection process. Based on the timing of expression, these genes are categorized as immediate early, early, late, and very late genes (17). These findings reveal the ability of baculoviruses to manipulate the antiviral responses of their host.
Our recent study revealed that host RNAi also plays a part in baculovirus-host interactions, where siRNAs are produced from viral transcripts as part of the host antiviral response (20). Silencing of Dicer-2 in host cells led to increased replication of Helicoverpa armigera single nucleopolyhedrovirus (HaSNPV) (20), suggesting that the host RNAi response limits replication of the virus. In addition, a similar RNAi response against another DNA virus (IIV6) was shown in Drosophila (21, 22). AcMNPV infection also affects host RNAi by overall suppression of cellular miRNAs and alteration of the abundance of the majority of Spodoptera frugiperda miRNAs following infection (23). In the present study, we explored the RNAi response of insect cells to AcMNPV and showed that a viral gene, p35, suppresses this response in insects as well as mammalian cells. p35 is well known for its antiapoptotic activity, suggesting a dual role for this protein in two major antiviral responses against the virus.
MATERIALS AND METHODS
Cells and virus.
The Spodoptera frugiperda cell line (Sf9) was maintained in SF900-II serum-free medium (Invitrogen) as a monolayer at 27°C. AcMNPV was amplified in Sf9 cells, and budded viruses that accumulated in the medium were used for inoculations. For AcMNPV infection, 2 × 106 cells were infected at a multiplicity of infection (MOI) of 5, as described previously (24). After 1 h of incubation at 27°C, fresh medium was added to cells, and the cells were incubated further at 27°C. Vero and NIH 3T3 cells were maintained as monolayers at 37°C in RPMI 1640 medium with 5% fetal bovine serum (FBS).
Cloning of p35.
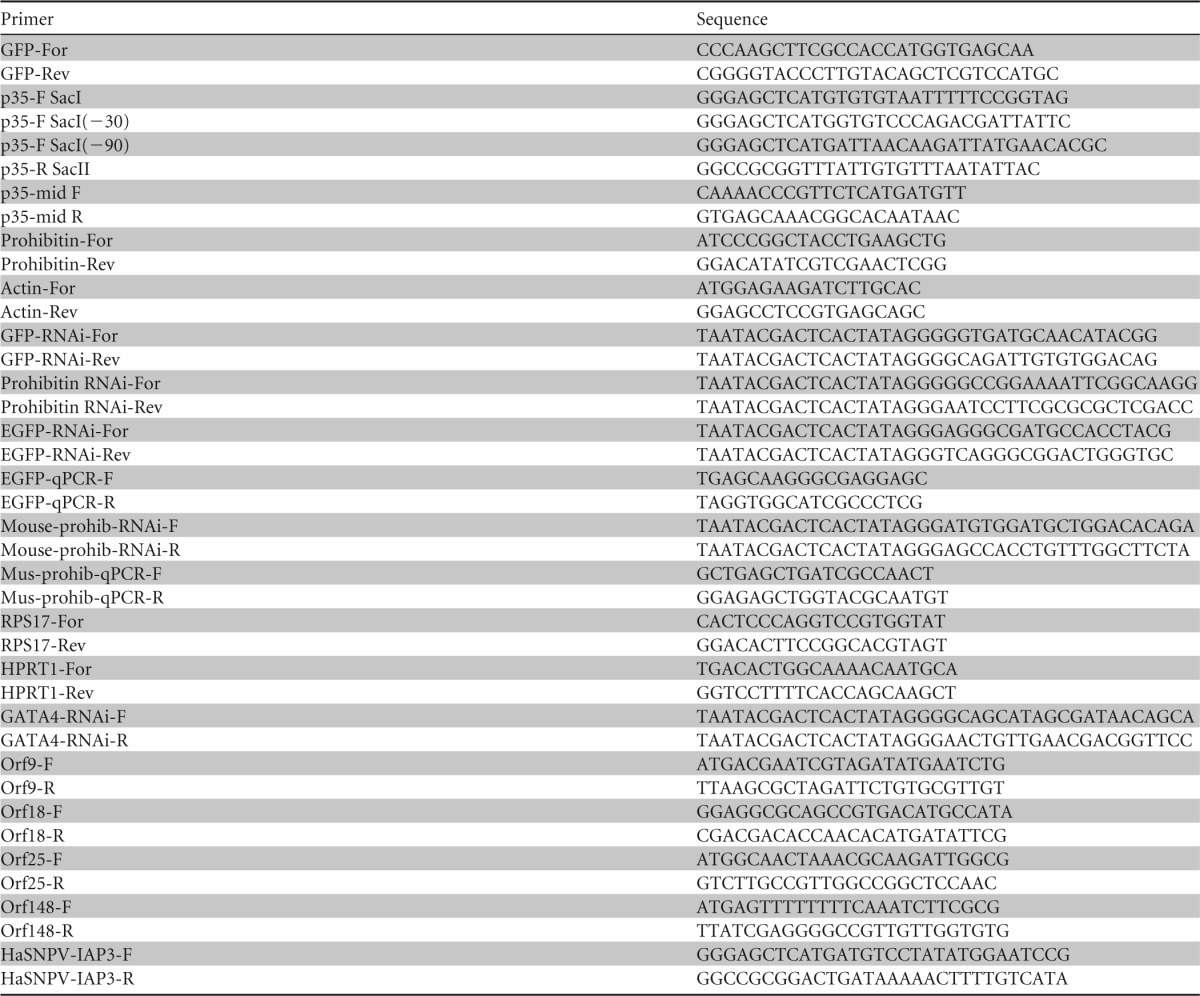
The p35 gene was amplified from AcMNPV-infected Sf9 cells by using specific forward and reverse primers bearing SacI and SacII restriction sites, respectively. The amplified p35 gene was then cloned into the pIZ/V5-His expression vector, resulting in pIZ/p35. To create p35 mutant genes, different truncations were made at both termini of the gene. The first mutant construct was made by truncation at the 3′ end (positions 654 to 900), using the SspI restriction enzyme on pIZ/p35 followed by self-ligation, producing the pIZ/p35Δ1 mutant construct. The other two p35 mutants were made by deletion of 30 nucleotides (nt) (pIZ/p35Δ2) and 90 nt (pIZ/p35Δ3) at the 5′ end, using specific forward primers bearing the SacI restriction site and the ATG start codon. The p35 ORF was also cloned into the mammalian expression vector pEGFP-N1, using BglII and XmaI restriction sites. Expression of p35 from the constructs was confirmed by reverse transcription-PCR (RT-PCR) using a pair of primers for the middle of p35 (p35-mid F and p35-mid R) (Table 1).
TABLE 1.
Primers used in this study
To generate the p35-V71P mutant, we exactly followed a protocol described previously (25). This approach involved amplification of the p35 gene by using overlap extension PCR, utilizing complementary primers (Table 1) in which the nucleotides coding for valine were changed to those for proline. The amplified fragment was cloned into the pIZ/V5-His vector, and mutation was confirmed by sequencing in both directions.
Small RNA deep sequencing and mapping of vsiRNAs.
Previously reported small RNA deep sequencing data utilized for the identification of miRNAs from Sf9 cells mock infected or infected with AcMNPV at 24 and 72 h postinfection (hpi) (23) were used in this study. Small RNA reads were filtered as previously described (23), mapped to the AcMNPV genome by using Bowtie mapping software, and visualized by using the Argo genome browser (http://www.broadinstitute.org/annotation/argo/). Bed files containing small RNAs that were mapped to the virus genome at 24 and 72 hpi are provided as Tables S1 and S2 in the supplemental material for fine visualization of mapping in a genome browser such as Argo.
Electrophoresis and Western blotting.
To visualize the levels of specific proteins (prohibitin, green fluorescent protein [GFP], and gp64), protein samples from whole cells (same number of cells) were run on a denaturing 12% SDS-PAGE gel, and Western blotting was carried out as previously described (26). The blot was blocked in TBST (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.05% Tween 20) containing 5% nonfat dry milk for 1 h, washed three times in TBST, and incubated in TBST–1% nonfat dry milk containing a primary antibody (prohibitin-2 polyclonal antibody [Abmart], GFP polyclonal antibody [Abcam], or gp64 monoclonal antibody [kind gift from Gary Blissard at Boyce Thompson Institute]) at a 1:10,000 dilution, followed by incubation with a secondary antibody (anti-rabbit or anti-mouse IgG antibody [Sigma] for polyclonal or monoclonal primary antibodies, respectively) conjugated with alkaline phosphatase (1:10,000) overnight at room temperature. The blot was washed and developed by using nitroblue tetrazolium chloride (NBT) and 5-bromo-4-chloro-3-indolylphosphate (BCIP) reagents.
Quantitative RT-PCR.
To determine the levels of viral DNA accumulation, total genomic DNA was extracted from cells by using a genomic DNA extraction kit (Invitrogen) and then subjected to quantitative PCR (qPCR) using primers specific to the ie-1 gene from the AcMNPV genome. DNA concentrations were measured by using a NanoDrop instrument, and 10 ng total genomic DNA was used for each qPCR using SYBR green (Invitrogen) with a Rotor-Gene 6000 instrument. Primers used for qPCR are shown in Table 1. PCR conditions were 50°C for 2 min and 95°C for 2 min, followed by 40 cycles of 95°C for 10s, 60°C for 10 s, and 72°C for 20 s and a final extension step at 72°C for 20 s.
Transcript levels of GFP, enhanced GFP (EGFP), and prohibitin were analyzed by RT-qPCR using gene-specific primers (Table 1), utilizing the actin and ribosomal protein S17 (RPS17) genes for insect cells and the hypoxanthine phosphoribosyltransferase 1 (HPRT1) gene for Vero and NIH 3T3 cells as references. For each experiment, three biological replicates with three technical replicates were analyzed by using a Rotor-Gene thermal cycler (Qiagen) under the following conditions: 95°C for 30 s and 40 cycles of 95°C for 10 s and 60°C for 45 s, followed by a melting curve (68°C to 95°C). Relative RNA levels in each sample were compared by using the Qgene template program.
Transcript levels of four AcMNPV ORFs (ORFs 9, 18, 25, and 148) were compared in wild-type AcMNPV-infected and p35-null AcMNPV (Δp35AcMNPV)-infected Sf9 cells (1.12 × 106 PFU/ml) at 16 and 24 hpi by RT-PCR using primers specific for the genes (Table 1).
Northern blot hybridization.
For monitoring the levels of dsRNA for GFP (dsGFP) in cells transfected with dsGFP and subsequently infected with AcMNPV, total RNA was extracted from cells at 4, 8, 16, and 24 hpi by using Tri reagent (Molecular Research Center Inc.). Northern blot detection was carried out by loading 10 μg of total RNA per sample onto a 1% agarose gel with 2.2 M formaldehyde. RNA was transferred onto a nylon membrane by using 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). Full-length PCR products of GFP were labeled with [α-32P]dCTP and used as the probe. Hybridization and membrane washes were carried out under high-stringency conditions at 65°C. Radioactive signaling was detected by phosphorimaging.
To detect dsGFP-derived siRNA (21 nt), 100 μg of total RNA was separated on a 15% polyacrylamide–7 M urea gel in 0.5× Tris-borate-EDTA (TBE) buffer, stained with ethidium bromide to visualize tRNA, and transferred onto a Hybond N+ membrane (Amersham) by electroblotting for 1 h. The membrane was cross-linked at 65°C for 2 h by using a chemical method (27). For the probe, a GFP PCR fragment was digested with NcoI, NdeI, and DdeI (internal restriction sites) and labeled with [α-32P]dATP by using a Random Primers DNA labeling kit (Life Technologies) according to the manufacturer's instructions. Hybridization and washes (twice for 15 min each with 2× SSC–0.1% SDS) were carried out at 50°C. After washing, radioactive signaling was detected by phosphorimaging.
Gene silencing.
To silence the genes of interest (GFP, EGFP, and prohibitin genes), we used RNAi by generating dsRNA synthesized in vitro. DNA fragments of ∼500 bp were amplified from the genes of interest by PCR. Forward and reverse primers contained a T7 promoter sequence (Table 1) at their 5′ end for in vitro RNA synthesis. dsRNA was then produced and purified from each fragment by using the MEGAScript kit according to the manufacturer's instructions (Ambion). Synthesis was confirmed by running dsRNA on an agarose gel, and the concentration of RNA was determined by measuring the absorbance at 260 nm.
To induce RNA silencing in vitro, cells were resuspended and equally added to individual wells of a 12-well plate. Once monolayers had formed (1 h), the medium was removed, and transfection medium was added. This medium consisted of 0.5 ml SF900-II, 8 μl Cellfectin (Invitrogen), and 2 μg dsRNA for either the prohibitin gene or GFP. Twenty-four hours later, each well was infected with 200 μl of the AcMNPV inoculum (MOI of 5). The plate was then incubated at 27°C for 48 h for analyses. For mammalian cells, the procedure was similar, except that Lipofectamine was used as a transfection reagent.
Small RNA sequencing data accession number.
The raw small RNA sequencing (RNA-seq) data have been submitted to the NCBI under GEO accession number GSE60064.
RESULTS
Deep sequencing reveals a large number of siRNAs that map to the AcMNPV genome.
We used deep sequencing to identify small RNAs from S. frugiperda Sf9 cells and their quantitative expression levels following AcMNPV infection. Three small RNA libraries were previously constructed from mock- and AcMNPV-infected Sf9 cells at 24 and 72 h postinfection (hpi). Analysis of the data showed the impact of virus infection on cellular miRNAs (23). By utilizing the deep sequencing data, a total of 2,366,928 reads were perfectly mapped to the AcMNPV genome. There was an insignificant number of reads (0.1%) that mapped to the AcMNPV genome in mock-infected cells, but the number of mapped reads vastly increased as infection progressed. When unique reads of <10 copies were excluded from the libraries, the total number of reads decreased to 1,378,285. This included 421,102 reads at 24 hpi and 957,183 reads at 72 hpi, which ranged in size from 16 to 26 nt, with the majority being 20 nt, which correspond to siRNAs as products of the RNAi response. To exclude miRNAs from the libraries, we used the miRDeep algorithm (28) to query the AcMNPV genome for any potential stem-loop structures (pre-miRNA secondary structures) at the positions that the small RNAs mapped to, in combination with other basic criteria, such as the ratio of miRNA/miRNA* (miRNA* is the complementary/passenger strand in the duplex that is usually degraded), which excluded any potential pre-miRNA. Based on previous studies of HaSNPV (20) and IIV6 (21, 22), the small RNAs were considered virus-derived small interfering RNAs (vsiRNAs).
Uneven mapping of vsiRNAs and hot spots in the AcMNPV genome.
The vsiRNAs detected by deep sequencing were found on both strands of the virus genome and were unevenly distributed along the AcMNPV genome, having areas with large numbers of mapped vsiRNAs (hot spots, such as polyhedrin, viral ubiquitin, the viral capsid-associated protein odv-e25, p6.9, gp64, and IE-1) and modest to low coverage at other regions (cold spots, such as orf18, orf25, lef-2, sod, DNA polymerase [Pol], lef-4, and ORFs 113 to 118) (Fig. 1A and B). The patterns of vsiRNA coverage across the viral genome were highly similar in the two AcMNPV-infected Sf9 cell libraries (i.e., 24- and 72-hpi libraries). However, the number of reads that mapped to the hot spots greatly increased between the two time points of viral infection.
FIG 1.

RNAi is an antiviral defense response against baculovirus infection in Spodoptera frugiperda (Sf9) cells. Shown is a schematic representation of 20-nt reads (blue lines) from Illumina deep sequencing mapped to the AcMNPV genome at 24 hpi (A) and 72 hpi (B). ORFs are shown with pink arrows. Some of the ORFs with hot spots are identified between the two panels and are indicated with open boxes in panel A. The location of the p35 gene is identified by a black circle. The white background indicates the positive strand, and the yellow background indicates the negative strand. The diagram is meant to show only the overall distribution of vsiRNAs across the genome length and not details. Bed files that can be used in a genome browser (e.g., Argo) to look at more detailed information are provided in the supplemental material.
AcMNPV infection suppresses the RNAi response in Sf9 cells.
Considering the antiviral RNAi response of the host against AcMNPV, we questioned whether the virus has evolved a strategy to modulate this antiviral response. To test the hypothesis that AcMNPV suppresses host RNAi, a pIZ/GFP plasmid construct expressing the green fluorescent protein (GFP) gene was cotransfected together with dsRNA for GFP (dsGFP) into Sf9 cells, which were subsequently infected with AcMNPV. While GFP was highly expressed in cells transfected with pIZ/GFP only, in cells cotransfected with dsGFP, no GFP expression was detected by Western blotting, confirming the efficient RNAi response in mock-infected Sf9 cells (Fig. 2). Twenty-four hours after cotransfection (pIZ/GFP plus dsGFP), cells were inoculated with AcMNPV and collected at different times postinfection to determine if virus infection impaired RNAi in the cells. Interestingly, the GFP protein was detected in similar quantities in infected cells transfected with pIZ/GFP only and in infected cells cotransfected with dsGFP (Fig. 2). These results suggested that AcMNPV infection of Sf9 cells hinders the host RNAi response.
FIG 2.
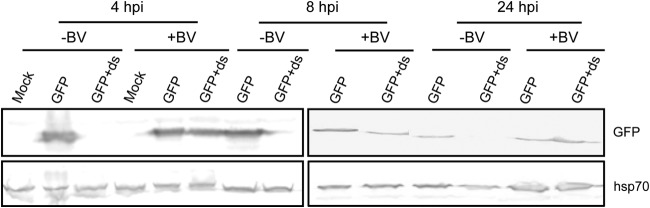
AcMNPV infection suppresses the host RNAi response. Shown is Western blot analysis of GFP expression in Sf9 cells transfected with the reporter plasmid encoding GFP with or without dsRNA targeting GFP (ds) and subsequently mock infected (−BV) or infected with AcMNPV (+BV) at 4, 8, and 24 hpi. Specific antibodies to GFP were used as the probe, and Hsp70 antibody was used to show equal loading of samples.
Baculovirus p35 is a broad suppressor of RNAi.
The AcMNPV-mediated suppression of RNAi in Sf9 cells implied that the virus might encode a viral suppressor of RNAi (VSR). To find out the potential gene coding for the VSR, we first queried the virus genome for those genes important in virus-host interactions, particularly genes responsible for virus defense against the host antiviral response. Of those genes, we focused on p35 (ORF135) due to its prominent antiapoptotic activity (29). Another reason for this selection was based on a previous study in which an inhibitor of apoptosis (transcriptional activator protein [TrAP]) from tomato leaf curl New Delhi virus was shown to have RNAi-suppressive activity (30). To investigate the potential VSR activity of p35, we inserted the gene coding for the protein into the pIZ/V5 vector (pIZ/p35) and transfected it into Sf9 cells, followed by transfection of the cells with pIZ/GFP and dsGFP after 48 h of initial transfection. Expression of p35 was confirmed by RT-PCR (data not shown). We found that in the presence of pIZ/p35, expression of GFP was restored. In contrast, in control cells transfected with the pIZ empty vector, GFP expression was much reduced or nearly undetectable (Fig. 3A). To ensure that this inhibition is due to the p35 protein and not the RNA transcript, a stop codon was introduced immediately after the p35 ATG start codon (Δp35), and the fragment was cloned into the pIZ vector. The construct was transfected into Sf9 cells with pIZ/GFP and dsGFP. Expression of the transcript was confirmed by RT-PCR (data not shown). While silencing of GFP was again suppressed by p35, this effect was not observed with pIZ/Δp35 (Fig. 3B). These findings suggested that p35 is able to suppress RNAi.
FIG 3.

The p35 gene of AcMNPV encodes a potent and broad-acting viral suppressor of RNAi. (A) Western blot analysis of Sf9 cells transfected with pIZ/p35 or the empty pIZ vector and then cotransfected with pIZ/GFP and dsGFP. (B) Western blot analysis of Sf9 cells transfected with pIZ/GFP and pIZ/p35 or pIZ/Δp35 with or without dsGFP. (C) Western blot analysis of Aag2 cells transfected with pIZ/p35 or the empty pIZ vector and then transfected with dsProhibitin. Antibodies specific to GFP (A and B) and prohibitin (C) were used as probes, and Hsp70 antibody was used to show equal loading of samples. (D) RT-qPCR analysis of RNA from Aag2 cells treated as described above for panel C, using primers specific to prohibitin. (E) RT-qPCR analysis of RNA from Sf9, S2, and HzFB cells treated as described above for panel A, using primers specific to the GFP gene. Error bars represent standard errors of averages from three biological replicates. Each biological replicate consisted of three technical replicates.
Since the GFP gene was an exogenous marker gene in Sf9 cells, we decided to test whether p35 also has suppressor activity in the case of RNAi against an endogenous gene. To examine this, we selected prohibitin, which is a multifunctional endogenous gene, and silenced it in Aedes aegypti Aag2 cells using dsRNA for prohibitin (dsProhibitin). In this case, we also observed that the expression of prohibitin, which was suppressed in the presence of dsProhibitin, was rescued in the presence of pIZ/p35 when examined by Western blot detection using polyclonal antibodies to the protein (Fig. 3C). This result was also confirmed at the RNA level by using RT-qPCR (Fig. 3D). These results had interesting implications: first, p35 was able to suppress the cellular RNAi machinery against both an exogenous gene (GFP) and an endogenous gene (prohibitin), and second, it suppressed RNAi not only in Sf9 cells derived from a lepidopteran, which is a natural and permissive host of AcMNPV, but also in Aag2 cells from a different insect order (Diptera) that is not considered a host for this virus. To expand this finding further, we analyzed GFP gene transcript levels in Sf9 cells, Drosophila S2 cells, and Helicoverpa zea fat body (HzFB) cells transfected with pIZ/GFP in the presence and absence of pIZ/p35 and dsGFP using RT-qPCR. We consistently observed that p35 displayed RNAi suppressor activity in different cell lines and restored the expression of the exogenous gene up to the levels of expression in the control cells (Fig. 3E). Together, these results showed broad RNAi suppressor activity of p35 in different insect cells.
In order to test whether p35 exhibits its RNAi suppressor activity in mammalian cells, we cloned the p35 gene into the pEGFP-N1 vector and used it for transfection of NIH 3T3 mouse cells as well as Vero monkey cells. The EGFP gene in the vector was used as a marker. EGFP gene expression was efficiently silenced in Vero monkey cells by using dsEGFP (Fig. 4A). However, transcript levels of the EGFP gene after transfection with dsEGFP and pEGFP-N1/P35 were recovered almost to the level of the control cells (Fig. 4A). GATA4 dsRNA was used for control transfection, which did not affect the expression of the EGFP gene (Fig. 4A). To further examine the p35 suppressor activity in mammalian cells, transcript levels of prohibitin were monitored in NIH 3T3 mouse cells transfected with dsProhibitin and the pEGFP-N1/p35 vector. Likewise, prohibitin expression in NIH 3T3 cells in the presence of dsProhibitin was rescued by p35 to the expression levels of mock-transfected cells (Fig. 4B), while prohibitin transcript levels in cells without p35 expression were significantly reduced (Fig. 4B).
FIG 4.

The p35 gene suppresses RNAi in mammal cells. (A) RT-qPCR analysis of RNA from Vero cells transfected with pEGFP-N1 and dsEGFP or dsGATA4 (as control) in the absence or presence of pEGFP-N1/p35 (p35), using primers specific to EGFP. (B) RT-qPCR analysis of RNA from NIH 3T3 cells transfected with pEGFP-N1/p35 with or without dsProhibitin, using primers specific to prohibitin. There are statistically significant differences between groups with different letters, at P values of <0.0001 (A) and <0.05 (B). Error bars represent standard errors of averages from three biological replicates. Each biological replicate consisted of three technical replicates.
Deletion of the p35 gene from the AcMNPV genome abolishes its VSR activity.
To strengthen our hypothesis that p35 is a VSR of AcMNPV, we used a mutant AcMNPV lacking the p35 gene (Δp35AcMNPV), which has previously been extensively characterized (31). For this, the pIZ/GFP construct and dsGFP were transfected into Sf9 cells, and the cells were subsequently infected with the wild-type virus and Δp35AcMNPV. The lack of p35 expression from this mutant virus was further confirmed by RT-PCR (data not shown). Sf9 cells were then collected at different time intervals postinfection to examine the mutant virus RNAi suppressor activity compared to that of the wild type. The results showed that Δp35AcMNPV did not suppress host RNAi, as very little to no GFP was detected in the mutant virus-infected cells up to 24 hpi, while infection with wild-type AcMNPV impaired host RNAi (Fig. 5A). In the control, the GFP protein was detected in cells transfected with pIZ/GFP only and subsequently infected with the mutant virus. Mutant virus infection and replication were also confirmed by detection of the virus surface protein gp64 (Fig. 5A). The results from this experiment confirmed the RNAi suppressor activity of p35.
FIG 5.
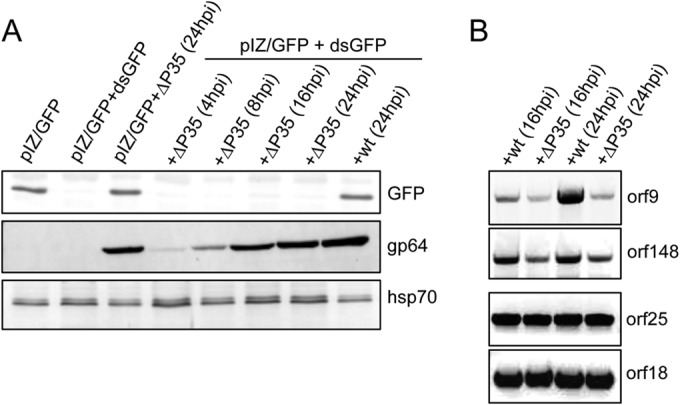
Deletion of the p35 gene from the AcMNPV genome abolishes virus-mediated RNAi suppressor activity. (A) Western blot analysis of Sf9 cells cotransfected with pIZ/GFP and dsGFP and then infected with an AcMNPV mutant lacking the p35 gene (Δp35AcMNPV) at various times after infection, using anti-GFP antibody as a probe. As a control, cells transfected with pIZ/GFP and dsGFP were infected with wild-type (wt) AcMNPV and analyzed at 24 hpi. Hsp70 antibody was used to show equal loading of samples, and anti-gp64 antibody was used to monitor virus replication. (B) RT-PCR analysis of RNA extracted from Sf9 cells infected with the same titers of wild-type AcMNPV and Δp35AcMNPV for 16 and 24 h. Primers specific to two hot spot genes (orf9 and orf148) and two cold spot genes (orf25 and orf18) were used.
Given that we demonstrated RNAi suppression by p35 against an endogenous gene (prohibitin) and an exogenous gene (GFP), we examined if this activity could also be shown in relation to viral genes, in particular those that are located in hot spots as targets of RNAi. For this, we infected Sf9 cells with the same titers of wild-type AcMNPV and Δp35AcMNPV for 16 and 24 h and assessed the transcript levels of two genes in the hot spots (orf9 and orf148) and two genes in the cold spots (orf18 and orf25). Consistently, we found that there were more transcripts of the hot spot genes in wild-type virus-infected cells than in p35-null virus-infected cells, but no change in the transcript levels of the cold spot genes was observed (Fig. 5B). This finding suggested that p35 is an effective VSR in protecting viral transcripts against the host RNAi machinery.
p35 does not block cleavage of dsRNA.
To find out if the VSR activity of p35 is due to an inhibition of dsRNA degradation by Dicer-2, we first transfected dsGFP into Sf9 cells and then infected the cells with AcMNPV. Using a specific probe, the presence of dsGFP and its levels in the cells were monitored by Northern blot analysis. We observed that AcMNPV infection did not block dsRNA cleavage, and in fact, lower dsGFP levels were found in AcMNPV-infected cells than in mock-infected Sf9 cells (Fig. 6A). In another experiment, the fate of dsGFP was monitored in Sf9 cells transfected with pIZ/p35 to find out if we could detect the same effect observed in AcMNPV-infected cells. The results revealed that p35 does not block cleavage of dsRNA (Fig. 6B). This was consistent with a small RNA Northern blot analysis of RNA extracted from Sf9 cells transfected with dsGFP together with pIZ and pIZ/p35. The results showed the presence of more 21-nt GFP siRNAs (siGFPs) in the presence of pIZ/p35 than in the presence of the empty vector (Fig. 6C). Our results suggest that p35 does not display its VSR activity by blocking dsRNA cleavage into siRNAs or inhibiting the accumulation of siRNAs.
FIG 6.

AcMNPV infection or p35 expression does not block degradation of dsRNA. (A) Northern blot analysis of dsGFP levels at times post-AcMNPV infection, using a probe specific to GFP. rRNA is shown as a loading control. (B) Northern blot analysis of dsGFP in Sf9 cells transfected with the pIZ empty vector and pIZ/p35. (C) Northern blot analysis of small RNAs from Sf9 cells cotransfected with dsGFP and the pIZ empty vector or pIZ/p35. The blot was rescanned only in the small RNA area (boxed) to enhance detection of siRNAs by phosphorimager analysis (shown by arrow). tRNAs are shown as a loading control. MW, molecular weight. (D) Western blot analysis of Sf9 cells transfected with pIZ/GFP only or transfected with pIZ/GFP and GFP siRNAs (siGFP) plus mock, the pIZ empty vector, or pIZ/p35. The blot was probed with an anti-GFP antibody and anti-Hsp70 to show equal loading of the samples.
Next, we examined whether p35 can still suppress RNAi in the presence of siRNAs. For this, siGFPs (21 nt) were transfected into Sf9 cells for 48 h, followed by cotransfection of cells with pIZ/GFP and the empty pIZ vector or pIZ/p35. Western blot analysis showed that siGFPs effectively silenced the GFP gene; however, in the presence of pIZ/p35, the expression of GFP was rescued (Fig. 6D). This finding further indicated that p35 blocks RNAi downstream in the RNAi pathway, perhaps by sequestering siRNAs, interfering with the loading of siRNAs into Ago-2, or blocking the activity of Ago-2 by protein-protein interactions. Interestingly, we found a GW/WG motif (240-YKLEFTTESSWGKSEKYNWKI-264) in the p35 protein that has been suggested to be important in the interactions of some VSRs with Ago proteins and to interfere with RNA silencing (32, 33). However, when we changed the WG residues to AC residues in p35, there was no change in siRNA production or VSR activity of the mutant (data not shown).
The VSR activity of p35 is not linked to its antiapoptotic activity.
To determine the functional region of the p35 gene in terms of its VSR activity, we produced three p35 mutants by making different size deletions in the gene. The first p35 mutant gene was made by a deletion at the 3′ end of the ORF (nucleotides 654 to 900) using the SspI restriction enzyme and then cloning the fragment into the pIZ vector (pIZ/p35Δ1). The other two p35 mutants were made by deletions of 30 nt (pIZ/p35Δ2) and 90 nt (pIZ/p35Δ3) from the 5′ end (Fig. 7A). These three p35 mutant constructs along with the complete p35 gene, as a control, were used for assessing the RNAi suppressor activity against GFP silencing in Sf9 cells using dsGFP. Expression of p35 from the constructs was confirmed by RT-PCR using primers specific to the middle of the p35 gene (data not shown; see Table 1 for primers), producing a 434-bp fragment. Based on Western blot analyses, GFP was efficiently silenced in cells by dsGFP, but silencing was inhibited by pIZ/p35, whereas in cells transfected with either of the deletion mutants, no GFP band was detected (Fig. 7B). These results suggested that both the N terminus as well as the C terminus are required for p35's VSR activity.
FIG 7.

Both the N and C termini of p35 are important for its VSR activity, and this activity is not linked to the protein's antiapoptotic activity. (A) Schematic diagram showing p35 deletion mutant constructs produced as described in Materials and Methods. (B) Western blot analysis of Sf9 cells cotransfected with pIZ/p35 or mutant constructs and dsGFP, using the anti-GFP antibody and anti-Hsp70 as a control. (C) Western blot analysis of Sf9 cells transfected with pIZ/GFP only or with pIZ/GFP and dsGFP plus pIZ/p35 or pIZ/p35-V71P. The blot was first probed with the anti-GFP antibody and subsequently probed with the anti-Hsp70 antibody as a control. (D) Same as described above for panel C except that pIZ/IAP3 was used for transfection instead of pIZ/p35-V71P.
To find out if the VSR activity of p35 is linked to its antiapoptotic activity or not, we mutated valine 71 to proline (p35-V71P). This single mutation was previously shown to disrupt the spatial configuration of the reactive loop structure in the protein and to completely abolish the antiapoptotic activity of p35 by failing to inhibit the caspase activity (25, 34). The amplified p35-V71P fragment was cloned into the pIZ vector, and the mutation was confirmed by sequencing. Sf9 cells were cotransfected with pIZ-GFP and dsGFP in the presence of pIZ/p35 or pIZ/p35-V71P. Cells were then analyzed by Western blotting using the anti-GFP antibody. This analysis showed that both p35 and p35-V71P equally suppressed the silencing of GFP (Fig. 7C). As an additional experiment to ensure that the observed RNAi suppression was not due to suppression of apoptosis, the full-length inhibitor of apoptosis 3 gene (IAP3) of HaSNPV was cloned into the pIZ vector by using the primers shown in Table 1. The identity and expression of the gene were confirmed by sequencing in both directions and RT-PCR, respectively (data not shown). HaSNPV IAP3 has been shown to inhibit apoptosis in Sf21 cells (35). The constructs were transfected into Sf9 cells with pIZ/GFP, with or without dsGFP. While silencing of GFP was suppressed by p35, this effect was not observed with pIZ/IAP3 (Fig. 7D). Overall, the suppression of RNAi by p35-V71P, which lacks antiapoptotic activity, and the finding that HaSNPV IAP3 did not suppress RNAi suggest that the VSR activity of p35 is not linked to its antiapoptotic activity.
DISCUSSION
Until recently, the antiviral RNAi response has been reported mainly for RNA viruses, due to their genome being RNA and a direct target of the RNAi response. However, host RNAi responses against dsDNA viruses (HaSNPV and IIV6) in insects were recently demonstrated (20–22). In addition, vsiRNAs of a single-stranded DNA (ssDNA) densovirus were detected in wild-caught Culex pipiens molestus mosquitoes, which implies the presence of an RNAi response against ssDNA insect viruses as well (36). In this study, we have shown cellular antiviral RNAi against a DNA virus, AcMNPV, which is a well-known baculovirus widely used for pest control, biological and biomedical research, and biotechnology. The vsiRNAs obtained from small RNA deep sequencing of virus-infected cells showed a large number of small RNAs that mapped to both strands of the AcMNPV genome, although there were hot and cold spots in terms of vsiRNA mapping.
With respect to RNAi against dsDNA viruses, an overlap of transcripts of adjacent genes on opposite strands and/or the presence of secondary structures in transcripts is assumed to be the reason for the production of dsRNAs. Recent findings have shed light on what appears to be one of the mechanisms of dsRNA generation in DNA virus infections (20–22). It has been shown that transcription from both the positive and negative strands of the genome produced complementary transcripts, which base paired and generated dsRNA (21). Consistently, we showed that vsiRNAs originated from both strands of the AcMNPV genome; therefore, it is likely that many transcripts with complementary sequences were produced, which formed virus-derived dsRNAs that could induce the antiviral RNAi response. Besides the antiviral role of vsiRNAs, it is possible that baculovirus-produced vsiRNAs autoregulate transcript levels of viral genes, thereby controlling virus replication in host cells to prevent rapid host cell death.
Host antiviral RNAi attenuates viral infections in hosts. In turn, some viruses have evolved VSRs to counter this host antiviral response. Suppression of RNAi by viruses has been reported for several RNA viruses infecting plants and animals. For example, CrPV infection of Drosophila S2 cells suppressed the host RNAi response and silencing of the firefly luciferase gene (37). Insect host infections with other RNA viruses (e.g., DCV and FHV) have also been shown to modulate the host RNAi response (11).
In this study, we functionally assessed RNAi in AcMNPV-infected cells and established that AcMNPV infection suppresses the RNAi response to dsGFP (an exogenous gene) and dsProhibitin (an endogenous gene). Subsequently, we discovered that the virus gene p35, which is a well-known antiapoptosis gene, has VSR activity. The VSR activity was detected as early as 4 hpi. p35 expression has been shown to start early in infection, which is required to suppress the host apoptosis response to viral infection (38). Ectopic expression of p35 independent of the virus in different insect cells using dsRNAs for exogenous and endogenous genes hampered the host RNAi response. The intensity of the RNAi suppressor activity in the presence of p35 was almost the same as that detected in AcMNPV-infected cells. In previous studies, dsRNAs have been used for silencing of AcMNPV genes (e.g., see references 39 and 40); however, in those studies, for efficient gene silencing, large amounts of dsRNAs (60 to 160 μg) were transfected into cells, compared to relatively small amount of dsRNAs (2 μg) used in this study, which were applied to about the same number of cells. Utilization of large quantities of dsRNA may overload the system, masking the VSR activity of p35. Similarly, dsRNA has been used for silencing of host genes in the presence of other viruses with known VSR activity (e.g., see references 41 and 42).
To further expand our findings, we tested a p35 knockout AcMNPV and found that it could not suppress the host RNAi response. Of note, considerably lower transcript levels of two genes selected from the hot spots were found in p35-null AcMNPV-infected Sf9 cells than in the wild-type-infected cells. These results together suggested that p35 is a VSR encoded by AcMNPV. Moreover, our preliminary study has shown that when p35 is ectopically expressed, replication of RNA viruses is enhanced (data not shown). Since RNAi is a major antiviral response in insect cells, logically, the suppression of this response by p35 leads to improved viral replication.
In mammals, it is not clear whether RNAi plays a role in antiviral defense. Early findings implied the existence of an RNAi response in mammalian cells, as reported for protection against influenza A virus infection (43) and HIV-1 infection (44, 45) in human cells. Very recently, studies on mammalian viruses provided more evidence to suggest that RNAi could be an antiviral response in mammalian cells (2, 46, 47). These reports demonstrated that RNAi acts as a functional and conserved antiviral response in diverse host cells. In order to examine the VSR activity of p35 in noninsect cells, the plasmid expressing p35 was transfected into two different mammalian cell lines (Vero and NIH 3T3), in which it showed RNAi suppressor activity when an exogenous (EGFP) or an endogenous (prohibitin) gene was targeted by the corresponding dsRNA. These results demonstrated that p35 is functional in different host cells as a potent VSR. p35 was the first antiapoptotic gene described for baculoviruses (48) and was subsequently shown to exert antiapoptotic activity in several different systems through caspase inhibition (49–51). Here, we report another important function of this protein, VSR activity, which is also functional in different systems.
To narrow down the functional domain of the p35 protein, we made different mutant constructs and found that both the C terminus and the N terminus are essential for the VSR activity. The N terminus of the protein also plays a significant role in the antiapoptotic activity of the protein, in particular the first 100 residues, which contain the charged regions CHR1 and CHR2 (52). However, the residues toward the end of the C terminus of the protein have also been shown to be important for its antiapoptotic function (38, 52). To exclude the possibility that the VSR activity of p35 is due to its antiapoptotic activity, we took advantage of a single amino acid mutation, valine 71 to proline, which was previously shown to completely abolish the antiapoptotic function of the protein (25, 34). Our results showed that the p35-V71P mutant was still able to suppress the RNAi activity, suggesting that the protein's VSR activity is not related to its antiapoptotic function. In addition, HaSNPV IAP3, a potent suppressor of apoptosis (35), did not suppress RNAi.
VSRs from different viruses use a variety of mechanisms to exert their activity, such as blocking the cleavage of dsRNA by Dicer, sequestering siRNA, interfering with RISC formation, or blocking Ago-2 activity. For example, the 1A protein of CrPV inhibits RNAi by binding to Ago-2 and inhibiting the RNAi machinery (37). Other reported VSRs from insect viruses, including the B2 protein and 1A protein of FHV and DCV, respectively, modulate RNAi through binding dsRNA and sequestering it from Dicer-2 access to produce vsiRNAs (11, 53). Our findings suggest that p35 does not inhibit the cleavage of dsRNA by sequestering dsRNA or interfering with Dicer-2 activity but displays its VSR activity downstream of the RNAi pathway, perhaps by sequestering siRNAs or blocking the activity of Ago-2 or the formation of the RISC. The detection of a large number of vsiRNAs following AcMNPV infection also confirmed that the cleavage of dsRNA was not blocked; however, suppression of RNAi through interfering with its downstream steps may benefit the virus by inhibiting the degradation of a population of mRNAs. p35 was also able to suppress RNAi when cells were transfected with siRNAs (21 nt), further suggesting that the VSR activity is exerted downstream of the RNAi pathway. Nevertheless, the exact mechanism of p35 VSR activity and the host RNAi protein(s) with which it may interact remain to be investigated.
In conclusion, we demonstrated that AcMNPV, a DNA virus, exhibits VSR activity against the host RNAi response. Notably, we identified the p35 gene of the virus as being responsible for the VSR activity, since deletion of this gene from the virus genome abolished the VSR activity, and ectopic expression of the protein in insect and mammalian cells, independent of the virus, suppressed the host RNAi response against exogenous as well as endogenous genes when dsRNAs specific to the genes were transfected into the cells. Our findings also suggest that p35 exerts its VSR activity by interfering with the downstream steps of the host RNAi response and that its VSR activity is independent of its antiapoptosis function. Finally, the outcomes provide evidence for the dual role of p35 in nullifying two central cellular antiviral responses, apoptosis and RNAi.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nicholas Davis-Pointer from The University of Queensland for providing NIH 3T3 cells and the pEGFP-N1 plasmid and Monique van Oers from Wageningen University for providing the p35-null AcMNPV constructed by Kai Yang. We also thank the Queensland Facility for Advanced Bioinformatics (QFAB) for their assistance in generating the bed files.
This work was supported by an Australian Research Council discovery grant to S.A. (DP110102112) and an ARC DECRA fellowship to M.H. (DE120101512).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00802-15.
REFERENCES
- 1.Ding S-W, Voinnet O. 2007. Antiviral immunity directed by small RNAs. Cell 130:413–426. doi: 10.1016/j.cell.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maillard PV, Ciaudo C, Marchais A, Li Y, Jay F, Ding SW, Voinnet O. 2013. Antiviral RNA interference in mammalian cells. Science 342:235–238. doi: 10.1126/science.1241930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siomi H, Siomi MC. 2009. On the road to reading the RNA-interference code. Nature 457:396–404. doi: 10.1038/nature07754. [DOI] [PubMed] [Google Scholar]
- 4.Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myles KM, Wiley MR, Morazzani EM, Adelman ZN. 2008. Alphavirus-derived small RNAs modulate pathogenesis in disease vector mosquitoes. Proc Natl Acad Sci U S A 105:19938–19943. doi: 10.1073/pnas.0803408105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brackney DE, Beane JE, Ebel GD. 2009. RNAi targeting of West Nile virus in mosquito midguts promotes virus diversification. PLoS Pathog 5:e1000502. doi: 10.1371/journal.ppat.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding S-W. 2010. RNA-based antiviral immunity. Nat Rev Immunol 10:632–644. doi: 10.1038/nri2824. [DOI] [PubMed] [Google Scholar]
- 8.Aliyari R, Wu Q, Li H-W, Wang X-H, Li F, Green LD, Han CS, Li W-X, Ding S-W. 2008. Mechanism of induction and suppression of antiviral immunity directed by virus-derived small RNAs in Drosophila. Cell Host Microbe 4:387–397. doi: 10.1016/j.chom.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flynt A, Liu N, Martin R, Lai EC. 2009. Dicing of viral replication intermediates during silencing of latent Drosophila viruses. Proc Natl Acad Sci U S A 106:5270–5275. doi: 10.1073/pnas.0813412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Q, Luo Y, Lu R, Lau N, Lai EC, Li W-X, Ding S-W. 2010. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc Natl Acad Sci U S A 107:1606–1611. doi: 10.1073/pnas.0911353107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chao JA, Lee JH, Chapados BR, Debler EW, Schneemann A, Williamson JR. 2005. Dual modes of RNA-silencing suppression by Flock House virus protein B2. Nat Struct Mol Biol 12:952–957. doi: 10.1038/nsmb1005. [DOI] [PubMed] [Google Scholar]
- 12.van Rij RP, Saleh M-C, Berry B, Foo C, Houk A, Antoniewski C, Andino R. 2006. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes Dev 20:2985–2995. doi: 10.1101/gad.1482006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Q, Wang X, Ding S-W. 2010. Viral suppressors of RNA-based viral immunity: host targets. Cell Host Microbe 8:12–15. doi: 10.1016/j.chom.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hussain M, Abraham AM, Asgari S. 2010. An ascovirus encoded ribonuclease III autoregulates its expression and suppresses RNAi-mediated gene silencing. J Virol 84:3624–3630. doi: 10.1128/JVI.02362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bronkhorst A, van Cleef K, Venselaar H, van Rij R. 2014. A dsRNA-binding protein of a complex invertebrate DNA virus suppresses the Drosophila RNAi response. Nucleic Acids Res 42:12237–12248. doi: 10.1093/nar/gku910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robalino J, Bartlett T, Chapman R, Gross P, Browdy C, Warr G. 2007. Double-stranded RNA and antiviral immunity in marine shrimp: inducible host mechanisms and evidence for the evolution of viral counter-responses. Dev Comp Immunol 31:539–547. doi: 10.1016/j.dci.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 17.Possee RD, Griffiths CM, Hitchman RB, Chambers A, Murguia-Meca F, Danquah J, Jeshtadi A, King LA. 2010. Baculoviruses: biology, replication and exploitation, p 35–57. In Asgari S, Johnson KN (ed), Insect virology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 18.Herniou EA, Olszewski JA, Cory JS, O'Reilly DR. 2003. The genome sequence and evolution of baculoviruses. Annu Rev Entomol 48:211–234. doi: 10.1146/annurev.ento.48.091801.112756. [DOI] [PubMed] [Google Scholar]
- 19.Clem RJ. 2007. Baculoviruses and apoptosis: a diversity of genes and responses. Curr Drug Targets 8:1069–1074. doi: 10.2174/138945007782151405. [DOI] [PubMed] [Google Scholar]
- 20.Jayachandran B, Hussain M, Asgari S. 2012. RNA interference as a cellular defense mechanism against the DNA virus baculovirus. J Virol 86:13729–13734. doi: 10.1128/JVI.02041-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bronkhorst AW, van Cleef KWR, Vodovar N, Ince IA, Blanc H, Vlak JM, Saleh MC, van Rij RP. 2012. The DNA virus invertebrate iridescent virus 6 is a target of the Drosophila RNAi machinery. Proc Natl Acad Sci U S A 109:E3604–E3613. doi: 10.1073/pnas.1207213109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kemp C, Mueller S, Goto A, Barbier V, Paro S, Bonnay F, Dostert C, Troxler L, Hetru C, Meignin C, Pfeffer S, Hoffman JA, Imler J-L. 2013. Broad RNA interference-mediated antiviral immunity and virus-specific inducible responses in Drosophila. J Immunol 190:650–658. doi: 10.4049/jimmunol.1102486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehrabadi M, Hussain M, Asgari S. 2013. MicroRNAome of Spodoptera frugiperda cells (Sf9) and its alteration following baculovirus infection. J Gen Virol 94:1385–1397. doi: 10.1099/vir.0.051060-0. [DOI] [PubMed] [Google Scholar]
- 24.King LA, Possee RD. 1992. The baculovirus expression system: a laboratory guide. Chapman & Hall, London, United Kingdom. [Google Scholar]
- 25.Fisher A, dela Cruz W, Zoog S, Schneider C, Friesen P. 1999. Crystal structure of baculovirus P35: role of a novel reactive site loop in apoptotic caspase inhibition. EMBO J 18:2031–2039. doi: 10.1093/emboj/18.8.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green MR, Sambrook J. 2012. Molecular cloning: a laboratory manual, 4th ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 27.Pall G, Hamilton A. 2008. Improved Northern blot method for enhanced detection of small RNA. Nat Protoc 3:1077–1084. doi: 10.1038/nprot.2008.67. [DOI] [PubMed] [Google Scholar]
- 28.Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. 2008. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 29.Clem RJ, Miller LK. 1994. Control of programmed cell death by the baculovirus genes p35 and iap. Mol Cell Biol 14:5212–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hussain M, Mansoor S, Iram S, Zafar Y, Briddon RW. 2007. The hypersensitive response to Tomato leaf curl New Delhi virus nuclear shuttle protein is inhibited by transcriptional activator protein. Mol Plant Microbe Interact 20:1581–1588. doi: 10.1094/MPMI-20-12-1581. [DOI] [PubMed] [Google Scholar]
- 31.Lin T, Yu M, Wu W, Yu Q, Weng Q, Yang K, Yuan M, Pang Y. 2010. Functional analysis of Spodoptera litura nucleopolyhedrovirus p49 gene during Autographa californica nucleopolyhedrovirus infection of SpLi-221 cells. Virus Genes 41:441–449. doi: 10.1007/s11262-010-0520-5. [DOI] [PubMed] [Google Scholar]
- 32.Azevedo J, Garcia D, Pontier D, Ohnesorge S, Yu A, Garcia S, Braun L, Bergdoll M, Hakimi MA, Lagrange T, Voinnet O. 2010. Argonaute quenching and global changes in Dicer homeostasis caused by a pathogen-encoded GW repeat protein. Genes Dev 24:904–915. doi: 10.1101/gad.1908710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin H, Zhu J-K. 2010. A viral suppressor protein inhibits host RNA silencing by hooking up with Argonautes. Genes Dev 24:853–856. doi: 10.1101/gad.1927310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.dela Cruz W, Friesen P, Fisher A. 2001. Crystal structure of baculovirus P35 reveals a novel conformational change in the reactive site loop after caspase cleavage. J Biol Chem 276:32933–32939. doi: 10.1074/jbc.M103930200. [DOI] [PubMed] [Google Scholar]
- 35.Liang C, de Lange J, Chen X, van Oers M, Vlak J, Westenberg M. 2012. Functional analysis of two inhibitor of apoptosis (iap) orthologs from Helicoverpa armigera nucleopolyhedrovirus. Virus Res 165:107–111. doi: 10.1016/j.virusres.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 36.Ma M, Huang Y, Gong Z, Zhuang L, Li C, Yang H, Tong Y, Liu W, Cao W. 2011. Discovery of DNA viruses in wild-caught mosquitoes using small RNA high throughput sequencing. PLoS One 6:e24758. doi: 10.1371/journal.pone.0024758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nayak A, Berry B, Tassetto M, Kunitomi M, Acevedo A, Deng C, Krutchinsky A, Gross J, Antoniewski C, Andino R. 2010. Cricket paralysis virus antagonizes Argonaute 2 to modulate antiviral defense in Drosophila. Nat Struct Mol Biol 17:547–554. doi: 10.1038/nsmb.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hershberger PA, LaCount DJ, Friesen PD. 1994. The apoptotic suppressor P35 is required early during baculovirus replication and is targeted to the cytosol of infected cells. J Virol 68:3467–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang N, Wu W, Yang K, Passarelli A, Rohrmann G, Clem R. 2011. Baculovirus infection induces a DNA damage response that is required for efficient viral replication. J Virol 85:12547–12556. doi: 10.1128/JVI.05766-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Means J, Muro I, Clem R. 2003. Silencing of the baculovirus Op-iap3 gene by RNA interference reveals that it is required for prevention of apoptosis during Orgyia pseudotsugata M nucleopolyhedrovirus infection of Ld652Y cells. J Virol 77:4481–4488. doi: 10.1128/JVI.77.8.4481-4488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hussain M, Torres S, Schnettler E, Funk A, Grundhoff A, Pijlman GP, Khromykh AA, Asgari S. 2012. West Nile virus encodes a microRNA-like small RNA in the 3′ untranslated region which upregulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res 40:2210–2223. doi: 10.1093/nar/gkr848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paradkar P, Trinidad L, Voysey R, Buchemin J-B, Walker PJ. 2012. Secreted Vago restricts West Nile virus infection in Culex mosquito cells by activating the Jak-STAT pathway. Proc Natl Acad Sci U S A 109:18915–18920. doi: 10.1073/pnas.1205231109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matskevich AA, Moelling K. 2007. Dicer is involved in protection against influenza A virus infection. J Gen Virol 88:2627–2635. doi: 10.1099/vir.0.83103-0. [DOI] [PubMed] [Google Scholar]
- 44.Bennasser Y, Le S-Y, Benkirane M, Jeang K-T. 2005. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 22:607–619. doi: 10.1016/j.immuni.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 45.Triboulet R, Mari B, Lin Y-L, Chable-Bessia C, Bennasser Y, Lebrigand K, Cardinaud B, Maurin T, Barbry P, Baillat V, Reynes J, Corbeau P, Jeang K-T, Benkirane M. 2007. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 315:1579–1582. doi: 10.1126/science.1136319. [DOI] [PubMed] [Google Scholar]
- 46.Kakumani PK, Ponia SS, S RK, Sood V, Chinnappan M, Banerjea AC, Medigeshi GR, Malhotra P, Mukherjee SK, Bhatnagar RK. 2013. Role of RNA interference (RNAi) in dengue virus replication and identification of NS4B as an RNAi suppressor. J Virol 87:8870–8883. doi: 10.1128/JVI.02774-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Lu J, Han Y, Fan X, Ding S-W. 2013. RNA interference functions as an antiviral immunity mechanism in mammals. Science 342:231–234. doi: 10.1126/science.1241911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clem R, Fechheimer M, Miller L. 1991. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 254:1388–1390. doi: 10.1126/science.1962198. [DOI] [PubMed] [Google Scholar]
- 49.Xue D, Horvitz HR. 1995. Inhibition of the Caenorhabditis elegans cell-death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature 377:248–251. doi: 10.1038/377248a0. [DOI] [PubMed] [Google Scholar]
- 50.Bump NJ, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P. 1995. Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science 269:1885–1888. doi: 10.1126/science.7569933. [DOI] [PubMed] [Google Scholar]
- 51.Xu G, Cirilli M, Huang Y, Rich RL, Myszka DG, Wu H. 2001. Covalent inhibition revealed by the crystal structure of the caspase-8/p35 complex. Nature 410:494–497. doi: 10.1038/35068604. [DOI] [PubMed] [Google Scholar]
- 52.Bertin J, Mendrysa SM, LaCount DJ, Gaur S, Krebs JF, Armstrong RC, Tomaselli KJ, Friesen PD. 1996. Apoptotic suppression by baculovirus P35 involves cleavage by and inhibition of a virus-induced CED-3/ICE-like protease. J Virol 70:6251–6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Rij RP, Andino R. 2006. The silent treatment: RNAi as a defense against virus infection in mammals. Trends Biotechnol 24:186–193. doi: 10.1016/j.tibtech.2006.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.