

Abstract
p300/cyclic AMP–responsive element binding protein–binding protein (CBP) are general coactivators for multiple transcription factors involved in various cellular processes. Several highly conserved domains of p300/CBP serve as interacting sites for transcription factors and regulatory proteins. Particularly, the intrinsic histone acetyltransferase (HAT) activity and transactivation domains (TAD) play essential roles for their coactivating function. Autoacetylation of p300/CBP is commonly observed in cell-free HAT assays and has been implicated in the regulation of their HAT activity. Here, we show that six lysine-rich regions in several highly conserved functional domains of p300 are targeted by p300HAT for acetylation in cell-free systems. We show that p300 is susceptible to acetylation in cultured tumor cells and that its acetylation status is affected by histone deacetylase inhibitor trichostatin A. We further show that either treatment with deacetylase inhibitors or coexpression of Gal4-p300HAT, which alone has no transactivation activity, stimulates the activity of the COOH-terminal TAD of p300 (p300C-TAD). We have defined the minimal p300C-TAD and show that it is sufficient to respond to deacetylase inhibitors and is a substrate for p300HAT. Finally, we show that acetylated p300 possesses enhanced ability to interact with p53. Taken together, our data suggest that acetylation regulates p300C-TAD and that acetylation of p300/CBP may contribute to the dynamic regulation of their complex formation with various interacting partners.
Introduction
p300 and cyclic AMP (cAMP)–responsive element binding protein (CREB)–binding protein (CBP) interact with and serve as coactivators for a variety of transcription factors, including MyoD, p53, CREB, signal transducers and activators of transcription (Stat) family, GATA, nuclear factor-κB, Smad family, E2F family (1), and hypoxia-inducible factor-1 (HIF-1; ref. 2). They are involved in multiple cellular processes, including proliferation, differentiation, growth arrest, apoptosis, angiogenesis, and cellular response to stress environments, such as hypoxia, inflammation, and exposure to chemotherapeutics (1, 3, 4). It has been proposed that p300/CBP serve as integrators of various signals and coordinately regulate the expression of multiple genes for a programmed cellular response to these signals. How p300/CBP distinguish and integrate the various signals and respond accordingly by selectively stimulating the expression of a specific group of genes is not completely understood.
It has been well documented that each molecule of p300 or CBP possesses two transactivation activities [NH2-terminal (N-TAD) and COOH-terminal (C-TAD) transactivation domains; refs. 1, 5–7] and an intrinsic histone acetyltransferase (HAT) activity (8, 9). These activities have been correlated with specific highly conserved domains of p300/CBP. Although all these activities may play essential roles in the regulation of gene expression, how these activities are regulated by various signaling pathways remains unclear. However, it has been well established that activation of signaling pathways may induce posttranslational modifications of either p300/CBP or their interacting partners. Posttranslational modifications of the latter, mostly transcription factors, may affect their interaction with p300/CBP, thus regulating the selective coactivating activity of p300/CBP and turning on a cooperative spectrum of genes to execute a programmed process (7, 10–15). Protein kinase A–mediated phosphorylation of CREB and DNA damage–induced phosphorylation of p53 enhance their interaction with p300/CBP and turn on genes involved in energy metabolism and DNA repair, respectively (16–18). The oxygen-dependent hydroxylation of HIF-α by factor inhibiting HIF-1 (FIH-1; ref. 19) effectively prevents the recruitment of p300 and subsequently inhibits the activation of HIF-1, a transcription complex that coordinates systemic response to hypoxic stress and controls glucose metabolism and angiogenesis (14, 15). Importantly, posttranslational modifications of p300/CBP also regulate p300/CBP functions. Both phosphorylation and methylation have been found to regulate p300 transactivation activity and its interaction with transcriptional factors, thus regulating gene expression (1, 20, 21).
The acetylation of nonhistone proteins by p300/CBP, including transcription factors, has been observed previously (1, 22, 23). Similarly, histone deacetylases (HDAC), which were originally named based on their ability to remove the acetyl group from histones, have been found to deacetylate nonhistone proteins (24–27). p300-catalyzed acetylation of transcription factors, such as p53, has been reported to enhance their ability to recruit p300/CBP, thus stimulating their transactivation activity (28). Autoacetylation of p300 was also observed in cell-free systems (29) and it has been reported that autoacetylation of the p300HAT region regulates p300HAT activity in cell-free systems (30). We recently reported that p300HAT acetylated p300CH1, which may be involved in the regulation of HIF-1 activity (31). However, whether other regions of p300 are subject to acetylation and the potential biological significance of such modifications have not been investigated.
HDAC inhibitors (HDAI) have been intensively explored as new therapeutics for cancers, neurodegenerative diseases, and hematologic disorders (32–35). Particularly, in cancer trials, HDAIs effectively repress tumor growth and angiogenesis. It has been found that HDAIs enhance the transactivation activity of a variety of transcription factors, but repress HIF-1 and HIF-2 (31), the master regulators of tumor angiogenesis. In line with that, FR901228 (also called romidepsin), an HDAI that is undergoing clinical phase II studies in treating patients who have relapsed or refractory multiple myeloma, has recently been shown to negatively affect the HIF-1 pathway (36). The potential effects of HDAIs on p300 or CBP function have not been well evaluated.
In this study, we investigated the p300HAT-catalyzed acetylation of p300. We report here that multiple domains of p300 are subject to p300HAT acetylation, and the acetylation status of p300 can be enhanced by HDAIs. We further show that the acetylation status of p300 regulates its transactivation activity and complex formation with p53 tumor suppressor.
Materials and Methods
Plasmids and DNA recombination
pGEX-hp53 and pGEX-pCAFaa352-832 were provided by Dr. S. Berger (Wistar Institute, Philadelphia, PA; refs. 32, 33). Glutathione S-transferase (GST)-FIH-1 was a kind gift from Dr. G. Semenza (Johns Hopkins University MD; ref. 19). GST-TFIIB was constructed by inserting the TFIIB coding fragment obtained from reverse transcription-PCR (RT-PCR) into pGEX-2T vector. GST-p300aa1-596, GST-p300aa744-1571, GST-p300aa1572-2376, and GST-p300aa1709-1915 (KR6) were provided by Dr. P.L. Puri (Burnham Institute, La Jolla, CA) or Dr. S. Bhattacharya (Oxford University, Oxford, United Kingdom; refs. 3, 34). Other GST-p300 constructs (GST-p300KR1aa230-430, GST-p300KR2aa560-660, GST-p300LK3aa954-1206, GST-p300KR4aa1196-1400, GST-p300KR5aa1401-1600, and GST-p300aa1000-1707) were made by PCR using wild-type p300 as template (primer information provided as Supplementary Materials). All PCR fragments were inserted into suitable pGEX vectors using compatible restriction sites to fuse with GST in frame.
pGal4-p300 constructs (full-length, amino acids 964–1,922, 1,737–1,922, and 1,737–2,414) were described previously (5, 20). pGal4-p300aa1-596 was generated by digesting pGal4-p300 with BamHI and NotI and followed by blunt-end religation. All other Gal4 fusion constructs of p300 were made by PCR amplification of corresponding coding fragments with designed primers (Supplementary Materials) that were synthesized in the Nucleic Acid Facility at Kimmel Cancer Center. PCR products were ligated into EcoRI and XbaI sites on pcDNA3-DNA binding domain (DBD; ref. 37). pcDNA3-flag-HDAC1 was provided by Dr. A. Giordano (Temple University, Philadelphia, PA; ref. 38). pcDNA3-Sirt2 was constructed by insertion of the coding human cDNA fragment (Genbank accession no. NM_012237), which was obtained by RT-PCR. All constructs were confirmed by automated sequencing analysis (Kimmel Cancer Center, Philadelphia, PA).
Special reagents
Trichostatin A (TSA) and sodium butyrate (NaB) were purchased from Biomol (Plymouth Meeting, PA) and Sigma-Aldrich (St. Louis, MO), respectively. Protein-A/G Sepharose beads were purchased from Pierce (Rockford, IL). HAT assay kits, including biotinylated histone H4 peptides and streptavidin beads, were purchased from Upstate Biotech (Lake Placid, NY).
GST fusion protein expression and purification
Plasmids expressing the indicated GST fusion proteins were transformed into competent Escherichia coli (strain BL21, Stratagene, La Jolla, CA) and the expression was induced by addition of isopropyl-l-thio-β-d-galactopyranoside (Promega, Madison, WI) to 0.1 mmol/L. The induction was done at 25°C with mild shaking for 3 to 6 h. The procedures used for purification were the same as described previously (39). Briefly, the induced cells were collected by centrifugation and lysed in NETN buffer (39) with sonication. After two rounds of centrifugation at 13,000 × g, the supernatants were collected and incubated with glutathione-coupled Sepharose beads (Amersham, Piscataway, NJ). The immobilized fusion proteins were washed four times with NETN buffer (39).
To release the expressed HAT from the GST beads, the protein amounts were first estimated by sampling on SDS-PAGE with serially diluted bovine serum albumin (BSA) standards. The fusion proteins were then equilibrated twice in 1× PBS. Thrombin or factor Xa (Sigma-Aldrich), whichever was applicable, was added in a ratio of 1:300 or 1:50, respectively, at 30°C for 5 h. The supernatant was collected by centrifugation at 13,000 × g for 5 min. Factor Xa was removed by immunoprecipitation, done with a removing kit obtained from Sigma. If needed, the supernatant was finally concentrated by microcentricons (Ambion, Austin, TX) by following the instructions provided by the manufacturer. The HAT activity was estimated with a HAT assay kit from Upstate Biotech, using biotinylated histone H4 peptides as substrates.
Cell lines and transfection
All cell lines were maintained at 37°C in a humidified incubator in an atmosphere of 5% CO2. HEK 293 (human embryonic kidney) and HeLa (human cervical cancer) cells were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 μg/mL; Invitrogen, Carlsbad, CA). Transient transfection was done with LipofectAMINE Plus or LipofectAMINE2000 reagents (Invitrogen) by following the low-serum protocol provided by the manufacturer. The amount of DNA used in transfection was described in each experiment. When needed, 0.5 μg of plasmid expressing β-galactosidase was cotransfected to normalize the transfection efficiency, as described previously (15). All transfected DNA amounts were balanced by filling up with empty expression vector. Twenty-four hours after transfection, the cells were trypsinized, pooled, and equally divided into 12-well-plate luciferase and β-galactosidase assays (at least in triplicate) or into 60-mm dishes for immunoblotting analysis.
Luciferase and β-galactosidase assays
All cell extracts were prepared and analyzed using the Luciferase Assay System purchased from Promega following the manufacturer's procedure. Luminescence was measured in a TD20/20 Luminometer (Promega), and the results were expressed as relative light units (RLU). Protein concentration was determined by the Bio-Rad method (Bio-Rad, Hercules, CA) when needed. β-Galactosidase assays were done in 1× Z buffer (40) with a β-galactosidase Assay kit from Promega.
Antibodies, immunoprecipitation, and immunoblotting analyses
Anti–acetyl-H3 antibody was purchased from Upstate Biotech. Anti-p300 and anti-p53 monoclonal antibodies were purchased from BD Biosciences, Pharmingen (San Jose, CA). Anti–acetyl lysine antibodies were purchased from Stressgen (Ann Arbor, MI) and Abcom (Cambridge, MA). Anti–acetyl-CBP/p300 was from Cell Signaling (Beverly, MA). Monoclonal anti-GAL4 DBD antibody was purchased from Clontech (Mountain View, CA). Horseradish peroxidase (HRP)–coupled anti-mouse IgG was purchased from Sigma. Monoclonal anti-E1A antibody (M73) and immunoprecipitations were described previously (40). Briefly, cells were lysed in lysis buffer [50 mmol/L Tris-HCl, 250 mmol/L NaCl, 1% Triton 100, 5 mmol/L EDTA, 50 mmol/L NaF, 0.1 mmol/L Na3VO4, 1 mmol/L phenylmethylsulfonyl fluoride, 1× protease inhibitor mix (pH 7.5); ref. 40]. Cell lysates were first incubated with nonspecific normal mouse serum and killed protein A–positive Staphylococcus aureus cells (Roche Diagnostics, Indianapolis, IN). The precleared lysates were incubated with 2 μg of monoclonal antibody on ice for 30 min followed by rocking with immobilized protein A/G (Pierce, Rockford, IL) at 4°C for 45 min. For immunoblotting analyses, samples were separated on a 4% to 20% gradient SDS-PAGE (Bio-Rad), if not specified, followed by electrotransfer onto polyvinylidene difluoride membrane. The membrane was blocked with 5% milk in TBST buffer (15), sequentially incubated with specific antibody, washed in TBST, and incubated with HRP-conjugated secondary antibody. The membranes were finally developed with the ECL plus system (Amersham).
Acetylation assays
Standard HAT assays were done as previously described (40). Briefly, for each assay, 250 μL of reaction mixture containing 2.5 mg/mL of BSA, 5 μL of [14C]acetyl-CoA (0.25 μCi, 4 nmol) were set up in 1× HAT buffer [50 mmol/L Tris (pH 7.5), 1 mmol/L EDTA, 1× protease inhibitor cocktail] and incubated at 30°C for 30 min. Fifty microliters of streptavidin-Sepharose beads were equilibrated in 1× HAT buffer first and rocked with the reaction mixture to collect biotinylated histone peptide at 4°C for 20 min. Following four washes with 500 μL radioimmunoprecipitation assay (RIPA) buffer, the beads were resuspended in 100 μL of RIPA buffer and transferred into vials for liquid scintillation counting. When immobilized samples were examined for HAT activity, the reactions were carried out with slow rocking at 30°C for 1 h followed by centrifugation. The supernatants were collected and rocked with streptavidin-Sepharose beads. When immobilized nonhistone proteins were used as substrates in acetylation assays, samples were rocked with mobilized recombinant p300HAT or pCAF in the presence of [14C]acetyl-CoA (Amersham) and BSA (2.5 mg/mL) in 300 μL of 1× HAT buffer at 30°C for 1 h. Immobilized protein samples were collected by centrifugation and followed by four washes with RIPA buffer. Finally, the samples were separated on SDS-PAGE gels followed by autoradiography.
Results
p300 and CBP possess six lysine-rich, highly conserved regions in their functional domains
It has been observed that p300 autoacetylates itself in acetylation assays in cell-free systems (29). Analysis of the primary structure of p300 revealed six lysine-rich regions (KR; Fig. 1A). These regions completely or partially overlap with several important functional domains of p300/CBP and have been shown to be essential for the coactivating activity of p300/CBP (1, 7, 29). KR1 (amino acids 243–423) overlaps with CH1 and p300N-TAD (1, 5). This region also interacts with multiple transcription factors (p53, HIF-1α, cited2, Ets-1, Stat2, etc.) and MDM2 (3, 4, 20). KR2 (amino acids 569–654) coincides with KIX, another domain that is important for protein complex formation and is responsible for cAMP-mediated signaling that regulates energy metabolism (1, 7, 16). KR3 (amino acids 970–1,180) overlaps the bromodomain, which is important for binding to the nucleosome and confers CtBP-mediated (41), as well as CtBP-independent repression (42). KR4 (amino acids 1,228–1,358) and KR5 (amino acids 1,407–1,592) are the NH2-terminal and COOH-terminal parts of the HAT domain (8, 9, 30). Finally, KR6 (amino acids 1,674–1,832) encompasses CH3, which is an interacting site for multiple transcriptional regulators (c-Fos, GATA-1, E2F, MyoD, p53, etc.) and tumor viral proteins (SV40 Tag, adenoviral E1A; reviewed in ref. 1). Further analysis shows that most of the lysyl residues are conserved among p300/CBP identified from multiple species, and some are among the most conserved residues in the p300/CBP primary structure (Fig. 1B), suggesting that they may be important for the functioning of these domains.
Figure 1.

Lysine-rich regions in the conserved domains of p300 and their alignment to p300 function. A, schematic structure of p300. Relative locations of KR regions are given, and known functions are assigned to corresponding regions. B, sequence alignments of the KR regions of p300/CBP. Alignments were done with the ClustalW software. Conserved residues are colored based on their classification: yellow, positive charge; purple, negative charge; green, polar; blue, nonpolar. *, highly conserved lysyl residues (K). Dm, Drosophila melanogaster; Ce, Caenorhabditis elegans; Hs, Homo sapiens; Mm, Mus musculus.
Multiple regions of p300 are susceptible to autoacetylation by p300HAT in cell-free systems
We first determined which region or regions were susceptible to acetylation by p300HAT. Initially, we expressed three large fragments that spanned the majority of full-length p300 as GST fusion proteins: GST-p300aa1-596, which covers KR1 and possesses the CH1 transactivation activity; GST-p300aa744-1571, which contains the bromodomain/KR3 and KR4 and KR5, and GST-p300aa1572-2376, which covers the KR6 and p300C-TAD (Fig. 2A). Acetylation assays in cell-free systems using a recombinant p300HAT (Upstate Biotech) showed that all these fusion proteins were susceptible to autoacetylation by p300HAT, whereas GST alone was not (data not shown), suggesting that multiple regions of p300 are potent substrates for p300HAT. To precisely map the acetylated regions, we inserted PCR fragments encoding each KR region of p300 into pGEX-4T and expressed the corresponding peptides as GST fusion proteins (Fig. 2A). These proteins were purified as immobilized proteins on beads and used as substrates for acetylation assays in cell-free systems. To facilitate further studies, we expressed p300HAT (amino acids 1,000–1,707) and pCAFHAT (amino acids 352–832), which were subsequently purified and confirmed to have satisfactory HAT activity by standard HAT assay with histone H4 peptides as substrates, whereas GST alone showed lack of HAT activity. Because acetylation of p300 has been implicated in repression of HIF-1, a confirmed HIF-1 repressor, FIH-1 (19) was also tested but revealed no HAT activity (Fig. 2B). Not surprisingly, we observed that all KR regions of p300 were acetylated by purified recombinant p300HAT (Fig. 2C). Purified recombinant pCAFHAT also acetylated some KR regions (data not shown). Because GST alone was neither acetylated by p300HAT nor by pCAFHAT, we concluded that these KR regions of p300 are susceptible to acetylation by p300 and pCAF. Neither GST-HIF-1αaa530-826 nor GST-FIH-1 was acetylated by p300HAT under identical experimental conditions (ref. 31 and data not shown), showing the substrate specificity of the acetylation of KR regions. Consistent with these data, the acetylation of CH1 (KR1; ref. 31), KR5 in the HAT domain (30), and KR3 (43) was recently reported. However, in our assays, the acetylation of KR5 was relatively weak (Fig. 2C).
Figure 2.

Mapping p300 regions susceptible to acetylation by p300HAT. A, schematic structure of the GST-p300 constructs. B, HAT activity of recombinant p300HAT and pCAFHAT. Top, structures of the recombinant constructs. The recombinant proteins were expressed in E. coli and purified by incubating the bacterial lysates with glutathione-Sepharose beads. Bottom, HAT activities. GST-FIH-1 was also tested for HAT activity. GST was used as negative control. Columns, average [14C]acetyl-CoA amounts incorporated in duplicate tests. C, KR regions were fused with GST and expressed in E. coli. After purification, the immobilized fusion proteins were used as substrates in p300HAT acetylation assays. Top, autoradiograph shows the incorporation of [14C]acetyl-CoA. *, relative position of each protein. Arrowhead, a band in all assays that is likely derived from p300HAT. Bottom, Coomassie blue staining. GST and GST-p53 were used as negative and positive controls, respectively. Note that the previously reported acetylated region (KR5) was acetylated weakly in our assays.
HDAIs enhance the acetylation and transactivation activity of p300 in cultured tumor cells
Protein acetylation is a common modification that regulates the transactivation activity of transcriptional factors and cofactors. Because both p300N-TAD and p300C-TAD could be acetylated by p300HAT, we investigated the effects of acetylation on p300 transactivation activity. In mammalian cells, the acetylation status of proteins is determined by a balance between acetylases and deacetylases. To examine if p300 is acetylated in cultured cells and to illustrate the effects of HDAIs on total levels and acetylation status of p300, HeLa cells were exposed to TSA for 3 h before lysate preparation. Although such treatment enhanced the acetylation of p300 and histone H3, endogenous p300 levels did not change significantly (Fig. 3A), confirming that TSA enhanced the acetylation of p300, but not p300 protein levels. To further test if TSA affects the transactivation activity of p300, we cotransfected a construct that expresses full-length p300 fused with the Gal4 DBD together with a firefly luciferase reporter gene into HeLa cells (38). In untreated cells, Gal4-p300 activated the expression of luciferase activity, and exposure of transfected cells to TSA significantly enhanced the expression of the reporter luciferase in a dose-responsive manner (Fig. 3B). This enhancement was not a consequence of elevated expression of Gal4-p300, because we used the Gal4-p300 under the control of the cytomegalovirus (CMV) enhancer-promoter, which is less affected by TSA compared with other common expressing promoter-enhancer combinations (31). More importantly, the relative luciferase units (RLU) presented here have been corrected against β-galactosidase activity expressed from cotransfected pCMV-LacZ, which has the same enhancer/promoter elements. In the absence of Gal4-p300, TSA showed no obvious effect on the reporter gene expression (not shown). These data suggest that TSA enhances luciferase reporter gene expression through Gal4-p300. Similar results were obtained when NaB was used. Because both TSA and NaB are inhibitors for the class I/II HDACs, the stimulatory effects of TSA on p300 transactivation activity suggest that it is repressed by a member of the class I/II HDACs. Consistent with this concept, it has been recently reported that p300 interacts with HDAC1 (38). We next examined the effect of overexpression of HDAC1 on p300 transactivation activity and observed that forced expression of exogenous HDAC1 efficiently repressed p300 activity (Fig. 3C). Overexpression of Sirt2, a member of class III HDACs, also repressed p300 transactivation activity, but to a lesser extend (Fig. 3C).
Figure 3.
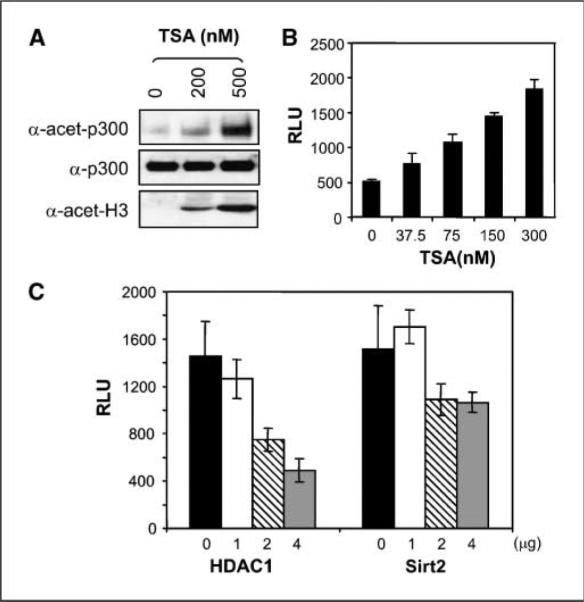
Transactivation activity of p300 is stimulated by TSA but repressed by HDAC1. A, TSA enhances p300 acetylation in cultured tumor cells. HeLa cells were exposed to 200 or 500 nmol/L TSA for 3 h before harvest. Cell lysates were prepared and resolved through 4% to 20% gradient SDS-PAGE. Immunoblotting assays were done with anti-p300, anti–acetyl-CBP/p300, and anti–acetyl-H3. B, TSA stimulates p300 transactivation activity. A plasmid expressing Gal4-p300 driven by a CMV promoter was cotransfected with pCMV-LacZ and pFRLuc reporter into HeLa cells. Transfected cells were exposed to increasing doses of TSA for 12 h before harvest. Luciferase activities were determined and normalized to β-galactosidase activity. C, repression of p300 activity by cotransfection of HDAC1 and Sirt2. The Gal4-p300 plasmid was cotransfected with increasing amounts of plasmids expressing either HDAC1 or Sirt2. Luciferase activities were determined and normalized to cotransfected control plasmid pCMV-LacZ. Bars, SD.
The C-TAD, but not the N-TAD of p300, is sensitive to HDAIs
p300N-TAD and p300C-TAD involve KR1 and KR6, respectively. In addition, KR5 in the HAT region is susceptible to acetylation (Fig. 2; ref. 30), which may indirectly affect the transactivation activity of p300. Effects of HDAIs on the transactivation activity of p300N-TAD and p300C-TAD were evaluated separately. We cotransfected either Gal4-p300N-TAD (amino acids 1–596) or Gal4-p300C-TAD (amino acids 1,737–2,414) with the luciferase reporter (15). Both Gal4-p300N-TAD and Gal4-p300C-TAD were shown to have stronger transactivation activity than the full-length p300 fused with Gal4 DBD (data not shown), consistent with previous reports (5, 6). The Gal4-p300aa964-1922, which carries the HAT activity and contains the region reported to interact with TFIIB (5, 6), showed no transactivation activity (not shown). Upon exposure of the transfected cells to TSA, the Gal4-p300N-TAD was only slightly stimulated (Fig. 4A), but Gal4-p300C-TAD was strikingly enhanced (Fig. 4B). Next, we cotransfected Gal4-p300C-TAD with either Gal4-p300aa964-1922 (bearing the HAT) or Gal4-p300aa1737-1922 (no HAT activity). We observed that coexpression of Gal4-p300aa964-1922 enhanced p300C-TAD activity ~3.5-fold in the absence of TSA (Fig. 4C). Gal4-p300 showed a similar effect, whereas transfection with Gal4-p300aa1737-1922, which lacks HAT activity, showed no effect (Fig. 4C). All combinations of cotransfections reached maximal activity (~13- to 20-fold compared with Gal4 DBD alone) when exposed to 300 nmol/L of TSA (overrange, not shown in chart), whereas 150 nmol/L of TSA markedly enhanced the transactivation and showed synergetic effects with coexpressed Gal4-p300aa964-1922 (HAT). These data suggest that an acetylation event enhances the activity of p300C-TAD.
Figure 4.
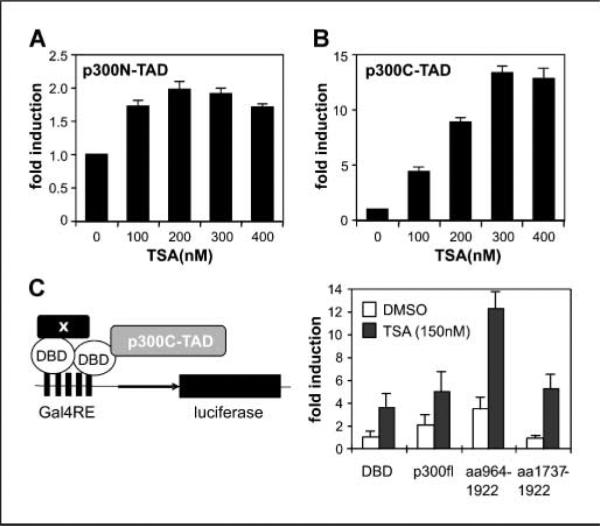
The p300C-TAD is affected by acetylation. A, effects of TSA on the transactivation potential of p300aa1-596. Gal4-p300N-TAD (amino acids 1–596) was cotransfected (3 μg) with the luciferase reporter (3 μg) and pCMV-LacZ (0.5 μg). Twenty-four hours after transfection, cells were split evenly into 15 wells of 12-well culture plates and treated with various doses of TSA in triplicate. RLUs were normalized to β-galactosidase activity and induction factors are given. B, effects of TSA on p300C-TAD. Experiments were done as in (A), except that Gal4-p300C-TAD (amino acids 1,737–2,414) was used in cotransfection. C, synergetic effects of p300HAT and TSA. The assay system is outlined on the left. Plasmid expressing Gal4-p300C-TAD (2 μg) was cotransfected with indicated plasmid (2 μg) in reporter assays. RLUs were normalized first to β-galactosidase activity and calculated into fold of enhancement for comparison. Data showed that corecruitment of p300 full-length (fl) or p300HAT domain to the Gal4 promoter enhanced the transactivation potential (open columns) and increased the sensitivity of Gal4-p300C-TAD to TSA (solid columns, 150 nmol/L). Bars, SD.
Defining the minimal p300C-TAD and the HDAI-responsive region
p300C-TAD has been generally described as amino acids 1,737 to 2,414, a fairly large region of 678 amino acids (5, 6). Although various constructs have been used to represent the p300C-TAD in different reports (5, 6, 20, 38), the minimal region for this activity has not been precisely defined. In addition, KR6/CH3 has been reported to interact with TBP, TFIIB (5, 6), and HDAC1 (38), whereas its role for the transactivation activity of p300C-TAD remains unclear. To explore the minimal domain requirement for p300C-TAD, its responsiveness to HDAIs and the role of KR6/CH3, we constructed a set of progressively truncated mutants of p300C-TAD fused with Gal4 (Fig. 5A). Cotransfection assays revealed that complete deletion of KR6/CH3 (C-TADe) did not impair, but enhanced, the transactivation activity, suggesting that KR6/CH3 is repressive for C-TAD transactivation activity (Fig. 5A). The enhancement caused by deletion of CH3 is consistent with the report that this region interacts with HDAC1 (38). Of note, immunoblotting assays showed similar levels of expression for all constructs used (not shown). Further investigation showed that deletion from the COOH-terminal side to amino acid 2,180 or from the NH2-terminal side to amino acid 1,910 did not impair p300C-TAD (p300C-TADf; Fig. 5B). However, deletion of the conserved I domain abolished the transactivation activity. Therefore, the amino acid 1,910 to 2,180 region is sufficient, and the I domain is necessary for p300C-TAD transactivation activity.
Figure 5.

Defining the minimal p300C-TAD and HDAI-responsive region. A, transactivation activity and sensitivity to TSA. Constructs (a–e) shown at left were used in luciferase reporter assays as in Fig. 4A. Twenty-four hours after transfection, cells were split evenly into 15 wells of 12-well culture plates, and treated with various doses of TSA in triplicate. RLUs were normalized to β-galactosidase activity. B, the amino acid 1,910 to 2,180 region is sufficient, and the I domain is necessary for p300C-TAD. Constructs (d–h) depicted at left were used in luciferase reporter assays, and RLUs were determined as in (A). Bars, SD. C, the minimal p300C-TAD is a substrate for p300HAT. GST fusion constructs of the minimal p300C-TAD, p300C-TADf, and p300C-TADh, respectively, were expressed in E. coli. These regions both contain three lysyl residues as indicated. Purified GST-p300C-TADf and GST-p300C-TADh were used in acetylation assays with p300HAT. GST alone and GST-p53 were included as negative and positive controls.
Interestingly, all the tested constructs, including the minimal transactivation domain (C-TADf and C-TADh), were stimulated by TSA (Fig. 5A and B), suggesting that the minimal C-TAD could be a direct target for p300HAT-catalyzed acetylation. To confirm this possibility, we expressed C-TADf and C-TADh as GST fusion proteins. Acetylation assays in cell-free systems with p300HAT showed that p300HAT acetylated both fusion proteins (Fig. 5C). Therefore, the minimal p300C-TAD is subject to autoacetylation by p300HAT.
Acetylation regulates the complex formation of p300
Because we observed that the major regions of p300 involved in protein-protein interactions are susceptible to acetylation, we examined if acetylation of p300 plays a regulatory role in the regulation of p300 complex formation with p53 as a model molecule. Indeed, TSA treatment enhanced the coprecipitation of p300 with exogenously expressed p53 in HEK 293 cells without affecting the endogenous p300 levels. Interestingly, complex formation of p300 with E1A, the adenoviral oncoprotein expressed in 293 cells, remained unchanged by TSA treatment, suggesting a specific role for HDAC inhibition in complex formation between p300 and p53 (Fig. 6A). Acetylation of Lys-382 of p53 has been reported to enhance the ability of p53 to recruit p300 (44, 45), making it difficult to determine the contribution of p300 acetylation in the regulation of p53-p300 interaction in cultured tumor cells. To distinguish the effect of p53 acetylation from that of p300 acetylation, we generated p53K382L mutant (Lys-382 substituted by leucine) and transfected it into HeLa cells. Coupled immunoprecipitation and immunoblotting assays revealed that TSA enhanced the coprecipitation of p300 with both wild type and p53K382L (Fig. 6B). Next, we expressed and purified p53 as GST fusion proteins from E. coli. Although exposure of cells to TSA and NaB did not affect the endogenous p300 levels, combined GST pull-down and immunoblotting assays showed that HDAIs enhanced the ability of p300 to bind bacterially expressed GST-p53 that lacks posttranslational modifications (Fig. 6C). HDAI treatment showed limited effect on p300 interaction with GST-TFIIB. Taken together, these data indicate that p300 acetylation functionally contributes to the regulation of p300-p53 complex formation.
Figure 6.

p300 acetylation contributes to the regulation of p53-p300 complex formation. A, HEK 293 cells were transfected with plasmids expressing wild-type p53, evenly split at 24 h posttransfection, and harvested 36 h after transfection. Immediately before harvest, cells were treated with DMSO (none), TSA (500 nmol/L), or NaB (5 mmol/L) for 3 h. Whole-cell lysates were prepared, separated directly on a SDS-PAGE gel, or subjected to immunoprecipitation (IP) with anti-p53, anti-E1A, or anti-p300 antibodies. Immunoblots were detected with anti-p300 antibody. B, plasmids expressing either wild-type p53 or p53K382L were transfected into HEK 293 cells. Thirty-six hours after transfection, cells were split evenly and treated with either DMSO or TSA (500 nmol/L) for 3 h. Whole-cell lysates (WCL) were prepared, and immunoprecipitation was done with anti-p53 antibody followed by immunoblotting to detect copurified p300. C, HeLa cells were treated with DMSO, TSA (500 nmol/L), or NaB (5 mmol/L) for 3 h before harvest and lysate preparation. GST-p53 and GST-TFIIB were purified from E. coli and incubated with the cell lysates in pull-down assays in the presence of TSA. The pull-down complexes were resolved through SDS-PAGE, followed by immunoblotting (IB) to detect the associated p300.
Discussion
Autoacetylation of full-length p300 has been observed since the first demonstration that p300 has intrinsic HAT activity (8). Our study revealed that six lysine-rich regions of p300 and the minimal p300C-TAD are susceptible to p300HAT-catalyzed autoacetylation. We also found that HDAIs, which have been shown to be effective in repressing tumor growth and angiogenesis, are able to induce p300 hyperacetylation in cultured tumor cells. Of note, KR1, KR2, and KR6 regions are important for p300 to form complexes with its interacting partners. We propose that the acetylation status of p300 might regulate p300 complex formation with interacting partners. As a proof of principle, we provided data to show that the acetylation status of p300 contributes to its interaction with p53. In addition, the interaction enhanced by p300 acetylation may explain a recent reported observation that HDAIs induce the function of mutant p53 and cause cytotoxicity preferentially for tumor cells with overexpressed mutant p53 (46).
Particularly, an acetylation event has been reported to be repressive to the transactivation activity of HIF-α, which depends on interactions with p300CH1/KR1 for activity (15, 31, 47). Because the C-TADs of HIF-α are not acetylated by p300HAT (31), it has been proposed that direct acetylation of CH1 or other p300KRs may be involved in HDAI-induced repression of HIF function and tumor angiogenesis (31, 48). We also studied if FIH-1 is involved in acetylation-mediated repression of HIF-1 function. We previously found that HDAIs have no effects on FIH-1 levels (31). We show that recombinant FIH-1 has no HAT activity (Fig. 2B), and p300HAT does not acetylate recombinant FIH-1 (not shown), suggesting that FIH-1 is not a mediator for HDAI-induced repression of HIF-α activity.
We noticed that the HAT domain is lysine rich (KR4 and KR5) and can be acetylated in cell-free systems. The acetylation of the KR5 region was recently reported to stimulate the HAT activity (30). However, the effect of such acetylation on the HAT activity was difficult to prove in our experimental setting. Immunoprecipitated p300 from HDAI-treated and nontreated cell lysates gave similar HAT activity, even with a very short incubation (10 min; data not shown). In addition, preincubation of p300HAT expressed in E. coli with cell lysates and acetyl-CoA failed to enhance its HAT activity. One of the possible explanations might be the rapid autoacetylation of p300HAT when used in HAT assays. It was also suggested that bacterially expressed recombinant p300HAT might have been autoacetylated in bacteria (30). However, because autoacetylation of p300HAT was clearly observed in acetylation assays in cell-free systems, the recombinant p300HAT should not have been completely, if at all, acetylated in bacteria.
Although it has been well demonstrated that p300HAT acetylates itself and several other domains of p300 in cell-free systems, and that HDAIs induce hyperacetylation of p300 in cultured cells, whether the p300 acetylation in cultured cell results from autoacetylation is difficult to determine. This is because p300 forms protein complexes with multiple partners, and some interacting partners such as pCAF, TAF1, and SRC also have HAT activity. The real acetylation status of p300 may be maintained by a combination of multiple HAT enzymes and counterbalanced by deacetylases. A recent report showed the interaction between Sirt1 and p300 and showed that acetyl-Lys-1020 and acetyl-Lys-1024 at p300KR3 are substrates for Sirt1 (43), suggesting that the acetylation status of p300 may be regulated by class III HDACs. Because p300 hyperacetylation can be induced by inhibitors of class I and II HDACs (Fig. 3A; ref. 31), the involvement of class I/II HDACs in regulation of p300 acetylation status is also suggested. The report of HDAC1 interacting directly with the p300KR6 region supports this possibility (38).
HDAIs are currently in clinical trials for cancers, neurodegenerative diseases, and hematologic disorders (17, 33–35). Our study showed that HDAIs induce hyperacetylation of p300 in cultured tumor cells. This hyperacetylation may involve multiple sites, including the six KR regions. Because these KR regions are essential for multiple functions, particularly for interaction with diverse proteins, alteration of the acetylation status of these regions may have complicated biological consequences. The diversity of p300 functions and the effects of HDAI-based therapies on p300/CBP functions suggest that further investigation of the HDAI effects on p300/CBP acetylation is essential to understand the molecular and biochemical mechanisms underlying the pharmacologic effects of HDAIs.
Supplementary Material
Acknowledgments
Grant support: National Cancer Institute, NIH, grant CA098809 and the W.W. Smith Charitable Trust C0505 (N. Sang).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Drs. J. Caro and R.H. Wenger for helpful discussion and critical review of the manuscript before submission; and Drs. A. Giordano, GL. Sanenza, P.L. Puri, S.L. Berger, and S. Bhattacharya for providing reagents for this research. N. Sang thanks Drs. A. Giordano and J. Caro the mentorship during his early career.
Footnotes
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–77. [PubMed] [Google Scholar]
- 2.Semenza GL. Physiology meets biophysics: visualizing the interaction of hypoxia-inducible factor 1a with p300 and CBP. Proc Natl Acad Sci U S A. 2002;99:11570–2. doi: 10.1073/pnas.192442299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arany Z, Huang LE, Eckner R, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci U S A. 1996;93:12969–73. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharya S, Michels CL, Leung MK, et al. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan W, Condorelli G, Caruso M, et al. Human p300 protein is a coactivator for the transcription factor MyoD. J Biol Chem. 1996;271:9009–13. doi: 10.1074/jbc.271.15.9009. [DOI] [PubMed] [Google Scholar]
- 6.Lee JS, Zhang X, Shi Y. Differential interactions of the CREB/ATF family of transcription factors with p300 and adenovirus E1A. J Biol Chem. 1996;271:17666–74. [PubMed] [Google Scholar]
- 7.Swope DL, Mueller CL, Chrivia JC. CREB-binding protein activates transcription through multiple domains. J Biol Chem. 1996;271:28138–45. doi: 10.1074/jbc.271.45.28138. [DOI] [PubMed] [Google Scholar]
- 8.Ogryzko VV, Schiltz RL, Russanova V, et al. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–9. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 9.Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–3. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 10.Puri PL, Avantaggiati ML, Balsano C, et al. p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J. 1997;16:369–83. doi: 10.1093/emboj/16.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sartorelli V, Huang J, Hamamori Y, et al. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol. 1997;17:1010–26. doi: 10.1128/mcb.17.2.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckner R, Yao TP, Oldread E, et al. Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes Dev. 1996;10:2478–90. doi: 10.1101/gad.10.19.2478. [DOI] [PubMed] [Google Scholar]
- 13.Yuan ZM, Huang Y, Ishiko T, et al. Role for p300 in stabilization of p53 in the response to DNA damage. J Biol Chem. 1999;274:1883–6. doi: 10.1074/jbc.274.4.1883. [DOI] [PubMed] [Google Scholar]
- 14.Lando D, Peet DJ, Whelan DA, et al. Asparagine hydroxylation of the HIF transactivation domain: a hypoxic switch. Science. 2002;295:858–61. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 15.Sang N, Fang J, Srinivas V, et al. Carboxyl-terminal transactivation activity of hypoxia-inducible factor 1 a is governed by a von Hippel-Lindau protein-independent, hydroxylation-regulated association with p300/CBP. Mol Cell Biol. 2002;22:2984–92. doi: 10.1128/MCB.22.9.2984-2992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chrivia JC, Kwok RP, Lamb N, et al. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–9. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 17.Ou YH, Chung PH, Sun TP, et al. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol Biol Cell. 2005;16:1684–95. doi: 10.1091/mbc.E04-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammond EM, Mandell DJ, Salim A, et al. Genome-wide analysis of p53 under hypoxic conditions. Mol Cell Biol. 2006;26:3492–504. doi: 10.1128/MCB.26.9.3492-3504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1a and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–86. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sang N, Stiehl DP, Bohensky J, et al. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem. 2003;278:14013–9. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu W, Chen H, Du K, et al. A transcriptional switch mediated by cofactor methylation. Science. 2001;294:2507–11. doi: 10.1126/science.1065961. [DOI] [PubMed] [Google Scholar]
- 22.Gay F, Calvo D, Lo MC, et al. Acetylation regulates subcellular localization of the Wnt signaling nuclear effector POP-1. Genes Dev. 2003;17:717–22. doi: 10.1101/gad.1042403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soutoglou E, Katrakili N, Talianidis I. Acetylation regulates transcription factor activity at multiple levels. Mol Cell. 2000;5:745–51. doi: 10.1016/s1097-2765(00)80253-1. [DOI] [PubMed] [Google Scholar]
- 24.Ng HH, Bird A. Histone deacetylases: silencers for hire. Trends Biochem Sci. 2000;25:121–6. doi: 10.1016/s0968-0004(00)01551-6. [DOI] [PubMed] [Google Scholar]
- 25.Verdin E, Dequiedt F, Kasler HG. Class II histone deacetylases: versatile regulators. Trends Genet. 2003;19:286–93. doi: 10.1016/S0168-9525(03)00073-8. [DOI] [PubMed] [Google Scholar]
- 26.Yang XJ, Gregoire S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol. 2005;25:2873–84. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. 2003;13:143–53. doi: 10.1016/s0959-437x(03)00015-7. [DOI] [PubMed] [Google Scholar]
- 28.Sakaguchi K, Herrera JE, Saito S, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–41. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraus WL, Manning ET, Kadonaga JT. Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol. 1999;19:8123–35. doi: 10.1128/mcb.19.12.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson PR, Wang D, Wang L, et al. Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol. 2004;11:308–15. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- 31.Fath DM, Kong X, Liang D, et al. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-a. J Biol Chem. 2006;281:13612–9. doi: 10.1074/jbc.M600456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marks PA, Richon VM, Breslow R, et al. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13:477–83. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 33.Sadri-Vakili G, Cha JH. Histone deacetylase inhibitors: a novel therapeutic approach to Huntington's disease (complex mechanism of neuronal death). Curr Alzheimer Res. 2006;3:403–8. doi: 10.2174/156720506778249407. [DOI] [PubMed] [Google Scholar]
- 34.Sartorelli V, Puri PL. The link between chromatin structure, protein acetylation and cellular differentiation. Front Biosci. 2001;6:D1024–47. doi: 10.2741/sartorel. [DOI] [PubMed] [Google Scholar]
- 35.Drummond DC, Marx C, Guo Z, et al. Enhanced pharmacodynamic and antitumor properties of a histone deacetylase inhibitor encapsulated in liposomes or ErbB2-targeted immunoliposomes. Clin Cancer Res. 2005;11:3392–401. doi: 10.1158/1078-0432.CCR-04-2445. [DOI] [PubMed] [Google Scholar]
- 36.Demidenko ZN, Rapisarda A, Garayoa M, et al. Accumulation of hypoxia-inducible factor-1a is limited by transcription-dependent depletion. Oncogene. 2005;24:4829–38. doi: 10.1038/sj.onc.1208636. [DOI] [PubMed] [Google Scholar]
- 37.Sang N, Avantaggiati ML, Giordano A. Roles of p300, pocket proteins, and hTBP in E1A-mediated transcriptional regulation and inhibition of p53 transactivation activity. J Cell Biochem. 1997;66:277–85. doi: 10.1002/(sici)1097-4644(19970901)66:3<277::aid-jcb1>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 38.Simone C, Stiegler P, Forcales SV, et al. Deacetylase recruitment by the C/H3 domain of the acetyltransferase p300. Oncogene. 2004;23:2177–87. doi: 10.1038/sj.onc.1207327. [DOI] [PubMed] [Google Scholar]
- 39.Sang N, Giordano A. Extreme N terminus of E1A oncoprotein specifically associates with a new set of cellular proteins. J Cell Physiol. 1997;170:182–91. doi: 10.1002/(SICI)1097-4652(199702)170:2<182::AID-JCP10>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 40.Sang N, Severino A, Russo P, et al. RACK1 interacts with E1A and rescues E1A-induced yeast growth inhibition and mammalian cell apoptosis. J Biol Chem. 2001;276:27026–33. doi: 10.1074/jbc.M010346200. [DOI] [PubMed] [Google Scholar]
- 41.Kim JH, Cho EJ, Kim ST, et al. CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat Struct Mol Biol. 2005;12:423–8. doi: 10.1038/nsmb924. [DOI] [PubMed] [Google Scholar]
- 42.Santoso B, Kadonaga JT. Reconstitution of chromatin transcription with purified components reveals a chromatin-specific repressive activity of p300. Nat Struct Mol Biol. 2006;13:131–9. doi: 10.1038/nsmb1048. [DOI] [PubMed] [Google Scholar]
- 43.Bouras T, Fu M, Sauve AA, et al. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem. 2005;280:10264–76. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- 44.Liu L, Scolnick DM, Trievel RC, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19:1202–9. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barlev NA, Liu L, Chehab NH, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243–54. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- 46.Blagosklonny MV, Trostel S, Kayastha G, et al. Depletion of mutant p53 and cytotoxicity of histone deacetylase inhibitors. Cancer Res. 2005;65:7386–92. doi: 10.1158/0008-5472.CAN-04-3433. [DOI] [PubMed] [Google Scholar]
- 47.Kung AL, Wang S, Klco JM, et al. Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat Med. 2000;6:1335–40. doi: 10.1038/82146. [DOI] [PubMed] [Google Scholar]
- 48.Liang D, Kong X, Sang N. Effects of histone deacetylase inhibitors on HIF-1. Cell Cycle. 2006;5:2430–5. doi: 10.4161/cc.5.21.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.