

ABSTRACT
The initiating events in autoimmune disease remain to be completely understood, but it is thought that genetic predisposition synergizes with “environmental” factors, including viral infection, leading to disease. One elegant animal model used to study the pathogenesis of multiple sclerosis that perfectly blends genetics and environmental components in the context of virus-induced autoimmunity is Theiler’s murine encephalitis virus-induced demyelinating disease (TMEV-IDD). TMEV-infected disease-susceptible SJL/J mice develop a persistent central nervous system (CNS) infection and later develop autoimmune demyelination, while disease-resistant C57BL/6 (B6) mice rapidly clear the infection and develop no autoimmune pathology. Mice of the (B6 × SJL/J)F1 cross between these two mouse strains are classified as intermediately susceptible. We employed this model to investigate if rapid virus clearance in B6 versus SJL/J mice was perhaps related to differences in the innate immune response in the CNS of the two strains in the first few days following intracerebral virus inoculation. Here we show that SJL/J mice lack, in addition to NK cells, a novel innate immune subset known as natural killer dendritic cells (NKDCs), which express phenotypic markers (CD11cint NK1.1+) and functional activity of both NK cells and DCs. These NKDCs are activated in the periphery and migrate into the infected CNS in a very late antigen 4 (VLA-4)-dependent fashion. Most significantly, NKDCs are critical for CNS clearance of TMEV, as transfer of NKDCs purified from B6 mice into TMEV-IDD-susceptible (B6 × SJL/J)F1 mice promotes viral clearance. Together the findings of this work show for the first time a link between NKDCs, viral infection, and CNS autoimmunity.
IMPORTANCE
Viral infection is an important cofactor, along with genetic susceptibility, in the initiation of a variety of organ-specific autoimmune diseases. Thus, in-depth understanding of how virus infections trigger autoimmunity may lead to novel ways to prevent or treat these diseases. Theiler’s murine encephalitis virus-induced demyelinating disease (TMEV-IDD) serves as an important model for the human T cell-mediated autoimmune demyelinating disease multiple sclerosis. Induction of TMEV-IDD is genetically controlled as SJL/J mice develop persistent central nervous system (CNS) infection leading to chronic autoimmune demyelination, while C57BL/6 mice rapidly clear virus and are disease resistant. We determined that, as opposed to resistant B6 mice, disease-susceptible SJL/J mice lacked a unique innate immune population, the natural killer dendritic cell (NKDC), which was shown to play a critical role in early CNS virus clearance via its ability to both present virus antigen to T cells and to lyse target cells.
INTRODUCTION
The underlying pathogenesis of autoimmune disease remains to be completely understood. While there is a strong genetic correlation (1, 2), genetics alone cannot completely explain the prevalence of autoimmunity. It is therefore thought that genetic predisposition combines with other “environmental” factors, including viral infection, which together culminate in disease initiation (3). There are numerous examples whereby infection correlates with autoimmune disease development. For example, development of multiple sclerosis (MS) is linked to previous infection with Epstein-Barr virus (EBV) (4) or human herpesvirus 6 (HHV-6) (5).
As the incidence of autoimmune disease continues to increase, there is a dire need to better understand the connection between viral infection and autoimmune disease development. One elegant model used to study MS-like pathogenesis that perfectly blends genetics and environmental components in the context of virus-induced autoimmunity is Theiler’s murine encephalitis virus-induced demyelinating disease (TMEV-IDD). Interestingly, as observed in humans, the transition from acute viral infection to chronic autoimmunity hinges on the genetic profile of the mouse strain infected and is linked to H-2 major histocompatibility complex class I (MHC-I) genes, specifically the H-2D locus (6–8). For example, infection of the susceptible H2-Ds SJL/J strain leads to the development of symptomatic TMEV-IDD (9, 10), while H2-Db C57BL/6 (B6) mice clear the infection before developing demyelination (7).
In SJL/J mice, infection with TMEV results in a chronic infection of the central nervous system (CNS). The establishment of chronic infection is a prerequisite for the transition from an immune response that is strictly antiviral in nature to one that involves pathological anti-myelin-specific autoimmune responses (11), a phenomenon known as epitope spreading (12). A number of studies have attempted to address the key differentiating factors involved in viral clearance, and thus the underlying factors that determine resistance versus susceptibility to chronic TMEV-IDD. The contrasting outcomes in these strains of mice have been correlated to the highly efficient antiviral capsid cytotoxic T cell (CTL) response seen in the B6 mice (13, 14). However, this remains controversial as independent studies have shown that SJL/J mice are capable of eliciting potent antiviral CTL responses (15), although CTL responses in the susceptible SJL/J mouse strain have been shown to be blunted by overabundant activation of CNS-resident regulatory T cells (Tregs) (16).
Despite previously described differences in CTL and Treg function between TMEV-IDD-susceptible SJL/J and TMEV-IDD-resistant B6 mice, potential differences in innate immune responses related to disease susceptibility have not been carefully examined. Biologically speaking, the primary roles for innate immunity include sensing pathogens, limiting viral expansion and spread, and finally stimulating effective adaptive immune responses. Within the sphere of innate immunity, we were particularly interested in investigating the potential differences in antigen-presenting cell (APC) function following TMEV infection. APCs, especially dendritic cells (DCs), are a critical bridge between the innate and adaptive immune responses and are the primary cells involved in sensing danger signals and collecting and presenting pathogen-associated antigens for initiation of appropriate adaptive immune responses. DCs have been shown to be crucial for virus-specific CTL priming, as mice that lack DCs are incapable of mounting a CTL response to lymphocytic choriomeningitis virus (LCMV) infection (17). In addition, a growing body of evidence indicates that specialized DC subsets can dictate the skewing of the adaptive response. Specifically in the context of autoimmunity, we have demonstrated that myeloid DCs of peripheral origin induce activation of inflammatory autoreactive Th17 cells in the CNS of SJL/J mice with experimental autoimmune encephalitis (EAE) (18). In contrast, we have shown that plasmacytoid DCs (pDCs) serve as a regulatory subset and limit the severity of EAE (19). Thus, we hypothesized that strain-linked differences between DC subsets found in the CNS during acute TMEV infection may contribute to outcome of viral clearance or lack thereof.
Here we show that SJL/J mice lack a novel innate immune cell subset, known as natural killer dendritic cells (NKDCs), that express phenotypic markers and functional activity of both NK cells and DCs (20). These peripherally derived cells penetrate the CNS of TMEV-infected B6 mice and are critical for viral clearance. Most importantly, infusion of NKDCs purified from B6 mice into the TMEV-IDD-susceptible (SJL/J × B6)F1 mice results in significant levels of viral clearance. Together this work shows for the first time a link between NKDCs, viral infection, and CNS autoimmunity.
RESULTS
Accumulation of a unique NK-like APC population in the brains of TMEV-infected B6 mice.
Viral titers within the brains of both susceptible SJL/J and resistant B6 mice peak at day 5 following intracerebral (i.c.) infection and result in a potent adaptive systemic immune response in both strains peaking 7 to 10 days postinfection (p.i.) (21). Given these kinetics, we examined the phenotype and numbers of various potential CD11c+ DC subsets that appeared within the CNS during the acute infection period. We found that both CD45+ CD11c+ Ly6c+ CD11b− plasmacytoid DCs and CD45+ CD11c+ Ly6c+ CD11bint/lo inflammatory monocytes infiltrated the CNS of both TMEV-infected SJL/J and B6 mice (Fig. 1A). Both of these DC subsets have been shown to be participants during CNS virus infection (22, 23). We were somewhat surprised, however, that there was no significant difference in the number of pDCs or inflammatory monocytes between infected B6 and SJL/J animals.
FIG 1 .

CNS viral infection results in accumulation of NK1.1-expressing dendritic cells in the CNS of TMEV-IDD-resistant B6 mice. Wild-type B6 and SJL/J mice were i.c. infected with 5 × 106 PFU of TMEV or sham infected. Brains were harvested during acute infection, and mononuclear cells were isolated and stained for flow cytometry. (A) Live CD45+ CD11c+ cells were analyzed for expression of Ly6c and CD11b. Representative flow plots from day 5 post-TMEV infection are shown. (B) Numbers of cells in the CNS expressing Ly6C and CD11b on days 3, 5, and 7 p.i. in TMEV- or sham-infected B6 and SJL/J mice are shown. (C) Live CD3− CD11c+ cells were analyzed for their expression of NK1.1 and DX5. Representative flow plots from day 3 post-TMEV infection of B6 and SJL/J mice are shown. (D) Quantification of CD3− CD11c+ DX5+ cell numbers on days 1, 2, and 3 p.i. in TMEV- or sham-infected B6 and SJL/J mice. (E) Quantification of CD3− CD45+ CD11c+ NK1.1+ DX5+ brain cells on days 1, 3, 5, and 7 p.i. in TMEV- or sham-infected B6 mice are shown. (F) Quantification of total CNS lymphocytes and NKDCs in sham-infected (black bars), UV-inactivated TMEV-infected (hatched bars), or WT TMEV-infected (black bars) B6 mice 3 days p.i. Data are representative of 2 to 3 independent experiments with 3 to 5 mice per group. Error bars show standard deviations. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
In contrast, a subset expressing the CD45+ CD11c+ Ly6C− CD11blo phenotype was found in 10-fold-greater numbers in the brains of infected B6 compared to SJL/J mice at days 3 to 7 p.i. (Fig. 1B; see Fig. S1 in the supplemental material). Upon further investigation, we found that CD45+ CD11c+ CD11blo DCs coexpressed proteins associated with NK cells, including DX5 and NK1.1, a phenotype akin to the recently described natural killer dendritic cells (NKDCs) (Fig. 1C). While very few of these cells were observed in SJL/J mice, within 48 h p.i., a significant accumulation of CD45+ CD11cint CD11blo NK1.1+ DX5+ NKDCs had occurred in the brains of infected B6 mice (Fig. 1D). Further temporal analysis showed that NKDCs comprised as much as 6% of the total leukocyte population, their numbers peaking between days 3 and 5 p.i. (Fig. 1E). In addition, CNS replicating virus was required for accumulation of NKDCs in the CNS of B6 animals as there was no increase in this population in the CNS of mice in mice receiving UV-inactivated TMEV (Fig. 1F). Collectively, these data support the hypothesis that differences in NKDC populations between B6 and SJL/J animals may be a key factor in determining resistance (i.e., viral clearance) versus susceptibility (i.e., chronic infection) and that the NKDCs are of peripheral origin.
NKDCs derive from a splenic precursor that migrates from the periphery into the infected brain in a VLA-4-dependent manner.
In the TMEV-IDD model, piercing of the blood-brain barrier (BBB) results from i.c. virus inoculation and results in peripheral viremia. We were thus interested in assessing how TMEV infection might influence peripherally located NKDCs. Activation was studied by examining proliferation of NKDCs as well as upregulation of key molecules associated with DC activation. Examination of NKDC kinetics in the spleen showed that the number and percentage of NKDCs in the spleen peaked at day 3 and returned to baseline levels at day 5 p.i. (Fig. 2A). Employing bromodeoxyuridine (BrdU) incorporation, we interestingly found that NKDCs, but not DCs or NK cells, underwent significant proliferation after virus infection (Fig. 2B; see Fig. S2 in the supplemental material), suggesting that the increase in the numbers of NKDCs in the spleen was due to active replication. Infection-induced NKDC activation was evidenced by upregulation of molecules associated with antiviral immunity, including MHC-II (I-Ab), CD40, CD80, and CD86. Interestingly, significant upregulation of these molecules was observed within 24 h p.i. compared to sham-infected animals (Fig. 2C and D). MHC class II expression was seen to kinetically increase on NKDCs in the CNS as well (not shown).
FIG 2 .

CNS TMEV infection induces proliferation and activation of splenic NKDCs. Wild-type B6 mice were i.c. infected with 5 × 106 PFU of TMEV or sham infected. Spleens were harvested during acute infection, and mononuclear cells were isolated and analyzed by flow cytometry. (A) Quantification of CD3− CD45+ CD11c+ NK1.1+ DX5+ spleen cells on days 1, 3, 5, and 7 p.i. in TMEV- or sham-infected B6 mice. (B) At 24 and 48 h p.i., mice were i.p. injected with 1 mg BrdU in 100 µl sterile PBS. At 96 h p.i., the percentage of BrdU-positive NKDCs from spleens of TMEV- or sham-infected mice was quantified. (C) Spleens were harvested from B6 mice at 24 and 72 h post-TMEV infection, and expression levels of I-Ab, CD40, CD86, and CD80 on CD3− CD45+ CD11c+ NK1.1+ DX5+ spleen cells were determined. (D) Quantification of I-Ab-, CD40-, CD86-, and CD80-positive CD3− CD45+ CD11c+ NK1.1+ DX5+ spleen cells on days 24 and 72 h p.i. in TMEV- or sham-infected B6 mice. For histogram overlays, the shaded portion represents the isotype control, and the black line represents antibody staining. Data are representative of 2 to 3 independent experiments with 5 mice per group. Error bars show standard deviations. **, P < 0.01; *, P < 0.05.
Given that accumulation of NKDCs in the CNS correlated with activation and proliferation of splenic NKDCs, we hypothesized that the spleen may serve as reservoir of NKDCs, which upon infection migrate out of the spleen and into the brain. To address this hypothesis, congenic B6 CD45.1 bulk splenocytes were adoptively transferred to wild-type (WT) CD45.2 B6 mice. After 2 days, mice were i.c. infected with TMEV, and then brains and spleens were analyzed on day 3 p.i. (Fig. 3A). We found that there was no difference in trafficking of CD45.1 NKDCs to the spleens of either sham- or TMEV-infected animals. However, NKDCs from the donor spleen only migrated to the brain in TMEV-infected animals (Fig. 3B and C). Interestingly, though the percentage of transferred cells was only a small percentage of CNS leukocytes, NKDCs made up a significant proportion of the transferred cells. These data highlight the fact that splenic NKDCs are capable of trafficking to the CNS after infection and are preferentially recruited during infection.
FIG 3 .

Peripheral NKDCs traffic to the CNS after TMEV infection in a VLA-4-dependent manner. (A) Scheme of adoptive transfer experimental design. Congenic B6 CD45.1 splenocytes were transferred intravenously (i.v.) into CD45.2 WT B6 mice 2 days prior to i.c. TMEV infection. Brains and spleens were harvested 3 days p.i. and analyzed for the presence of CD45.1 donor cells. (B) Representative flow plots of CD11c and NK1.1 expression on CD45.1+ cells in the brain and spleen of TMEV- or sham-infected mice. (C) The percentage of transferred donor CD45.1+ cells and CD45.1+ CD11c+ NK1.1+ is quantitated. (D) Quantification of total CNS lymphocytes and NKDCs (CD3− CD45+ CD11c+ NK1.1+ DX5+) 3 days p.i. in TMEV-infected B6 mice treated with the isotype control (black bars) or anti-VLA-4 (white bars) 6 h prior to infection. Data are representative of 2 independent experiments with 5 mice per group. Error bars show standard deviations. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
The lack of NKDCs in the CNS of normal mice suggests that NKDCs likely derive from a circulating population; however, it is also possible that TMEV infection may induce the differentiation of a CNS-resident CD11c population to express NK cell-associated proteins. To attempt to differentiate between these two possibilities, we treated B6 mice with 500 µg of antibody specific for very late antigen 4 (VLA-4) to inhibit leukocyte trafficking into the brain 6 h prior to TMEV infection (24). VLA-4 inhibition resulted in a broad reduction in the total number leukocytes in the brain at 3 days p.i., with the number of NKDCs being significantly reduced compared to IgG control antibody (Ab) treatment (Fig. 3D). Collectively, these data strongly suggest that NKDCs are recruited from the periphery during TMEV infection and do not differentiate from local CD11c-expressing populations.
NKDCs act as both DCs and cytolytic NK cells.
Based on both the accumulation of NKDCs in the CNS and their upregulation of classical APC molecules following TMEV infection, we hypothesized that NKDCs may functionally serve as APCs, playing a potential role in the initiation of the antiviral adaptive immune response. To assess their APC capacity in comparison to conventional DCs, fluorescence-activated cell sorter (FACS)-sorted NKDCs (CD11c+ NK1.1+) and DCs (CD11c+ NK1.1−) from the spleens of B6 animals 24 h post-TMEV infection were cocultured with carboxyfluorescein succinimidyl ester (CFSE)-labeled T cells isolated from naive B6 animals primed with either VP425-38 or VP2121-130, which are the dominant TMEV CD4+ and CD8+ T cell epitopes, respectively. While DCs were considerably more efficient at inducing proliferation of both CD4+ and CD8+ T cells, NKDCs were still however able to induce T cell proliferation, as assessed by CFSE dilution (Fig. 4A). In terms of activation marker (CD44 and CD69) expression on the CD4+ and CD8+ T cells (Fig. 4B), there was a significant background in the cultures lacking peptide as the cells came from primed mice and the DCs and NKDCs likely were presenting endogenous viral peptides as they were purified from TMEV-infected B6 mice. Upregulation of CD44 on CD4+ T cells cocultured with VP425-28 was observed with both NKDCs (60%) and DCs (64%) as stimulators. However, there was no CD44hi population in cultures with NKDCs. Interestingly, the percentages of CD69+ CD4+ T cells were greater after culture with NKDCS (62.5%) versus DCs (50.8%). In both cases, most of the CD69+ CD4+ T cells were also CD44+.
FIG 4 .

TMEV-activated NKDCs are poor APCs but efficient killers. FACS-purified NK cells, DCs, and NKDCs from TMEV-infected B6 mice were tested for APCs and killing function cocultured with target cells (T cells or YAC-1 cells). (A and B) Splenic DCs and NKDCs were FACS sorted from B6 mice 24 h post-TMEV infection and cocultured at a 1:1 ratio with CFSE-labeled pan-splenic T cells from VP425-38- or VP2121-130-primed mice ± 2 µM cognate peptide. Seventy-two hours later, CD3+ T cells were analyzed for proliferation via CFSE dilution (A) and activation marker expression via expression of CD44 and CD69 (B). (C) YAC-1 target cells were cocultured with FACS-purified NK cells, DCs, and NKDCs from TMEV-infected B6 mice at the indicated effector-to-target ratios for 4 h and assayed for LDH release as a marker of cytotoxicity. Data are representative of 3 independent experiments.
The most notable differences in T cell activation by NKDCs versus DCs were in cultures stimulated with the CD8 cognate VP2121-130 peptide. In cultures where NKDCs were used as stimulators, the majority of the T cells did not upregulate either activation marker compared to the control with no peptide added at the time point assayed. DCs, on the other hand, were quite capable of inducing upregulation of activation markers on peptide-stimulated CD8+ T cells. Twenty-five percent were CD69+ CD44lo, and 28% were single positive for CD69. Overall the results indicate that NKDCs are capable of reactivating peptide-specific T cells, CD4+ more efficiently than CD8+, although to a significantly lesser extent than DCs. Thus, presentation of antigen is a legitimate function that NKDCs could be eliciting in the context of infection.
We next asked if NKDCs from TMEV-infected mice were able to kill a classic NK-susceptible target cell. In order to evade detection by the immune system, viruses have evolved mechanisms to downregulate MHC class I, which then allows NK cells to carry out cellular killing via the NKG2D activating receptor (25). Therefore, we compared the efficiencies of killing by NKDCs compared to those of both NK cells and DCs. FACS-sorted splenic NKDCs, NK cells, and DCs from TMEV-infected B6 mice were cocultured with YAC-1 cells in a classical NK killing assay. At higher effector-to-target cell (E:T) ratios, TMEV-activated NKDCs were able to kill YAC-1 target cells slightly more efficiently than NK cells, whereas no killing was seen from DCs (Fig. 4C).
Repopulation of TMEV-susceptible (SJL/J × B6)F1 mice with B6 NKDCs induces CNS virus clearance.
While the data to this point suggest that NKDCs function similarly to NK cells after infection, the limitations of the in vitro assays employed do not allow us to determine their importance in limiting viral titers. However, because they possess no single unique phenotypic surface marker, specific depletion of NKDCs is not possible. In addition, attempted transfer of B6 NKDCs into TMEV-susceptible SJL/J mice is not possible due to MHC incompatibility. Therefore, we turned to the (B6 × SJL/J)F1 model of mice that are moderately susceptible to induction of TMEV-IDD. Due to their susceptibility to TMEV-IDD, we hypothesized that these F1 mice may also exhibit a deficiency in NKDC expression.
Interestingly, analysis of the spleens of F1 mice showed that unlike SJL/J mice, they had a large population of NK cells (data not shown). Upon examination of their NKDC population, we observed they possessed significantly fewer NKDCs than B6 animals and also expressed much lower levels of NK1.1 (Fig. 5A). We thus hypothesized that F1 NKDCs were not as effective as B6 NKDCs given these constraints. In order to address our hypothesis, we tested if infusion of purified NKDCs from B6 donors would lead to CNS virus clearance in (B6 × SJL/J)F1 mice infected with TMEV. Viral titers were quantified using plaque assay on brain homogenates from day 7-infected F1 mice. Strikingly, F1 mice that had received an infusion of B6 NKDCs prior to infection had significantly less virus than noninfused F1 infected mice (Fig. 5B). These data therefore indicate that strain-specific generation of NKDCs is a critical factor in determining the capacity to effectively clear TMEV from the CNS.
FIG 5 .
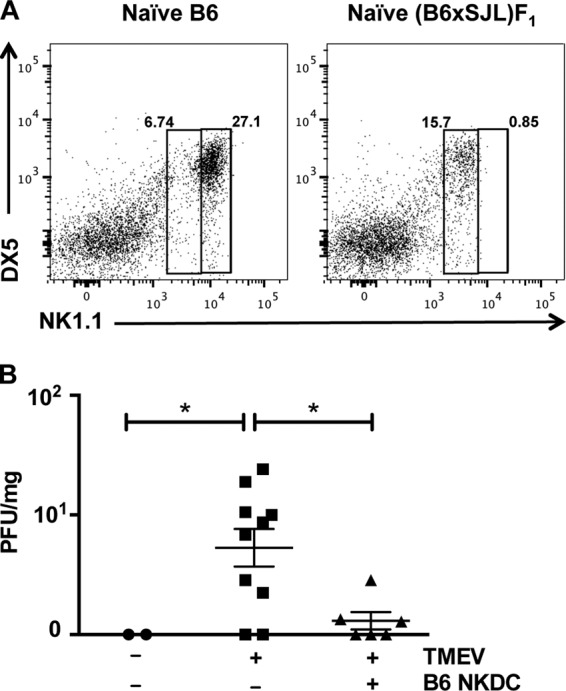
Repopulation of TMEV-susceptible (B6 × SJL/J)F1 mice with B6 NKDCs induces CNS virus clearance. Spleens from naive WT (B6 × SJL/J)F1 mice were examined for the presence of NKDCs in comparison to WT B6 mice. (A) CD45+ CD11c+ cells were analyzed for expression of NK1.1 and DX5. (B) Six to 10 (B6 × SJL/J)F1 mice per group with our without adoptively transferred FACS-purified NKDCs from WT B6 mice were infected with TMEV or sham infected. The number of PFU in brain tissue was determined by plaque assay. *, P < 0.05.
DISCUSSION
A large body of epidemiological evidence indicates that both genetics and environmental factors contribute to MS susceptibility (26). We hypothesized that differences in DCs involved in the very early aspects of CNS virus clearance could represent a factor that predicted disease outcome in the TMEV-IDD model of MS. To test this hypothesis, we employed the TMEV mouse model, which seamlessly blends both genetic and environmental factors for initiation of a late-phase autoimmune demyelinating disease (12). We demonstrated, for the first time, that peripheral NKDCs are recruited to the brain in TMEV-IDD-resistant B6 mice but are lacking in TMEV-susceptible SJL/J mice after CNS TMEV infection. Therefore, NKDCs are a strain-specific DC subset that plays a critical role in virus clearance and thus in inhibiting development of autoimmunity.
The experiments that led us to pursue the use of unconventional phenotypic markers, revolved around our observation that the Ly6c− CD11bint/lo DC population was present in high numbers and percentages in the CNS of TMEV-infected B6 mice. The unlikely possibility existed that these were merely tissue-resident monocytes, which proliferated and differentiated as a result of virus-induced inflammatory signals. The largest monocyte/macrophage-like population in the CNS is microglia, and we have previously shown that they do not proliferate to a great extent after TMEV infection (27). Therefore, we concluded that the Ly6c− CD11bint/lo population represented a novel infiltrating population. Examination of expression of the phenotypic markers B220 and CD8α did not help to precisely identify this subset (unpublished observations); therefore, we explored several nontraditional markers to help define it.
NK-associated cell surface markers have been recently described to be present on a subset of CD11c+ DCs, known as NKDCs (20, 28). While this subset has been defined using a myriad of markers, the gross characterization consists of CD11cint/+ cells that also express NK1.1 and DX5. Given that SJL/J mice are NK cell deficient (29), we reasoned that this DC population accumulating in the CNS of TMEV-infected B6 but not SJL/J mice may consist of NKDCs. While further studies are needed, temporal splenic NKDC cell numbers and activation kinetics suggest that initial TMEV exposure occurs within the spleen, culminating in activation and subsequent splenic egress within 24 to 48 h. Indeed, we showed that the numbers of NKDCs increased selectively in the CNS of TMEV-infected B6 mice migrating from the peripheral immune system in a VLA-4-dependent manner. In addition, we observed that NKDC numbers peak by day 5 and are diminished by day 7. Interestingly, day 5 represents the peak viral load in the CNS as measured by TMEV RNA transcripts (21). This is also before the CTL response occurs in B6 animals (30), bolstering the idea that NKDCs contribute significantly toward viral clearance and resistance to TMEV-IDD.
We also observed that NKDC accumulation in the CNS required live, replicating virus, indicating that activation of the innate immune system by pathogen-associated molecular patterns (PAMPs) and Toll-like receptors (TLRs) is important for recruitment of NKDCs to the CNS and the ultimate clearance of TMEV. In fact, we have observed B6 TLR3-deficient mice develop a mild form of TMEV-IDD and harbor titers of persistent virus (D. Duncan and S. D. Miller, unpublished data). Although Jin et al. reported no clinical symptoms when they infected TLR3 knockout (KO) mice, they did observe increased levels of virus in the CNS mice (KO) mice, indicating the importance of TLR3 in limiting viral replication and persistence (31). NKDCs have been reported to respond to TLR9 (32), and therefore it is likely that they are primarily activated by virus via activation of TLRs 3, 7, 8, and 9.
As NKDCs are not found in the CNS of naive or sham-infected animals, it is likely that they infiltrate the infected CNS from peripheral sources. We found that peripheral stores of NKDCs are present in the spleen, lymph nodes, and bone marrow (see Fig. S3 in the supplemental material). As they constitute 1 to 2% of splenic cells, they represent a unique and significant DC population. Interestingly, we saw that after CNS TMEV infection, splenic NKDCs but not DCs or NK cells (see Fig. S2 in the supplemental material) expanded significantly in response to peripheral virus, as confirmed by BrdU incorporation. We confirmed their ability to traffic to the CNS using adoptive transfer and blockade with anti-VLA-4. Although transferred NKDCs trafficked to the spleen in both sham- and TMEV-infected animals, strikingly NKDCs trafficked to the CNS only in TMEV-infected animals. While it is not surprising that CNS TMEV infection results in systemic viremia as a result of the break in the BBB following the needle stab, it would be interesting to examine whether NKDCs were recruited to the CNS in the natural setting of infection (i.e., via retrograde transport of virus to the CNS from the gut following TMEV infection).
Consistent with the published literature regarding NKDC function (32–35), we showed that NKDCs from TMEV-infected B6 mice were capable of activating both TMEV-specific CD4+ and CD8+ T cells, although to a lesser extent than traditional CD11chi DCs, and also quite capable of killing YAC-1 cells, a conventional NK cell target that lacks MHC-I expression. Both of these functions (i.e., presentation of virus antigens to CD4+ and CD8+ T cells and killing of virus-infected cells that have downregulated MHC-I) would contribute to the antiviral effects of NKDCs in the B6 strain leading to early virus clearance.
Our results demonstrate for the first time that NKDCs are required for CNS virus clearance in TMEV-IDD-susceptible (B6 × SJL/J)F1 mice. These mice are intermediately susceptibility to development of the chronic autoimmune phase of TMEV-IDD compared to the highly susceptible SJL/J strain. Interestingly, F1 mice have a population of CD11cint DCs that coexpress NK1.1 at levels much reduced compared to those of TMEV-resistant B6 mice, while SJL/J mice fail to express NK1.1 (Fig. 1). Thus, in these parental and F1 strain combinations, the expression level of NK1.1 on CD11cint DCs correlates with functional in vivo NKDC activity, as evidenced by the ability of transferred NKDCs from resistant B6 mice to lead to CNS virus clearance in (B6 × SJL/J)F1 recipients (Fig. 5) and ultimately resistance to TMEV-IDD.
These results are consistent with the idea that NKDCs in the B6 mouse CNS initially limit viral loads, allowing for the eventual clearance of virus by antigen-specific CTLs. In contrast, SJL/J mice that are deficient in NKDCs, and thus do not possess the capability to limit virus replication during the initial hours to days following TMEV infection, ultimately fail to clear virus, resulting in persistent CNS infection leading to the development of the late-phase autoimmune demyelinating disease via epitope spreading (12). Similar defects in NKDC function may play a role in strain differences observed in experimental autoimmune models, including EAE, and should also be addressed in genetically susceptible humans that develop disease secondary to a viral infection.
MATERIALS AND METHODS
Mice.
Wild-type C57BL/6 (B6) and (B6 × SJL/J)F1 mice were purchased from Jackson Laboratories, Bar Harbor, ME. Wild-type SJL/J mice were purchased from Harlan Laboratories, Indianapolis, IN. All mice were housed in the Center for Comparative Medicine, Northwestern University, Chicago, IL, under the guidelines of the Animal Care and Use Committee (ACUC).
TMEV infection.
Anesthetized mice were infected by intracranial inoculation in the right cerebral hemisphere with 5 × 106 PFU of the BeAn 8386 strain of TMEV in 30 µl of serum-free Dulbecco’s modified Eagle’s medium (DMEM). Sham infection involved a needle stab only. For UV-inactivated virus, virus was kept on ice under UV light for 30 min.
CNS lymphocyte isolation.
Mice were anesthetized with 50 mg/kg Nembutal and perfused with 30 ml of ice-cold phosphate-buffered saline (PBS) through the left ventricle. Brains were harvested and mechanically separated with the blunt plunger of a syringe and incubated in Accutase (Millipore scr005) for 30 min at 37°C. Samples were pushed through a 100-mesh stainless steel screen to create single-cell suspensions. To isolate mononuclear cells, cells were resuspended in 40% Percoll (Sigma Aldrich, St. Louis, MO) gradient and centrifuged at 1,000 × g for 30 min at room temperature.
Flow cytometry.
Cells were blocked with anti-CD16/32 (eBioscience) and then stained with 0.5 µg antibody per 106 cells. CD45 (30-F11), CD11b (M1/70), CD11c (N418), NK1.1 (PK136), DX5, B220 (RA3-6B2), CD8a (53-6.7), I-Ab (AF6-120.1), CD80 (16-10A1), CD86 (GL1), and CD45.1 (A20) were obtained from eBioscience. Samples were run on the Canto II flow cytometer with FACS Diva Software (Becton, Dickinson) and analyzed using FlowJo software (Treestar FlowJo).
VLA-4 blockade.
Mice were treated intraperitoneally (i.p.) with 500 µg control rat Ig or anti-VLA-4/CD49d antibody (BioLegend, San Diego, CA) in a total volume of 100 µl 6 h prior to infection with TMEV.
BrdU proliferation.
For BrdU labeling and staining, the fluorescein isothiocyanate (FITC) BrdU flow kit (BD Pharmingen) was used according to the manufacturer’s instructions. Briefly, animals were injected i.p. with 1 mg of BrdU at 48 and 72 h p.i. Splenocytes were harvested at 96 h, stained extracellularly for NKDC markers, and then fixed, permeabilized, and DNase treated prior to anti-BrdU staining.
NK/DC purification.
NKDCs, DCs, and NK cells were purified from TMEV-infected B6 mice. Spleens from infected mice were injected and incubated with 400 U/ml collagenase D (Fisher Scientific 50-720-3639) for 30 min at 37°C. They were then passed through a stainless steel screen to create single-cell suspensions. To enrich for populations, cells were depleted of B and T cells with complement treatment. Finally, cells were stained with phycoerythrin (PE)-labeled anti-CD11c and APC-labeled anti-NK1.1 and sorted on a FACS Aria with a purity of 99%.
T cell proliferation.
B6 mice were primed with 50 µg of the TMEV peptides VP2121–130 (FHAGSLLVFM) or VP425-38 (YSNQYQNSIDLSAS) in complete Freund’s adjuvant (CFA). After 7 days, T cells were isolated with magnetic beads using the Pan T cell isolation kit II (Miltenyi Biotec). T cells were stained with CFSE at 37°C prior to coculture with NKDCs or DCs and 2 µM peptide.
Cell-mediated cytotoxicity assay.
The CytoTox 96 nonradioactive cytotoxicity assay kit (Promega) was used to measure cytotoxicity. Briefly, YAC-1 (ATCC TIB-160) target cells were cocultured with effector cells (NKDCs, NK cells, or DCs) for 4 h at 37°C. Supernatants from cultures were removed, lactate dehydrogenase (LDH) concentrations were measured in an enzymatic reaction, resulting in visible color change of wells, and the absorbance was read on an enzyme-linked immunosorbent assay (ELISA) plate reader at 490 nm. Controls for lysis included target cells alone, effector cells alone, and maximum target lysis. The percentage of cytotoxicity was calculated using the following ratio: [(experimental − effector spontaneous − target spontaneous)/(target maximum − target spontaneous)] × 100%.
Virus plaque assay.
Brains were harvested from sham- and TMEV-infected (B6 × SJL/J)F1 mice, weighed and homogenized in plain DMEM. Homogenates were added to culture plates (Nunc, Fisher Scientific 12-565-020) of confluent BHK-21 (ATCC CCL-10) cells for 1 h at room temperature with gentle rocking. The cells were then covered with a layer of 1% Difco agar noble (Fisher Scientific) and 2× DMEM. Plates were incubated for 5 days at 34°C, at which time agar was removed, cells were fixed with methanol, and plaques were visualized by staining with crystal violet. Results are shown as PFU per milligram of brain tissue.
SUPPLEMENTAL MATERIAL
NKDC gating strategy. A typical gating strategy was used to identify NKDCs, DCs, and NK cells. After singlet selection, lymphocytes were gated on a forward scatter/side scatter (FSC/SSC) profile. CD3− cells and CD45+ cells were then selected. Finally, expression of CD11c versus NK1.1 was used to analyze NKDCs, DCs, and NK cells. Download
CNS TMEV infection induces proliferation of splenic NKDCs. Wild-type B6 mice were i.c. infected with 5 × 106 PFU of TMEV or sham infected. Spleens were harvested during acute infection, and mononuclear cells were isolated and analyzed by flow cytometry. At 24 and 48 h p.i., mice were i.p. injected with 1 mg BrdU in 100 µl sterile PBS. At 96 h p.i., the percentages of BrdU-positive NKDCs, DCs, and NK cells from spleens of TMEV- or sham-infected mice were quantified. Data are representative of 2 to 3 independent experiments with 5 mice per group. Error bars show standard deviations. **, P < 0.01. Download
Resident NKDCs across lymphoid tissues. Spleen, lymph nodes, and bone marrow from naive B6 animals were analyzed via flow cytometry, and the percentage of NKDCs (CD45+ CD11c+ NK1.1+) was quantified. Error bars represent standard deviations from n = 3 animals. Download
ACKNOWLEDGMENTS
We thank the other members of the Miller lab for helpful discussions.
This work was supported by NIH research grant NS-062365. E.M.L.C. was supported by NIH T32 AI-0007476. S.D.M. is the Judy E. Gugenheim Research Professor at Northwestern University.
Footnotes
Citation Chastain EML, Getts DR, Miller SD. 2015. Deficient natural killer dendritic cell responses underlay the induction of Theiler’s virus-induced autoimmunity. mBio 6(4):e01175-15. doi:10.1128/mBio.01175-15.
REFERENCES
- 1.Sadovnick AD, Ebers GC. 1993. Epidemiology of multiple sclerosis: a critical overview. Can J Neurol Sci 20:17–29. [DOI] [PubMed] [Google Scholar]
- 2.Sadovnick AD, Ebers GC, Dyment DA, Risch NJ. 1996. Evidence for genetic basis of multiple sclerosis. The Canadian Collaborative Study Group. Lancet 347:1728–1730. doi: 10.1016/S0140-6736(96)90807-7. [DOI] [PubMed] [Google Scholar]
- 3.Münz C, Lünemann JD, Getts MT, Miller SD. 2009. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol 9:246–258. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salvetti M, Giovannoni G, Aloisi F. 2009. Epstein-Barr virus and multiple sclerosis. Curr Opin Neurol 22:201–206. doi: 10.1097/WCO.0b013e32832b4c8d. [DOI] [PubMed] [Google Scholar]
- 5.Voumvourakis KI, Kitsos DK, Tsiodras S, Petrikkos G, Stamboulis E. 2010. Human herpesvirus 6 infection as a trigger of multiple sclerosis. Mayo Clin Proc 85:1023–1030. doi: 10.4065/mcp.2010.0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin X, Pease LR, Rodriguez M. 1997. Differential generation of class I H-2D- versus H-2K-restricted cytotoxicity against a demyelinating virus following central nervous system infection. Eur J Immunol 27:963–970. doi: 10.1002/eji.1830270424. [DOI] [PubMed] [Google Scholar]
- 7.Azoulay-Cayla A, Syan S, Brahic M, Bureau JF. 2001. Roles of the H-2D(b) and H-K(b) genes in resistance to persistent Theiler’s murine encephalomyelitis virus infection of the central nervous system. J Gen Virol 82:1043–1047. [DOI] [PubMed] [Google Scholar]
- 8.Lipton HL, Melvold R. 1984. Genetic analysis of susceptibility to Theiler’s virus-induced demyelinating disease in mice. J Immunol 132:1821–1825. [PubMed] [Google Scholar]
- 9.Bureau JF, Montagutelli X, Lefebvre S, Guénet JL, Pla M, Brahic M. 1992. The interaction of two groups of murine genes determines the persistence of Theiler’s virus in the central nervous system. J Virol 66:4698–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bureau JF, Montagutelli X, Bihl F, Lefebvre S, Guénet JL, Brahic M. 1993. Mapping loci influencing the persistence of Theiler’s virus in the murine central nervous system. Nat Genet 5:87–91. doi: 10.1038/ng0993-87. [DOI] [PubMed] [Google Scholar]
- 11.Neville KL, Padilla J, Miller SD. 2002. Myelin-specific tolerance attenuates the progression of a virus-induced demyelinating disease: implications for the treatment of MS. J Neuroimmunol 123:18–29. doi: 10.1016/S0165-5728(01)00479-9. [DOI] [PubMed] [Google Scholar]
- 12.Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, Katz-Levy Y, Carrizosa A, Kim BS. 1997. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat Med 3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- 13.Lyman MA, Lee HG, Kang BS, Kang HK, Kim BS. 2002. Capsid-specific cytotoxic T lymphocytes recognize three distinct H-2D(b)-restricted regions of the BeAn strain of Theiler’s virus and exhibit different cytokine profiles. J Virol 76:3125–3134. doi: 10.1128/JVI.76.7.3125-3134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Getts MT, Richards MH, Miller SD. 2010. A critical role for virus-specific CD8(+) CTLs in protection from Theiler’s virus-induced demyelination in disease-susceptible SJL mice. Virology 402:102–111. doi: 10.1016/j.virol.2010.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang BS, Lyman MA, Kim BS. 2002. The majority of infiltrating CD8+ T cells in the central nervous system of susceptible SJL/J mice infected with Theiler’s virus are virus specific and fully functional. J Virol 76:6577–6585. doi: 10.1128/JVI.76.13.6577-6585.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards MH, Getts MT, Podojil JR, Jin Y-H, Kim BS, Miller SD. 2011. Virus expanded regulatory T cells control disease severity in the Theiler’s virus mouse model of MS. J Autoimmun 36:142–154. doi: 10.1016/j.jaut.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Probst HC, van den Broek M. 2005. Priming of CTLs by lymphocytic choriomeningitis virus depends on dendritic cells. J Immunol 174:3920–3924. doi: 10.4049/jimmunol.174.7.3920. [DOI] [PubMed] [Google Scholar]
- 18.Bailey SL, Schreiner B, McMahon EJ, Miller SD. 2007. CNS myeloid DCs presenting endogenous myelin peptides “preferentially” polarize CD4(+) T(H)-17 cells in relapsing EAE. Nat Immunol 8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- 19.Bailey-Bucktrout SL, Caulkins SC, Goings G, Fischer JA, Dzionek A, Miller SD. 2008. Cutting edge. CNS plasmacytoid dendritic cells regulate the severity of relapsing experimental autoimmune encephalomyelitis. J Immunol 180:6457–6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Josien R, Heslan M, Soulillou JP, Cuturi MC. 1997. Rat spleen dendritic cells express natural killer cell receptor protein 1 (NKR-P1) and have cytotoxic activity to select targets via a Ca2+-dependent mechanism. J Exp Med 186:467–472. doi: 10.1084/jem.186.3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trottier M, Schlitt BP, Kung AY, Lipton HL. 2004. Transition from acute to persistent Theiler’s virus infection requires active viral replication that drives proinflammatory cytokine expression and chronic demyelinating disease. J Virol 78:12480–12488. doi: 10.1128/JVI.78.22.12480-12488.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Getts DR, Terry RL, Getts MT, Müller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. 2008. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med 205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swiecki M, Colonna M. 2010. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol Rev 234:142–162. doi: 10.1111/j.0105-2896.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Getts DR, Terry RL, Getts MT, Müller M, Rana S, Deffrasnes C, Ashhurst TM, Radford J, Hofer M, Thomas S, Campbell IL, King NJ. 2012. Targeted blockade in lethal west Nile virus encephalitis indicates a crucial role for very late antigen (VLA)-4-dependent recruitment of nitric oxide-producing macrophages. J Neuroinflammation 9:246. doi: 10.1186/1742-2094-9-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. 2013. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol 31:413–441. doi: 10.1146/annurev-immunol-032712-095951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Getts DR, Chastain EM, Terry RL, Miller SD. 2013. Virus infection, antiviral immunity, and autoimmunity. Immunol Rev 255:197–209. doi: 10.1111/imr.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duncan DS, Miller SD. 2011. CNS expression of B7-H1 regulates pro-inflammatory cytokine production and alters severity of Theiler’s virus-induced demyelinating disease. PLoS One 6:e18548. doi: 10.1371/journal.pone.0018548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Homann D, Jahreis A, Wolfe T, Hughes A, Coon B, van Stipdonk MJ, Prilliman KR, Schoenberger SP, von Herrath MG. 2002. CD40L blockade prevents autoimmune diabetes by induction of bitypic NK/DC regulatory cells. Immunity 16:403–415. doi: 10.1016/S1074-7613(02)00290-X. [DOI] [PubMed] [Google Scholar]
- 29.Kaminsky SG, Nakamura I, Cudkowicz G. 1985. Genetic control of the natural killer cell activity in SJL and other strains of mice. J Immunol 135:665–671. [PubMed] [Google Scholar]
- 30.Lyman MA, Myoung J, Mohindru M, Kim BS. 2004. Quantitative, not qualitative, differences in CD8(+) T cell responses to Theiler’s murine encephalomyelitis virus between resistant C57BL/6 and susceptible SJL/J mice. Eur J Immunol 34:2730–2739. doi: 10.1002/eji.200324811. [DOI] [PubMed] [Google Scholar]
- 31.Jin YH, Kaneyama T, Kang MH, Kang HS, Koh CS, Kim BS. 2011. TLR3 signaling is either protective or pathogenic for the development of Theiler’s virus-induced demyelinating disease depending on the time of viral infection. J Neuroinflammation 8:178. doi: 10.1186/1742-2094-8-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan CW, Crafton E, Fan HN, Flook J, Yoshimura K, Skarica M, Brockstedt D, Dubensky TW, Stins MF, Lanier LL, Pardoll DM, Housseau F. 2006. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat Med 12:207–213. doi: 10.1038/nm1352. [DOI] [PubMed] [Google Scholar]
- 33.Pillarisetty VG, Katz SC, Bleier JI, Shah AB, Dematteo RP. 2005. Natural killer dendritic cells have both antigen presenting and lytic function and in response to CpG produce IFN-gamma via autocrine IL-12. J Immunol 174:2612–2618. doi: 10.4049/jimmunol.174.5.2612. [DOI] [PubMed] [Google Scholar]
- 34.Chaudhry UI, Plitas G, Burt BM, Kingham TP, Raab JR, DeMatteo RP. 2007. NK dendritic cells expanded in IL-15 exhibit antitumor responses in vivo. J Immunol 179:4654–4660. doi: 10.4049/jimmunol.179.7.4654. [DOI] [PubMed] [Google Scholar]
- 35.GeurtsvanKessel CH, Bergen IM, Muskens F, Boon L, Hoogsteden HC, Osterhaus AD, Rimmelzwaan GF, Lambrecht BN. 2009. Both conventional and interferon killer dendritic cells have antigen-presenting capacity during influenza virus infection. PLoS One 4:e7187. doi: 10.1371/journal.pone.0007187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NKDC gating strategy. A typical gating strategy was used to identify NKDCs, DCs, and NK cells. After singlet selection, lymphocytes were gated on a forward scatter/side scatter (FSC/SSC) profile. CD3− cells and CD45+ cells were then selected. Finally, expression of CD11c versus NK1.1 was used to analyze NKDCs, DCs, and NK cells. Download
CNS TMEV infection induces proliferation of splenic NKDCs. Wild-type B6 mice were i.c. infected with 5 × 106 PFU of TMEV or sham infected. Spleens were harvested during acute infection, and mononuclear cells were isolated and analyzed by flow cytometry. At 24 and 48 h p.i., mice were i.p. injected with 1 mg BrdU in 100 µl sterile PBS. At 96 h p.i., the percentages of BrdU-positive NKDCs, DCs, and NK cells from spleens of TMEV- or sham-infected mice were quantified. Data are representative of 2 to 3 independent experiments with 5 mice per group. Error bars show standard deviations. **, P < 0.01. Download
Resident NKDCs across lymphoid tissues. Spleen, lymph nodes, and bone marrow from naive B6 animals were analyzed via flow cytometry, and the percentage of NKDCs (CD45+ CD11c+ NK1.1+) was quantified. Error bars represent standard deviations from n = 3 animals. Download