

Abstract
The literature provides compelling evidence pointing to tight metabolic interactions between retinoids and xenobiotics. These are extensive and important for understanding xenobiotic actions in the body. Within the body, retinoids affect xenobiotic metabolism and actions and conversely, xenobiotics affect retinoid metabolism and actions. This article summarizes data that establish the importance of retinoid-dependent metabolic pathways for sustaining the body’s responses to xenobiotic exposure, including the roles of all-trans- and 9-cis-retinoic acid for protecting mammals from harmful xenobiotic effects and for ensuring xenobiotic elimination from the body. This review will also consider molecular mechanisms underlying xenobiotic toxicity focusing on how this may contribute to retinoid deficiency and disruption of normal retinoid homeostasis. Special attention is paid to xenobiotic molecular targets (nuclear receptors, regulatory proteins, enzymes, and transporters) which affect retinoid metabolism and signaling.
Keywords: Retinol, retinoic acid, retinyl ester, retinoid disruption, tetrachlorodibenzo-p-dioxin, polychlorinated biphenyls, cytochrome P450
Introduction
Many different linkages between retinoid metabolism and actions and xenobiotic metabolism and actions have been described in the literature. This literature provides compelling evidence that these interactions are extensive and important for understanding xenobiotic actions in the body. It is clear from this literature that retinoids affect xenobiotic metabolism and actions and that conversely xenobiotics affect retinoid metabolism and actions (Figure 1). Many different xenobiotics interact with retinoids including environmental pollutants (toxicants), pharmacologic agents/drugs, and experimental agents used in the study of disease. These will all be considered below. The primary focus of this review will be on interactions in the liver but the reader needs to be aware that these interactions are very important elsewhere in the body, especially in the developing embryo where they contribute to impaired embryogenesis.
Figure 1.
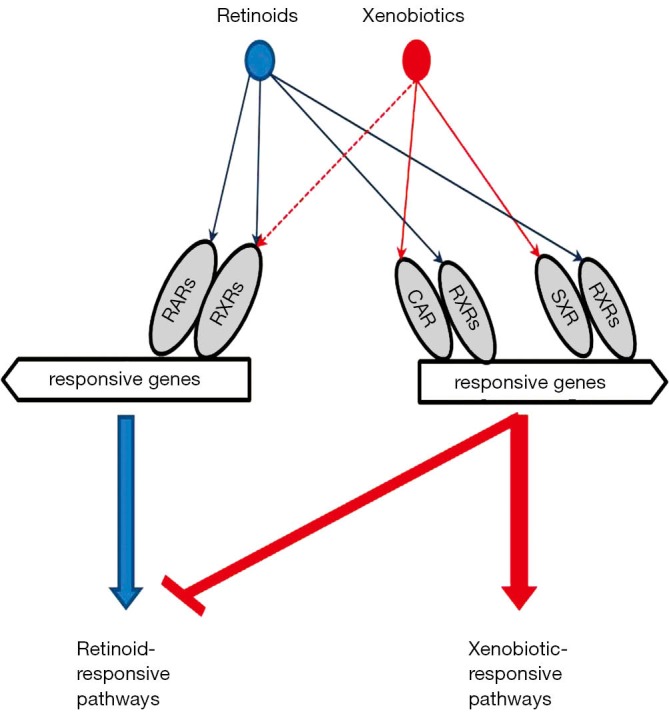
A schematic diagram representing retinoid-xenobiotic interaction. Retinoids (all-trans-retinoic acid and 9-cis-retinoic acid) are ligands for canonical retinoic acid receptors (RARs) and retinoid X receptors (RXRs). RXRs are common heterodimerization partners for xenobiotic receptors (CAR and SXR). The activation of xenobiotic-responsive pathways, like the CAR/RXR or SXR/RXR pathways, alters the balance and consequently the functioning of canonical retinoid-responsive pathways (RAR/RXR pathways).
Retinoid effects on xenobiotic metabolism and toxicity
Metabolic pathways that provide protection to living organisms from the harmful effects of xenobiotic and assure their elimination from the body are functionally combined into an elaborate three-phase system referred to as the xenobiotic detoxification system (1). The phase I reactions are catalyzed by mixed function oxidases and dehydrogenases and increase xenobiotic hydrophilicity through oxidation, reduction, and hydroxylation. The phase II reactions result in the conjugation of phase I metabolites and are catalyzed by a number of transferases. The phase III reactions involve transmembrane transporters which actively pump the conjugated xenobiotics out of the cell (1).
A large amount of published data suggests that the optimal functioning of the xenobiotic detoxification system requires retinoid actions for sensing, detoxifying and eliminating xenobiotics. These responses to xenobiotic exposure can be mediated either directly or indirectly. Direct responses involve the actions of the retinoic acid nuclear receptors (both retinoic acid receptors (RARs) and retinoid X receptors (RXRs) in regulating gene expression, while indirect responses arise through crosstalk with other transcription factors.
Xeno-sensors and their target genes
Several transcriptional regulatory proteins have been identified to be direct targets for xenobiotics and these are referred to as xenobiotic receptors or xeno-sensors (1-3). These include the aryl hydrocarbon receptor (AhR) and a number of members of the nuclear hormone receptor superfamily including human orphan steroid and xenobiotic receptor (SXR) and its rodent ortholog pregnane X receptor (PXR), constitutive androstane receptor (CAR), peroxisome proliferator-activated receptors (PPARs), and nuclear factor erythroid 2-related factor 2 (NRF2) (4,5). These will be discussed below.
The AhR is a ligand-inducible transcription factor that belongs to the basic helix-loop-helix/Per-Arnt-Sim (bHLH/PAS) family. However, because of its similarities in function, it is often considered along with the members of the nuclear hormone receptor superfamily (6). The unliganded inactive form of AhR exists in a multiprotein complex residing in the cytoplasm and containing one molecule of AhR, two molecules of hsp90, a protein termed p23, and an immunophilin-like protein variously named XAP, AIP, or ARA9 (7). Upon ligand binding, AhR translocates to the nucleus, where it loses its cytoplasmic protein partners and binds to a nuclear transcription factor—the aryl hydrocarbon receptor nuclear translocator (ARNT) (8). The heterodimer of AhR and ARNT binds to specific sequences containing the reversed substituted intolerant core sequence (5'-GCGTG-3') in the regulatory domains of the target genes, so called dioxin- or xenobiotic-response elements (referred to as DREs or XREs) (9). AhR binding to a DRE within a responsive gene stimulates transcription of the gene (4,6,10). A number of environmental contaminants including polyhalogenated aromatic hydrocarbons (PHAHs), polychlorinated biphenyls (PCBs), polycyclic aromatic hydrocarbons (PAHs) and polyhalogenated dioxins are known agonists of AhR (Figure 2). 2',3',7,8'-Tetrachlorodibenzo-p-dioxin (TCDD) is a prototypic activator of AhR (4) and is able to increase AhR expression as well (11). Other well-known AhR agonist species include benzo[a]pyrene (B[a]P), 3-methylcholanthrene, β-naphthoflavone, omeprazole (12).
Figure 2.
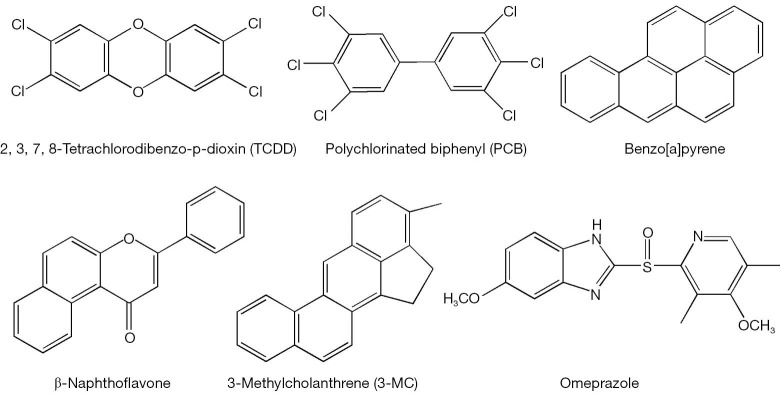
Chemical structures for some aryl hydrocarbon receptor (AhR) ligands.
It has been proposed that aside from functioning as a xeno-sensor, the AhR has an important physiological role in regulating fundamental metabolic processes (13). Naturally occurring agonists of AhR are reported to include indole-containing compounds such as indole-3-carbinol, tryptophan photoproducts, tetrapyroles such as bilirubin and biliverdin, and possibly, arachidonic acid metabolites, however the physiological relevance of each of these remains unclear (12). 2-(1'H-indole-3'-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) is generally accepted to be an endogenous ligand for AhR (14,15).
On binding to one of its ligands, the AhR can regulate expression of a variety of genes including primarily those encoding cytochrome P450 (CYP) enzymes. CYP1A1, CYP1A2, CYP1B1 and CYP2S1 are well-studied AhR target genes which are induced upon AhR agonist exposure (2,7,12). Recent studies have revealed that at least a dozen additional genes encoding xenobiotic metabolizing enzymes, including phase I and II enzymes, and phase III transporters, collectively referred to as the “AhR gene battery”, are specifically induced as a result of AhR activation upon ligand binding. These include isoforms of aldehyde dehydrogenases (Aldh7a1), glutathione transferases (Gstm3), sulfotransferases (Sult5a1), UDP-glucuronosyltransferases (Ugt1a1, Ugt1a6, Ugt1a9, Ugt2b35), solute carrier (Slcs) and ATP binding cassette (Mrp4) transporter proteins (4,5,12,16-18). Flavin-containing monooxygenases (Fmo2 and Fmo3) that were earlier thought to be uninducible, have now been convincingly shown to be highly induced in an AhR-dependent manner (19,20).
The nuclear orphan receptor CAR, which normally does not require a ligand to be transcriptionally active (3), can be activated to induce CYP2B gene expression upon binding to phenobarbital and phenobarbital-type inducers (i.e., 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene, TCPOBOP). In this instance, the receptor forms a heterodimer with the retinoid X receptor (RXR) and binds to a conserved enhancer element known as NR1, thus activating transcription of responsive genes (21,22). Recently, several polychlorinated biphenyl congeners also have been identified to be CAR agonists (23). CAR has been shown to control expression of AhR, suggesting a tight cooperation between these two xeno-sensors (4). CAR-specific genes, in addition to Cyp2b10 and AhR also include aldehyde dehydrogenases (Aldh1a1, Aldh1a7), glutathione transferases (Gstt1, Gsta1, Gsta4, Gstm1-4), sulfotransferases (Sult1e1, Sult3a1, Sult5a1, Sult1d1), UDP-glucuronosyltransferases (Ugt1a1, Ugt2b34), 3'-phosphoadenosine 5'-phosphosulfate synthase 2 (Papps2), esterase 1 (Pon1), hepatic flavin-containing monooxygenase 5 (Fmo5), betaine-homocysteine methyltransferase (Bhmt), and multi-drug resistance-associated proteins (Mrp2-4) (3-5).
The human orphan receptor SXR and its rodent ortholog PXR initially were isolated as candidate xenobiotic receptors that were postulated to regulate CYP3A gene expression (2). This receptor is activated by rifampicin, dexamethasone, PCBs and PAHs (Figure 3) (23,24). The importance of CYP3A in xenobiotic metabolism is underscored by its high abundance in human liver (~30% of all CYPs expressed in the liver) and its estimated role in the catalyzing the metabolism of about 50% of pharmaceuticals encountered by the liver (4). SXR and PXR mediate CYP3A expression via binding to direct repeats of the hexameric response element (AGGTCA) separated by three nucleotides (referred to as a DR-3) or an inverted repeat sequence with six nucleotide spacing (referred to as an IR-6) located in the promoter region of the gene (25,26). These SXR elements have also been identified in the genes encoding phase I and phase II enzymes including CYP2A, CYP2C, CYP2E, and UDP-glucuronosyltransferase, raising the potential of broad SXR and PXR actions in the body’s responses to xenobiotic exposure (2). Gene expression signatures specific to PXR include induction of carboxylesterase 2 (Ces2) and 3 (Ces3), UDP-glucuronosyltransferase 1a (Ugt1a1, Ugt1a5, Ugt1a9), glutathione transferases (Gsta1, Gstm1-m3), multi-drug resistance proteins 1 (Mdr1a, Mdr1b, Mrp3), organic anion transporter polypeptide (Oatp02, Oatp1a4), and ATP-binding cassette transporter (Abcb9) (3-5,27).
Figure 3.

Chemical structures for some constitutive androstane receptor (CAR) and pregnane X receptor (PXR) ligands.
PPARs are also members of the nuclear receptor superfamily of transcription factors and are widely studied for their central roles in regulating fatty acid metabolism and glucose homeostasis. Three isoforms, designated PPARα, PPARβ/δ and PPARγ, are expressed in different tissues and at different times of development (3). Each of the isoforms is highly specific with respect to ligands, target genes and physiological function. Gene expression profiling aimed at investigating molecular mechanisms associated with toxic effects resulting from PPAR activation has been carried out to a large extent for PPARα and to lesser degrees for PPARγ and PPARβ/δ (4). Studies performed on PPARα knockout mice revealed Cyp4a14, Aldh1a1, mGst3, Gstm4, and Mrp4 as PPARα target genes involved in xenobiotic metabolism (5). There is much less information regarding the roles for PPARγ and PPARβ/δ in xenobiotic metabolism compared to other receptors mentioned above. However, this does not detract from their potential role in the response to xenobiotic exposure that has to be detected in the future.
NRF2 is a cap’n’collar (CNC) basic-region leucine zipper (bZIP) transcription factor, which plays a major role in protecting cells from pro-oxidants and electrophiles through regulation of basal and inducible expression of genes containing a core sequence (RTGACnnnGC) in their promoters. This element is referred to as the antioxidant response element (ARE) (28). NRF2-target genes include those encoding antioxidant and detoxication enzymes such as aldo-keto reductase (Akr1e2), aldehyde dehydrogenases (Aldh1a1), heme oxygenase-1 (Hmox1), glutathione S-transferase (Gsta1, Gsta4, Gstm1-m4, mGst3), glutamatecysteine ligase (Gclm), NADP(H):quinone oxidoreductase-1 (Nqo1) and multi-drug resistance proteins (Mrp3 and Mrp4) (5).
The xeno-sensors usually show either synergism or antagonism with regards to how they overlap in their ligands, as well as with respect to their target genes. For instance, CAR and PXR mediate induction of common phase I and II enzymes and transporters. CAR and PXR agonists have been shown to be able to induce expression of AhR (4). AhR activation upon binding to 2,3,7,8-tetrachlorodibenzo-p-dioxin or B[a]P results in CAR and NRF2 upregulation in hepatocytes (29). AhR can induce Nrf2 mRNA expression via binding to DRE-like elements in the mouse Nrf2 promoter, and the induction of Nqo1 by TCDD is dependent on the presence of both AhR and NRF2 in cells (18). AhR and PPARα possess antagonistic effects on CYP1A, CYP2B, CYP3A and CYP4A gene expression, but display an additive inhibitory effect on CYP2C11 gene expression (30). Suppression of PPARα pathways also has a common effect on both CAR and PXR activation (2,4). PPARβ/δ has been shown to be involved in sustaining optimal AhR-mediated signaling in keratinocytes (31).
Retinoid involvement in xeno-sensors and their target genes function
RXRs are common heterodimerization partners for most xenobiotic receptors, including all those mentioned above except AhR. Consequently, the RXRs function as master regulators for genes encoding enzymes and transporters involved in xenobiotic metabolism (32,33). Studies involving the hepatocyte-specific disruption of the mouse Rxrα gene revealed decreased basal expression of phase I enzymes, including CYP2A, CYP2B, CYP3A, CYP4A, and CYP7A (33-35). The decrease in CYP3A expression due to RXR ablation was confirmed experimentally when Rxr mRNA degradation was induced by microRNA (36) and by arsenic-induced RXR proteasomal degradation (37). Similarly, CYP genes including CYP2A5, CYP2B10, CYP3A1, but not CYP2E1 or CYP2D6, have been shown to be RXRα target genes in vivo (38). RXRα-deficiency alters homeostasis of glutathione (GSH), a common conjugate employed in phase II conjugation reactions. RXRα-deficiency results in reduced hepatic levels of GSH and decreased expression of glutathione-metabolizing enzymes, including glutamate-cysteine ligase catalytic subunit (GCLC), glutathione-S-transferase µ, and glutathione peroxidase 1 (39,40). On the other hand, RXRα functions as a repressor of NRF2-mediated gene expression. RNAi-mediated knockdown of RXRα was shown to increase both basal ARE-driven gene expression and induction of ARE-driven genes by the NRF2 activator tert-butylhydroquinone (tBHQ), while RXRα overexpression leads to decreased ARE-driven gene expression (41). RXRα has been shown to interact physically with NRF2 in many cell types, including cancer cells, murine small intestine and liver cells, and to bind to ARE sequences in the promoters of NRF2-regulated genes (41).
Consistent with the findings described above are published data showing that treatment of rats with the RXR selective agonist, bexarotene (LGD1069/Targretin), results in increased hepatic protein levels of CYP2B1/2, CYP2C11, CYP3A and CYP4A (42). In vitro studies employing transient transfection assays carried out in human HepG2 hepatoma cells and in primary mouse hepatocytes revealed that retinoids are capable of activating RXR/SXR- and RXR/CAR-mediated pathways leading to CYP3A4 induction (43,44). Acidic retinoids, including 9-cis-(9CRA), 13-cis-(13CRA) and all-trans-retinoic acids (ATRA) and retinaldehyde (9-cis-retinal) were found to be among the most potent agonists for activating the RXRα/SXR-mediated pathway as well as for inducing endogenous CYP3A4 activity. These retinoid species were shown to exhibit either a greater or an equivalent potency to that of the classic inducer of CYP3A4 rifampicin, which binds to SXR directly (43). On the other hand, synthetic rexinoids (selective ligands for the RXR) are only weak activators of SXR, but they can antagonize SXR activation by rifampicin, suppressing rifampicin-induced expression of SXR target genes and reducing the binding of SXR/RXR to SXR response elements both in vivo and in vitro (45). This effect involves the induction of both SXR and RXR degradation (45). ATRA and 9CRA were also found to be more effective for inducing CYP3A4 than the CAR xenobiotic agonist TCBOPOP. Addition of ATRA or 9CRA together with TCBOPOP was found to further enhance the induction of responsive genes through a process mediated by RXRα/CAR and RXRγ/CAR heterodimers (44). Studies performed by Ruhl et al., employing full-length PXR receptor protein and a PXR-responsive reporter gene showed that carotenoids and retinol both have concentration-dependent effects on reporter gene activity in HepG2 cells. This effect is comparable in magnitude to that of the PXR agonist rifampicin. However, apo-carotenals and lycopene were found to exert significantly less or no PXR activation. Exposure of these cells to carotenoids and retinol was reported to increase gene expression levels of PXR responsive genes including CYP3A4/CYP3A7, CYP3A5, MDR-1 and MRP-2 at comparable or higher levels than those induced by rifampicin (46). However, these effects were not observed for all genes involved in xenobiotic metabolism. Dietary β-carotene supplementation of rats (15 days using a diet containing 300 mg β-carotene/kg diet) failed to induce hepatic CYP1A1 or 1A2 in vivo. But this in vivo study revealed that β-apo-8'-carotenal and two other dietary carotenoids canthaxanthin and astaxanthin may act as strong inducers of these AhR downstream targets (47).
Retinoic acid receptor (RAR) activation was found to diminish CYP concentrations in livers of rats, leading to decreased microsomal xenobiotic metabolism (42). Ligands with RAR agonist activity have been shown to decrease hepatic levels for CYP1A2, CYP2B1/2, CYP3A (42). However, in human HL-60 promyelocytic leukemic cells, ATRA treatment leads to an upregulation of CYP1B1 expression (48). Treatment with 9CRA or ATRA also represses CYP2B induction in primary mouse and rat hepatocytes and in human HepG2 cells. This is proposed to occur due to increased heterodimerization of RAR with RXR, leading to a reduction of ligand-free RXR available for heterodimerization with CAR. This results in the repression of CAR activity required for regulating CYP2B expression (21,49). Studies of human and rat primary hepatocytes and HepG2 cells revealed that RARα is able to transactivate the human, but not the rat CAR gene, by binding to the CAR promoter region, located at +1453/+1469 within intron 1 of the gene (50). ATRA and other RARα agonists have also been shown to antagonize expression of NRF2 target genes through an interaction with RARα, whereas treatment with RARα antagonists or knockdown of RARα augments NRF2 effects on CYP expression (51).
To date, functional retinoic acid responsive elements (RAREs) have been identified in several genes encoding phase I and phase II xenobiotic-metabolizing enzymes and phase III transporters. This underscores the connection between xenobiotic and retinoid actions. A canonical RARE in the CYP1A1 gene initially was shown to be activated by RARα and thyroid hormone nuclear receptor (TRα) in an in vitro system; however, further in vivo and in vitro studies, using RAR agonists and antagonists have now established that the CYP1A1 RARE is suppressive and that endogenous retinoids may negatively modulate CYP1A1 induction by xenobiotics (52-54). Along the same lines, studies of RARα-null mice have shown that the absence of RARα leads to an induction of CYP1A1 deethylase activity towards ethoxyresorufin (55). ATRA, at physiological concentrations (1-100 nM), has also been shown to reduce AhR-mediated CYP1A1 transcription due to an inhibition in the recruitment of ARNT to XREs (56). More in depth analysis employing human Caco-2 enterocytes revealed that treatment with ATRA represses AhR actions within these cells through physical interactions of AhR with the RAR-corepressor SMRT, leading to a decrease in AhR-induced CYP1A1 expression (57). In undifferentiated keratinocytes, continuous treatment with 1 µM ATRA inhibited TCDD-induced AhR and ARNT mRNA upregulation and transcription of their downstream target CYP1A1, even though RARγ and RXRα mRNA levels were unaffected (58-60). In rat lungs, ATRA administration was shown to attenuate hyperoxia-induced CYP1A1 upregulation (61). A RARE has also been identified in the promoter regions of the genes encoding the drug-metabolizing CYP2C family members (62), which in humans are reported to metabolize more than 60 clinically relevant drugs (63).
As for phase II detoxification enzymes, retinoids, including ATRA, 9CRA, as well as the RAR panagonist 4-[(E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl]benzoic acid (TTNPB, arotinoid acid) have been shown to suppress, in a concentration-dependent manner, UDP-glucuronosyltransferase 2B7 isoform (UGT2B7) mRNA expression in Caco-2 cells (64). This regulation may represent an example of a negative feedback relationship between retinoids and xenobiotics, taking into consideration that UGT2B7 was shown to be the only UGT isoform capable of catalyzing glucuronidation of ATRA and its oxidized metabolites, including 4-OH-ATRA, 4-oxo-ATRA and 5,6-epoxy-ATRA (65). The formation of these glucuronides renders the retinoids water soluble and results in their elimination in the urine. Surprisingly, this effect was not observed either in HepG2 cells or for other UGT isoforms, specifically UGT2B15 and UGT1A6, demonstrating the specificity of the inhibitory effect (64). The authors of this study however were unable to verify that UGT2B7 mRNA expression is regulated through canonical RAREs; however the observation that the RAR agonist TTNPB showed the highest potency for suppressing UGT2B7 gene expression suggests that this regulation probably occurs at the transcriptional level.
Human Pi-class glutathione S-transferase (GSTP1) expression has been shown to be negatively modulated by ATRA in the presence of hRARβ. This is proposed to result through decreased transcription of the GSTP1 gene (66). Subsequent investigations showed that RXR is not involved in repressing GSTP1 expression upon ATRA treatment and does not augment RAR action. Interestingly, it was also established that RAR-mediated ATRA repression of GSTP1 promoter activity does not involve the binding of an RAR-ATRA complex to the promoter of this gene, rather it arises because the RAR-ATRA complex blocks transcription factor activator protein-1 (AP1) or AP1-like proteins from binding to the promoter and regulating GSTP1 gene expression (67).
In vitro studies employing bovine airway epithelial cells showed that ATRA treatment attenuates hydrocortisone-stimulated mRNA expression of phenol sulfotransferase (PST). Doses of ATRA as low as 0.05 nM inhibited PST expression by as much as 50% as evidenced by dose-response measurements of both PST enzyme activity and mRNA levels. This was further confirmed by immunoblot analyses (68).
Among the phase III xenobiotic transporter proteins, the promoter of the gene encoding organic solute transporter-β (OSTβ) has been identified as possessing a functional RARE. ATRA treatment of either Huh7 or HepG2 cells resulted in a dose-dependent induction of OSTβ expression due to the presence of this RARE in the OSTβ gene. It was further established that this element binds either RARα or CAR heterodimerized with RXRα (69). Another phase III transporter protein, breast cancer resistance protein (BCRP), an ABC-transporter known to be involved in B[a]P-3-sulfate excretion, has also been shown to be induced upon ATRA treatment via the RAR/RXR signaling pathway, resulting in more effective removal of B[a]P metabolites from human Caco-2 intestinal cells (70). Pretreatment of Caco-2 cells either with ATRA, RAR agonists or RXR agonists resulted in enhanced apical excretion of B[a]P-3-sulfate, a phase II metabolite of B[a]P initially formed from B[a]P by CYP1A1 (11).
Xenobiotic effects on retinoid metabolism and action
The literature provides considerable evidence for significant retinoid-xenobiotic interactions within the body, ones which result in disruptions of normal retinoid metabolism. These alterations ultimately result in disrupted retinoid signaling leading to developmental, metabolic and other functional impairments. For instance, TCDD-treated animals exhibit toxicity symptoms, including wasting syndrome, like those of observed in retinoid-deficient animals (71). It has been proposed that some of the adverse effects of xenobiotic intoxication arise through disruptions of normal retinoid homeostasis and normal retinoic acid signaling pathways (72-75). Experimental observations reported by many groups and involving different experimental models are conclusive that a number of the common adverse effects observed upon xenobiotic exposure are associated with decreased tissue, especially hepatic, retinoid levels. These are caused either by increased retinol mobilization from retinyl ester stores or impairments in retinol esterification affecting retinyl ester accrual, or a redistribution of retinoids amongst tissues, and/or through an increase in retinoic acid catabolic turnover and increased elimination of oxidized retinoid catabolic products (Figure 4).
Figure 4.

A schematic diagram summarizing retinoid metabolism within mammals. The red arrows indicate the direction of metabolic fluxes observed following xenobiotic exposure. Hepatic retinoid is stored primarily as retinyl ester. Following retinyl ester hydrolysis, the retinol released can undergo a 2-step enzymatic conversion into retinoic acid (all-trans-retinoic acid and 9-cis-retinoic acid). These acidic retinoids act as ligands for their cognate nuclear hormone receptors (RARs and RXRs), affecting expression of retinoic acid-responsive genes. Retinol is mobilized from the liver into the circulation in a complex with retinol-binding protein (RBP) which delivers the retinol to peripheral tissues. Retinoic acid can be inactivated through cytochrome P450-mediated oxidation, possibly followed by glucuronidation, allowing for the elimination of retinoic acid from cells and tissues.
Direct disruption of retinoic acid signaling
The disruption of retinoid homeostasis by xenobiotics is a process that can take place at multiple metabolic and functional levels. The simplest way for understanding the actions of xenobiotics on retinoid physiology is to propose that they possess agonist or antagonist activities towards either RAR- or RXR-mediated transcription. Indeed, eighty-five of 543 environmental toxins screened, including 16 organochlorine pesticides, 14 styrene dimers, 9 monoalkylphenols and 6 parabens, were found to possess RAR agonist activity in reporter assays (76). The diverse chemical structures for some of these compounds are displayed in Figure 5. Organochlorine pesticides, including chlordane, dieldrin, aldrin, endrin, and endosulfan, can activate RARβ and RARγ, but not RXR isoforms, and are able to induce RAR-mediated transcription of the gene encoding CYP26A1. Induction of CYP26A1 expression will clearly disrupt normal retinoid homeostasis and signaling since this CYP species catalyzes retinoic acid oxidation, a first catabolic step contributing to retinoid elimination from the body (77). Methoxy-methoprene acid, a metabolite of the insect juvenile hormone methoprene, has been shown to be a ligand for RXRs and is capable of activating transcription through RAR/RXR response elements in transfected CV-1 and F9 cells (78). Using an in vitro screening assay employing two-hybrid yeast strains cotransformed with plasmids expressing human RXRβ, a RXR coactivator (GRIP1) and a reporter 9CRA-responsive β-galactosidase gene, it was shown that 2-t-butylphenol, 2-isopropylphenol, 2,4-dichlorophenol (2,4-DCP), 3,4-dichlorophenol (3,4-DCP), 4-tert-octylphenol (4-t-OP) and hexachlorobenzene (HCB) all possess RXR agonist activity. This study also revealed that bisphenol A (BPA) brings about a high level of induction of RXR transcriptional activity and also possesses a high binding affinity towards recombinant human RXR (79). This finding was later confirmed and expanded upon by in silico molecular docking studies (80). This was unlike r-hexachlorocyclohexane (HCH), p,p'-dichlorodiphenyltrichloroethane (p,p'-DDT) and 2,4-DCP, which show a marked inhibition of RXR signaling in these yeast two-hybrid studies (79).
Figure 5.
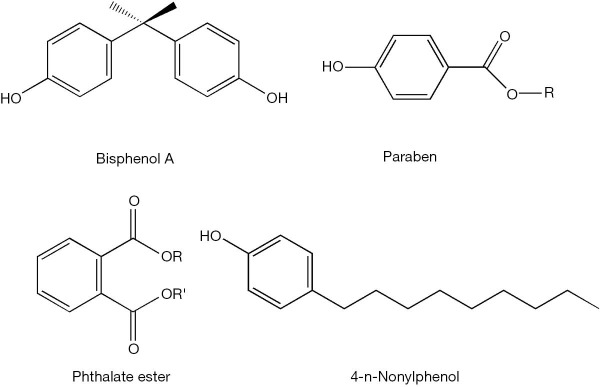
Chemical structures for some xenobiotics that show agonist activities towards RARs and RXRs.
Other studies have identified xenobiotic effects in blocking known retinoic acid-dependent processes. Through reporter assays carried out in P19/A15 cells, it was found that PAHs and their N-heterocyclic analogs are able to interfere with retinoid signaling through modulation of ATRA-mediated cellular responses, including cellular processes controlling cell pluripotency and differentiation. Benz[a]anthracene and benz[c]acridine significantly upregulate ATRA-mediated responses upon their coadministration with ATRA over a range of ATRA concentrations, while 1,7-phenanthroline was found to downregulate ATRA-mediated responses (81). Studies of mouse pluripotent P19 cells established that expression of the retinoic acid-inducible Hoxa1 gene expression is disrupted by several common xenobiotics including citral, disulfiram, nitrofen and bisdiamine, and by common xenoestrogens including BPA, diethylstilbestrol, 4-n-nonylphenol, genistein and phthalate esters (dibutyl phthalate and dipentyl phthalate) (82). In vitro studies of mouse embryonic stem (ES) cell differentiation have shown that BPA administration upregulates expression of the meiotic entry gene Stra8, thus interfering with normal retinoic acid signaling in ES cells (83).
Consistent with the in vitro findings discussed above, exposure to BPA in utero, at 0.02 µg/kg/day, a concentration that is 1% of BPA concentrations that are sometimes encountered in the environment, was shown in developing mouse embryos to increase mRNA expression of RARα and RXRα simultaneously, along with an increase in AhR mRNA expression (84,85). In a standard in vivo uterotrophic assay in rats, an inhibitory effect of ATRA (provided at 5 mg/kg once a day for 3 days) on the estrogenic activity of BPA (dosed at 100 mg/kg once a day for 3 days) was observed (86). Thus, in utero both ATRA and BPA are mutually able to antagonize the actions of the other.
Retinyl ester hydrolysis and synthesis
The complex interactions involving xenobiotics and retinoids also may give rise to impairments in retinoid homeostasis and signaling through a direct disruption of hepatic retinoid metabolism. The first metabolic site where xenobiotic-induced disruption of hepatic retinoid homeostasis may take place is in the hydrolysis of retinyl ester stores, the first enzymatic step needed either to mobilize stored hepatic retinoids into the circulation or for generating retinoic acid within the liver. Hepatic retinyl ester stores plummet rapidly upon administration of dioxin to rats (74) or PCBs to rats and mice (72,73,87). Hepatic retinyl ester levels are also reported to decrease upon 3-methylcholanthrene, 7,12-dimethylbenzanthracene, and B[a]P administration to rats (88). Likewise, administration of several therapeutic drugs, including methyldopa, prednisone, hydrocortisone, methadone, phenothiazine, phenytoin, cyclosporin A, flupenthixol and cefotiam to human subjects, as well as to experimental animals, has been shown to induce hepatic retinyl ester depletion (89-91).
The most complex, and to date the best studied, metabolic interactions between retinoids and xenobiotics, ones with consequences for disrupting retinoid stores in the liver, involve the PHAHs. Most of these toxic effects of PHAHs are mediated by the AhR (92,93). The prototypic AhR agonist, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), possesses a planar molecular conformation with lateral chlorine substitutions. The generally accepted understanding of the actions of TCDD and related compounds are as prototypic agonists of AhR. Thus, it is thought that TCDD disruption of retinoid homeostasis is mediated through activation of this xeno-sensor with the subsequent upregulation of downstream CYP-mediated pathways. Indeed, this notion is strongly supported by findings from studies employing AhR-deficient mice, which are protected from TCDD toxicity and which fail to display decreases in hepatic retinoid levels after TCDD treatment (94). The initial signs of toxicity upon TCDD administration to rats are seen as early as 24 hours after administration of a single dose of 1 µg TCDD/kg body weight (95). Systemic TCDD-induced effects on retinoid metabolism as well as effects on hepatic retinoid metabolism are seen at this early time point upon administration of a single dose of 10 µg TCDD/kg body weight. After 24 hours, for this dose of TCDD administered to rats, hepatic ATRA levels increase, while hepatic retinyl ester and retinol levels are not yet changed (74). Three days following administration of a single 10 µg/kg dose, a decrease in hepatic retinyl ester levels can be detected (74). Repeated TCDD administration to rats for more than 28 days, at a daily dose 10 µg TCDD/kg body weight, is accompanied by an almost 50% decrease in hepatic retinyl ester concentrations (96). Continuous TCDD exposure, even at lower doses, is characterized by decreases in all major hepatic retinoid species including retinyl esters, retinol and ATRA (97). Continuous TCDD administration to rats lasting for several weeks has been shown to disrupt retinoid homeostasis even for doses as low as 1 ng TCDD/kg body weight per day (71,74,97). In rats, the consumption of a dioxin-containing diet for 13 weeks, including for a low dose of 14 ng TCDD/kg body weight per day, resulted in a strong reduction in hepatic retinol and retinyl palmitate levels to ones that were respectively 56% and 20% of those of controls. At the highest dose used, 13 weeks after the start of administration almost no hepatic retinoid could be detected (98).
The retinoid disrupting effects of TCDD exposure also have been shown in a number of other rodent species during experimentally induced dioxin toxicity (71,97,99,100). Moreover, these same effects are observed in the wild upon environmental exposure of marine mammals and reptiles residing in TCDD-contaminated areas (101).
Coplanar PCBs (non- and mono-ortho chlorinated biphenyl congeners with chlorine substituents in both para and at least two meta positions) are approximate isostereomers of TCDD (see Figures 2, 6, and 7 for chemical structures). These include 3,3',4,4'-tetrachlorobiphenyl (PCB77), 3,3',4,4',5-pentachlorobiphenyl (PCB126) and 3,3',4,4',5,5'-hexachlorobiphenyl (PCB169) and are referred to as dioxin-like. Each of these compounds elicits similar toxic effects and influences the same spectrum of metabolic responses due to the ability of each to bind to AhR. These coplanar PCBs are able to competitively displace [3H]TCDD from the cytosolic AhR, and these compounds all exhibit the properties of AhR agonists including induction of CYP1A1 and CYP1A2 (102). The retinoid disrupting effects observed upon coplanar PCB administration are very similar to those observed for TCDD exposure as described above.
Other PCB congeners (substituted with more than two para and ortho substituents), referred to as non-dioxin-like [including 2,3',4,4',5-pentachlorobiphenyl (PCB118); 2,2',4,4',5,5'-hexachlorobiphenyl (PCB153), and 2,2',3,4,4',5,5'-heptachlorobiphenyl (PCB180)] (see Figures 6 and 7 for chemical structures) also can have adverse effects on retinoid homeostasis (103), however these PCB congeners do not exhibit AhR agonist activity. It was first suggested and then confirmed experimentally that these compounds are able to activate CAR and SXR/PXR receptors, behaving as phenobarbital-like or dexamethasone-like inducers, thus resulting in an induction of CYP2B1 and CYP3A1 respectively (92,103,104). Fully chlorinated, non-coplanar biphenyl 2,2',3,3',4,4',5,5',6,6'-decachlorobiphenyl (PCB 209) does not exhibit the human health hazards common to coplanar PCBs; it does not elicit either CYP enzyme induction or genotoxicity, nor does it produce alterations consistent with endocrine-modulating activity (105). Thus, the chlorine substituent positions on the PCB molecule must determine the metabolic behavior of PCBs in affecting retinoid metabolism.
Figure 6.
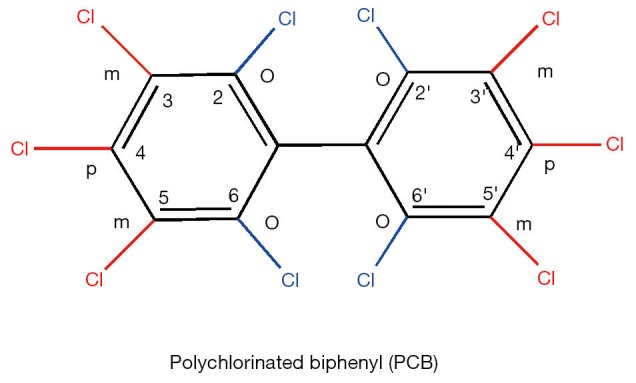
Generic chemical structure of polychlorinated biphenyl (PCB) congeners. Note: o, m, p denote ortho, meta, and para positions respectively.
Figure 7.

A schematic diagram showing polychlorinated biphenyl (PCB) congener metabolism within mammals. Coplanar PCBs (dioxin-like) induce AhR transcriptional activity. Non-coplanar PCBs (referred to as non-dioxin-like, phenobarbital-like or dexamethasone-like) induce CAR and SXR transcriptional activity. PCBs undergo metabolic transformation, involving cytochrome P450 (CYP)-dependent oxidation giving rise to the formation of hydroxylated metabolites (OH-PCBs). Subsequent metabolism of the OH-PCBs occurs via conjugation reactions resulting in formation of conjugates and sulfur-containing metabolites, which can be directly eliminated from cells and tissues.
Despite differences in the nuclear receptor which is affected and the downstream target genes affected, exposure to dioxin-like and non-dioxin like PCB congeners leads to a dose-dependent reduction in hepatic and serum retinoid concentrations (102). A reduction in hepatic retinyl ester and retinol levels, as well as a significant decrease in serum retinol level accompanied by a lowering in serum retinol-binding protein (RBP) levels, has been observed in rats after exposure to either coplanar (PCB77 or PCB126) or non-coplanar (PCB153 or PCB180) congeners or a mixture of PCB species (Arochlor 1254) (73,87,106-108). However the magnitude of reductions in hepatic and serum retinoid levels varies between these congeners, being more pronounced for dioxin-like PCBs. The effect of non-dioxin-like PCBs is quantitatively less strong by an order of magnitude and requires higher amounts to be administered in order to obtain responses similar to those of coplanar PCBs (73,87,103,106,107). Importantly, this phenomenon has been confirmed by studies employing both AhR-responsive and non-responsive mouse strains.
Dose dependent decreases in hepatic and serum retinoid levels as great as 50% of control levels were observed after both chronic and acute exposure to the dioxin-like PCB77 congener in an AhR-responsive mouse strain (C57Bl/Rij) (72,109-111). However, similar pronounced effects were not observed in AhR-non-responsive DBA/2 mice, suggesting that functional AhR is required for a maximal response to be observed upon dioxin-like PCB exposure. Although hepatic retinoid levels are unaffected in the AhR-non-responsive DBA/2 mouse strain, even at PCB77 concentrations 50-times higher than those that are needed to bring about a hepatic retinoid reduction in the C57Bl/Rij mice, serum retinol levels were observed to dramatically decrease (72,109,110).
A mechanistic explanation that might account for the observed reductions in hepatic retinyl ester levels upon xenobiotic exposure could involve either diminished rates of retinol esterification or accelerated retinyl ester hydrolysis. However, studies aimed at directly proving this hypothesis have failed to confirm this seemingly obvious and simple mechanism. The decrease in hepatic and serum retinoid levels upon exposure of rats to dioxin-like coplanar PCBs was surprisingly reported to be accompanied by reduced hepatic retinyl ester hydrolase (REH) activity (87,112). mRNA expression of carboxylesterase 3 (CES3, ES10), a known REH, was shown to be 2-fold downregulated in the liver 24 hours after a single dose of 40 µg TCDD/kg body weight orally administered to rats. Expression of CES3 remained low even 7 days after exposure. Likewise, expression of esterase 2 (ES2), another REH, was decreased by 5-fold at day 7 after TCDD administration (113). A pronounced decline in hepatic retinyl ester concentrations has been observed upon either flupenthixol or cefotiam administration to rats but, unlike TCDD administration, this was accompanied by an upregulation in hepatic REH activity (91).
Alterations in retinyl ester formation after TCDD exposure of rats has been shown to be accompanied by diminished hepatic lecithin:retinol acyltrasferase (LRAT) activity (114), the predominant enzyme responsible for retinol esterification in mammals. However, the relevance of this observation in the context of the rapid and dramatic hepatic retinoid decline arising several hours or a few days after TCDD administration remains controversial.
A complicating factor that makes gaining insights into the relationship between xenobiotic exposure and altered retinyl ester hydrolysis/synthesis complex is the topological separation and compartmentalization of different cellular sites involved in hepatic retinoid metabolism. Although upon administration, the majority of TCDD and dioxin-like PCBs accumulate in the liver (115,116), primarily within hepatocytes (117). However, hepatic stellate cells are the cellular site where the majority of hepatic retinoid is found, but these cells are unresponsive to TCDD administration due to an absence of detectable amounts of the AhR protein as well as its target CYP1A1 in hepatic stellate cells (118). Hepatic stellate cell activation however develops as a later event in xenobiotic-induced injury, secondary to hepatocyte injury. This was established by showing that hepatic stellate cells are unaffected even 25 days after exposure of rats with a single dose of 10 µg TCDD/kg body weight (119). In mice, the features of hepatic stellate cell activation, specifically elevated collagen 1A1 (Col1A1) and α-smooth muscle actin (αSMA) expression, cannot be detected earlier than 21 days after TCDD administration even when the compound is given in doses as great as 10 or 25 µg TCDD/kg body weight (120,121). An increase of hepatic stellate cell activation in rats, as evidenced in the acquisition of a myofibroblast-like phenotype, has been reported to occur as early as 7 days after administration of a very large dose of 3,3',4,4'-TCB (given at 87 mg/kg of body weight (122).
It is clear that the cellular compartmentalization of retinoid storage and metabolism in the liver has not been studied in depth in the context of xenobiotic exposure. However, from the existing data it is possible to conclude that it is unlikely that there is a direct influence of xenobiotics on retinyl ester hydrolyzing and/or esterifying enzymes per se that directly affect hepatic retinoid stores. The underlying events leading to the observed effects of xenobiotics on hepatic retinyl ester stores must occur downstream of the hydrolysis/esterification pathway and involve more complex systemic affects. Specifically, retinol liberated from hepatic retinyl ester stores is either redistributed to extrahepatic tissues and/or channeled towards ATRA and 9CRA formation, which are needed to provide sufficient transcriptional signaling for maintaining the normal health of cells, tissues and the organism. However, based on the literature, it would appear that the retinoid metabolic flux triggered by increased ATRA and 9CRA demand does not reach saturation, suggesting that the loss of hepatic retinoids upon xenobiotic exposure must also involve both enhanced retinoid secretion and redistribution from the liver, as well as increased catabolism of ATRA and 9CRA and elimination of the resulting catabolic products from the body.
Xenobiotic-induced retinoid redistribution from the liver to extrahepatic tissues
The literature clearly establishes that xenobiotic exposure causes a loss of retinyl esters and retinol from hepatic stores. However, alterations in tissue retinoid levels are also observed in other tissues, especially the kidney but also the lung and other tissues. It is important to ask whether this redistribution of tissue retinoids within the body represents a protective response aimed at shielding the body from the toxic effects of xenobiotics or whether this is simply a toxic manifestation of xenobiotic exposure. At present, this is not known.
The decrease in hepatic retinoid levels observed upon TCDD administration is accompanied by a more than 10-fold increase in kidney retinoid levels (102). Other investigators reported that the observed TCDD-induced loss of hepatic total retinol (retinol + retinyl ester) was accompanied by a significant dose-dependent increase in plasma and renal retinol and ATRA concentrations (96-98). However, the observed changes in retinoid levels in different organs do not simply reflect a redistribution of retinoids between the organs. Rather, the actual amount of retinoid detected in TCDD-treated kidneys is far lower than the amount lost by the liver. The increase in kidney retinoid levels accounts for only approximately 1/40 and 1/60, respectively, of the retinoid lost from the liver upon dioxin-like PCB77 or TCDD administration (102,111). The increase in retinyl ester levels observed in rat kidneys was accompanied by an increase in renal LRAT mRNA levels and activity (96,123). It has been reported that the increase in renal LRAT activity in TCDD-exposed rats, followed temporally by an increase in renal retinyl esters, correlates well with increased renal ATRA levels (96).
A 40% decline in hepatic total retinol content upon dioxin-like PCB77 administration was accompanied by 3-fold increase in renal total retinol levels (111). PCB77, PCB126 or PCB153 administration was found to lead to an increase in total retinol in kidney, but was also associated with a decrease in pulmonary total retinol levels in rats (87,106).
After exposure to a mixture of PCBs (Arochlor 1254), pregnant rats showed a dose-dependent long-term reduction in hepatic and serum retinoid levels for both the dams and offspring (108). TCDD exposure of pregnant rats, aside from its effects on the dam, leads to reduced hepatic and pulmonary total retinol levels and a persistently impaired ability of the developing fetuses and neonates to accumulate and store hepatic total retinol that is accompanied by increased retinoid accumulation in kidneys (124,125). The cellular processes responsible for this in utero effect are not known.
Disruption of retinoid transport in the circulation by xenobiotics
For retinol to be redistributed from hepatic retinyl ester stores to the kidney or other tissues requires the actions of RBP. RBP is the sole protein able to mobilize retinol that is formed upon retinyl ester hydrolysis from hepatic stores. Except for PCB toxicity elicited directly by agonist activities towards AhR and AhR-independent mechanisms, PCB biotransformation products such as the hydroxylated-PCBs (OH-PCB) (see Figure 7) are thought to be responsible for a majority of the disrupting effects on hepatic and extrahepatic retinoid homeostasis. Upon entering the body, PCBs first undergo cytochrome P450 (CYP)-dependent oxidation resulting in the formation of highly reactive arene oxide or other radical intermediates that are capable of forming covalent adducts with cellular components. Non-enzymatic rearrangements of oxidized PCBs give rise to phenolic metabolites or OH-PCBs. These phenolic metabolites can undergo further oxidative metabolism resulting in the formation of potentially cytotoxic dihydroxylated- or quinoid-PCB metabolites (126). When the hydroxyl moiety is located para to the phenyl-phenyl bond and has adjacent chlorine atoms, the structure resembles that of the prohormone thyroxine (T4). This structural similarity allows the OH-PCBs to bind with high affinity to the thyroxine-binding protein transthyretin (TTR). The importance of TTR for sustaining retinoid homeostasis is that this serum protein forms a complex with serum RBP (forming the TTR-RBP complex), thus preventing renal filtration of the relatively small RBP by the glomeruli (127). OH-PCB binding to TTR results in a lessened affinity of TTR for RBP and dissociation of the TTR-RBP complex, enhancing renal filtration of RBP together with its bound retinol. This lessens blood levels of retinol-RBP and diminishes the total amount of retinoid present in the body. OH-PCB was also shown to decrease circulating levels of thyroid hormone forms in rats through competitive binding to transthyretin, inhibiting the formation of the serum transport protein complex carrying both retinol and thyroxin (103,128). A pronounced decrease in plasma retinol levels is observed upon administration of single dose of dioxin-like PCB congeners, including for the mixture Aroclor 1254. However, no significant changes in plasma retinol levels are observed after the exposure to approximately the same amount of the non-dioxin-like PCB, PCB 153, or a mixture of these as Aroclor 1260 (103).
Administration of acetaminophen, carbon tetrachloride and D-galactosamine to rats also has been shown to be accompanied by a decrease in plasma retinol and RBP concentrations (129-131). Based on these findings, it was proposed that RBP can serve as a biomarker of drug-induced hepatotoxicity (129-131). Reduction in serum and tissue retinoic acid levels also has been reported to occur after phenobarbital and 3-methylcholantrene treatments (132). Fungicide administration, including administration of propiconazole, triadimefon, or myclobutanil, also is reported to markedly decrease hepatic ATRA levels, with a reduction ranging from 45-67% (133).
It is our view that the dramatic loss of hepatic total retinol upon xenobiotic exposure cannot be explained solely by retinol being mobilized from the liver and redistributed to extrahepatic tissue or being lost from the circulation due to enhanced renal filtration of retinol-RBP. Rather, the disruptive effects of xenobiotics that are discussed below must also be considered a major cause of hepatic retinoid loss.
Retinoic acid metabolism and actions: effects of xenobiotics
The demand for ATRA and 9CRA to act as transcription regulators in response to xenobiotic exposure is underscored by the studies discussed above (see the text under “Retinoid Effects on Xenobiotic Metabolism and Toxicity”). The functional importance of ATRA and 9CRA as triggering agents for activating xenobiotic metabolism is underscored by studies showing that retinol mobilization from hepatic stellate cells and retinol oxidation to ATRA or 9CRA occurs more quickly than most other signs of xenobiotic intoxication, often preceding even CYP upregulation (134).
TCDD-mediated toxicity relies on the maintenance of proper retinoid signaling. During mouse development, ATRA has been shown to control AhR expression via a RARγ-dependent process (135). In adult mouse liver, the retinoic acid-responsive RAR isoform, RARβ, was demonstrated to be responsible for TCDD-induced effects on retinoid homeostasis, since RARβ-deficient mice show no toxic responses upon dioxin administration. TCDD exposure leads to a 2-fold induction of RXRγ in rat liver 6 hours after exposure to a large dose of TCDD (40 µg/kg) (113) and increases both RARγ and RXRα mRNA levels in normal human keratinocytes exposed to TCDDs at a media concentration of 10−8 M (136). However, genetic ablation of only one RXRα allele or both alleles for RARα, RXRβ or RXRγ does not change the responses observed in mice to TCDD exposure (55). Hepatocyte RXRα-deficiency was shown to protect mice from TCPOBOP-induced hepatomegaly and other morphological changes (38) as well as from acetaminophen-induced hepatotoxicity (39). Moreover, the absence of sufficient retinoid stores was shown to prevent the development of thioacetamide-induced hepatotoxicity in mice (137). Collectively, these findings strongly point to roles for ATRA and 9CRA, acting through RARγ and RXRα, in facilitating xenobiotic metabolism.
Studies of retinoid-related gene knock-out mice indicate an important role for cellular retinol-binding protein I (CRBPI) in the prevention of dioxin-induced hepatic retinoid depletion. Mice lacking CRBPI and both cellular retinoic acid-binding protein I (CRABP I) and II (CRABP II), but not individual knockouts of CRABP I or CRABP II, show an almost complete depletion of hepatic retinyl ester, retinol and ATRA, 28 days after TCDD exposure when given at a dose of 50 µg TCDD/kg of body weight. This was accompanied by an increased induction of CYP1A1 activity in this triple knockout strain (CRBPI-/-/CRABPI-/-/CRABPII-/- mice), which was 2-times higher than observed in matched wild type mice (55).
Tight functional connections between the retinoid and xenobiotic metabolic pathways are further evidenced by studies showing that AhR-/- mice compared to matched wild type mice display a 3-fold increase in hepatic concentrations of retinyl palmitate and retinol, and an elevation in ATRA levels (138). Several TCDD-independent AhR-mediated effects on selected retinoid metabolizing enzymes and transport proteins were reported. The absence of functional AhR in mice due to its genetic ablation leads to a more than 6-fold upregulation of hepatic CRBPI mRNA expression (19) and a marked reduction (by 8-10-fold) in the expression of mRNAs for aldehyde dehydrogenase 1 (ALDH1) and 2 (ALDH2), enzymes involved in ATRA biosynthesis (138). However, in wild type mice, expression of these genes is not affected following TCDD-induced AhR activation (19,138).
Based on the data available in the literature, one must surmise that either competitive utilization of ATRA and 9CRA for regulating expression of inducible xenobiotic metabolizing enzymes and/or accelerated catabolism of these transcriptionally active retinoid species contribute to the hepatotoxicity that is induced by xenobiotic exposure. Each of these possibilities would lead to a relative lack of ATRA and 9CRA, negatively affecting normal retinoic acid-dependent signaling pathways, further contributing to the toxic manifestation of xenobiotic exposure. This could be very important for leading to the development of symptoms of xenobiotic toxicity since retinoid metabolism is known to be regulated by ATRA, acting in a negative feedback mode. Suppression of retinoic acid-responsive genes involved in retinoid metabolism and actions upon TCDD administration has been reported. Diminished hepatic LRAT activity observed after TCDD exposure of rats is a representative example of this, since the expression level of LRAT is highly dependent on ATRA availability (139). Similarly, for murine embryonic palate mesenchymal cells, TCDD administration inhibited both RARβ and CRABP II expression, other known targets of ATRA (140). These data clearly point to xenobiotic effects on ATRA/9CRA signaling and/or levels upon xenobiotic exposure.
CYP involvement in modulating retinoid-xenobiotic interactions
What is the molecular basis for the observed interactions between retinoids and xenobiotics in the metabolism of ATRA and 9CRA? The effects of natural and synthetic retinoid agonists and antagonists on cytochrome P450 enzymes have attracted considerable research interest. These enzymes are modulated both by endogenous and exogenous factors, and in turn, they influence the tissue levels and biological actions of environmentally-derived compounds (141). Compounds that are strong CYP enzyme inducers such as glucocorticoids, phenobarbital and rifampicin can dramatically affect the clearance of co-consumed drugs or other compounds that are substrates for the enzymes (2). Retinoids are not an exception to this observation. There are many examples where PAH exposure either enhances the clearance and/or alters the tissue disposition of drugs. In both animal models and cell lines, it has been shown that PAHs not only induce CYP1A isoforms, but they also can alter the expression of other CYPs, including CYP1B1/2 and CYP2E1, certain phase II enzymes, some phase III xenobiotic transport proteins, levels of plasma proteins, and liver mass. Modifications of any of these parameters can lead to changes in the biological disposition of drugs, altering their concentrations in blood and tissues (142). As noted below, each of these CYP species has been reported to metabolize retinoids and thus it seems likely that retinoids too are being acted upon by CYPs that are induced upon PAH exposure.
A number of hepatic CYPs are known to be directly regulated at the transcriptional level by ATRA, including CYP26A1, CYP26B1 and CYP26C1 (32,143). These CYPs are widely accepted as being responsible for retinoic acid catabolism in the body (144-147). As summarized in Table 1, other CYPs too have been shown to metabolize ATRA in vitro and in vivo including CYP2E1, CYP2S1, CYP2C22, CYP2C39, CYP2C8, CYP2C9 and CYP3A4 (62,147). Thus, many CYPs that can be induced by retinoids are able to contribute either to increased retinoic acid production or to its catabolic elimination.
Table 1. A summary of CYPs reported in the literature to be involved in retinoid oxidation.
| Metabolic transformation | Specific CYPs | Non-specific CYPs |
|---|---|---|
| Retinol → Retinal | − | CYP1A1 (148) |
| CYP1A2 (148) | ||
| CYP1B1 (148,149) | ||
| CYP3A4 (148) | ||
| Retinal → Retinoic acid | − | CYP1A1 (150,151) |
| CYP1A2 (148,151) | ||
| CYP1B1 (149,151) | ||
| CYP3A4 (148,151) | ||
| Retinoic acid → Oxidized metabolites | CYP26A1 (147); CYP26B1 (152); CYP26C1 (146) | CYP1A1 (153) |
| CYP1A2 (154) | ||
| CYP2A6 (153) | ||
| CYP2B4 (154) | ||
| CYP2B6 (153) | ||
| CYP2C8 (154-156) | ||
| CYP2C9 (154-156) | ||
| CYP2C39 (155) | ||
| CYP3A4 (154-156) | ||
| CYP3A7 (153) |
Although it is generally accepted that oxidation of retinol to retinal and subsequently to retinoic acid in biological systems is catalyzed by known dehydrogenases (144) (Figure 4), it has been known for many years that CYPs are also capable of catalyzing these reactions. Tomita et al. reported that the synthesis of ATRA from all-trans-retinal by microsomes of 3-methylcholanthrene-treated rats can be catalyzed by a CYP-linked monooxygenase system, that when purified and characterized, was identified to be CYP1A1 (150). Exploring the effects of selective inhibitors on human liver microsome-catalyzed oxidation of all-trans-retinol to all-trans-retinal and human cDNA expressed CYP enzymes Chen et al. showed that CYP1A2 and CYP3A4 are able to contribute to retinoic acid formation from retinol. Moreover, based on catalytic activities, low Km values, and high inducibility by environmental chemicals and xenobiotics, it was proposed that both CYP1A1 and CYP1B1 may play important roles in ATRA biosynthesis from all-trans-retinol by human extrahepatic and fetal tissues, but not in hepatic synthesis of ATRA (148). Subsequently, Choudhary et al. reported that both human and mouse CYP1B1 are able to oxidize all-trans-retinol to all-trans-retinal and all-trans-retinal to ATRA, however CYP1B1 was not able to oxidize ATRA forming hydroxylated or other ATRA catabolic products (149).
Using a series of individual CYPs (including CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, 3A41 + cytochrome b5, 3A5, and 4A11) expressed from cDNAs in insect cells together with NADPH-P450 reductase, Zhang et al. investigated the role of these human CYPs in the oxidation of all-trans- and 9-cis-retinal to the corresponding retinoic acid isomers (151). Among the CYP species tested only CYP1A1, 1A2, 1B1, and 3A41 + b5 converted all-trans-retinal (provided at 20 mM) to ATRA and only CYP1A2, CYP1A1, 2C19, and 3A41+ b5 oxidized 9-cis-retinal to 9CRA. CYP1A2 was reported to be the most efficient for 9CRA formation. Despite characterizing these reactions with regards to turnover numbers, kinetic constants, including Vmax and Km values, the physiological importance of these enzymes in retinoid metabolism is not generally accepted. This is because the concentrations of substrates employed in these in vitro studies were 1,000-5,000-times greater that the concentrations present in vivo, raising obvious questions regarding their physiologic relevance. However, Zhang et al. proposed that preferential disposition of protein-bound retinals into the endoplasmic reticulum rather than the cytoplasm in vivo, would favor a role for CYPs in retinal oxidation (151). Generally though, this suggestion seems to be outside of the mainstream of present understanding.
Upon xenobiotic-induced CYP activation, a condition of retinoid wasting is often observed in vivo, suggesting that increased catabolism of ATRA and 9CRA is occurring. In addition to the three CYP26 species, many CYP isoforms have been shown to contribute to retinoic acid catabolism. Roberts et al. reported that purified rabbit CYP1A2, as well as phenobarbital-inducible CYP2B4, show high activities for catalyzing the 4-hydroxylation of retinoic acid, retinol and retinal (154). Mouse CYP2C39 has been shown to catalyze 4-hydroxylation of ATRA in the liver. Moreover, CYP2C39 down-regulation has been hypothesized to be responsible for high ATRA levels observed in AhR-null mice (155).
In vitro studies employing recombinant human CYPs identified CYP2C8, CYP2C9 and CYP3A4, but not CYP1A1 or CYP1A2, as enzymes able to catalyze 4-hydroxylation of ATRA. Based on Km values and inhibition studies, it was suggested that CYP2C8 is a major contributor to 4-hydroxylation of ATRA in human liver and that CYP3A subfamily members may also be minor participants in this enzymatic process (156).
Other studies, making use of phenotyped human liver microsomes, allowed for the identification of CYP2B6, CYP2C8, CYP3A4/5, and CYP2A6 as being able to catalyze the formation of both 4-OH-ATRA and 4-oxo-ATRA. CYP2B6, CYP2C8, and CYP2A6 activities also contributed to the formation of 18-OH-ATRA, whereas CYP2A6, CYP2B6, and CYP3A4/5 activities correlated with 5,6-epoxy-ATRA formation. Another study involving the use of recombinant human CYPs expressed in lymphoblast microsomes, showed that the formation of 4-OH-ATRA by CYP3A7 > CYP3A5 > CYP2C18 > CYP2C8 > CYP3A4 > CYP2C9, whereas 18-OH-ATRA formation involved CYPs 4A11 > 3A7 > 1A1 > 2C9 > 2C8 > 3A5 > 3A4 > 2C18. Kinetic studies have identified CYP3A7 as the most active CYP for catalyzing the formation of 4-OH-, 4-oxo- and 18-OH-ATRA. The use of embryonic kidney cells stably transfected with human cytochrome P450 cDNAs confirmed the involvement of CYP3A7, CYP1A1, and CYP2C8 in the oxidation of ATRA (157). To a lesser extent, CYP1A2, CYP2C9, and CYP3A4 also are able to catalyze ATRA oxidation (153). Through the use of inhibition studies and correlation analysis, McSorley & Daly concluded that CYP2C8 is the major CYP present in human liver microsomes that is involved in the formation of 4-OH-ATRA from ATRA. These investigators reported that CYP3A4 and, to a lesser extent CYP2C9, also contribute to 4-OH-ATRA formation. Based on their data, these investigators concluded that CYP2C8, CYP2C9 and CYP3A4 together with the CYP26 species likely contribute to ATRA oxidation in the liver when ATRA levels are high (158). Thus, based on these data, it appears that xenobiotic-induced CYPs are able directly to affect tissue steady state levels of ATRA and 9CRA by influencing both their formation and catabolism.
Consistent with the biochemical data, xenobiotic exposure increases ATRA turnover rates dramatically, both in vivo and in vitro, leading to diminished levels of ATRA and affecting ATRA-dependent signaling. Using Pxr-/-, wild-type, and PXR-humanized transgenic mouse models, it was established that pregnenolone 16α-carbonitrile and several clinically relevant PXR ligands (rifampicin and dexamethasone) each increased ATRA metabolism both in vitro and in vivo. This effect was shown to be PXR-dependent and up-regulation of Cyp3a expression was proposed to be the major factor responsible for this observation. Induction of the Mdr1a, Mrp3, and Oatp2 genes was also observed. The results of this study suggest that administration of PXR ligands can increase ATRA metabolism through activation of the PXR-CYP3A pathway. This is proposed to be one of the mechanisms accounting for ATRA loss upon xenobiotic exposure (159). Administration of conazoles, fungicides used in agriculture and as pharmaceuticals, including propiconazole, triadimefon, and myclobutanil, increases hepatic microsomal ATRA metabolism in mice, resulting in the formation of 4-OH- and 4-oxo-ATRA species. CYP26A1, CYP2B, and CYP3A, but not CYP1A1 proteins have been identified as being responsible for more than 50% of the observed decrease in hepatic levels of ATRA in vivo (133). In rats, a single intraperitoneal injection of 20 mg 3,3',4,4',5,5'-hexabromobiphenyl (a brominated analog of coplanar PCB169)/kg body weight, which results in a decline in hepatic retinol and retinyl palmitate levels to 23% and 21% respectively, compared to corresponding control values, is proposed to be responsible for the significant elevation of ATRA hydroxylation catalyzed via the CYP system. For this study, increased rates of 4-OH- and 4-oxo-ATRA formation were associated with increased CYP1A1 activity (157).
Xenobiotic exposure has been shown to increase not only retinoic acid oxidation rates, but also retinoic acid isomerization and glucuronidation as well. 3-Methylcholanthrene (a TCDD-like CYP1A inducer) administration to rats increased microsomal glucuronidation of both 13CRA and ATRA and enhanced formation of 13-cis-retinoyl-β-glucuronide by up to 7-fold in rat microsomes obtained from treated animals. However, the rates of glucuronidation by control or phenobarbital-induced rat microsomes differed only slightly. In addition to glucuronides of 13CRA and ATRA, 9CRA and its glucuronide were also detected, although these were less abundant (160). Increased UDP-glucuronyltransferase activity was also observed upon 3,3',4,4',5,5'-hexabromobiphenyl administration to rats (157).
The physiological importance of increased CYP expression and activity and the degree to which this accounts for increased ATRA catabolism upon xenobiotic exposure is a key issue that remains to be resolved. Two of the central questions that need to be addressed are: To what extent do particular CYP isoforms contribute to diminished ATRA and 9CRA levels in vivo? And, do other CYP isoforms or oxygenases catalyze retinoid catabolism; CYPs that have not yet been identified in the literature as doing so?
Effects of retinoid supplementation on xenobiotic toxicity
Attempts to replenish retinoid stores as a means of minimizing the toxic manifestations of xenobiotic exposure have generally not been successful. But this is not universally been the case. Most attempts have not resulted in restored retinoid homeostasis and, often have resulted in an aggravation or potentiation of xenobiotic toxicity. However, some investigators have provided data showing that retinol administration can protect mice from xenobiotic-induced hepatic toxicity.
In studies of CCl4-induced liver injury (161,162), pretreatment of rats with relatively large (pharmacological) doses of retinol prior to xenobiotic exposure has repeatedly been shown to potentiate liver injury induced by CCl4. The same conclusion was reached when either allyl alcohol, acetaminophen or galactosamine treatment were used to induce hepatic injury in mice (163-165). In rats, dietary studies involving feeding diets containing high levels of retinol (a daily dose of 250,000 IU/kg body weight) for different time periods prior to administration of a single intraperitoneal dose of CCl4 showed for all treatments equivalent potentiation of CCl4 hepatotoxicity. Although retinol feeding was found to increase concentrations of retinyl palmitate in the liver, a linear correlation was not seen between concentrations of retinyl palmitate and the extent of retinol-potentiated CCl4 hepatotoxicity. Retinol feeding also has been reported to potentiate hepatotoxicity of even minimally toxic doses of acetaminophen, allyl alcohol, and endotoxin. Because each of these agents produces hepatic injury through different mechanisms, it was concluded that retinol potentiates hepatic injury by altering a shared process needed for the progression of hepatocellular injury (166). As an example of this, treatment of mice orally with 3,000 IU retinyl acetate, four times at 12 h intervals, after administration of the hepatotoxin thioacetamide aggravated many parameters that are known to contribute to acute hepatotoxicity (137).
Other studies also have reached a similar conclusion that retinoid supplementation enhances xenobiotic toxicity. In C57BL/6N mice, simultaneous administration, as a single dose consisting of up to 18 µg TCDD/kg body weight and 200 mg ATRA/kg body weight to pregnant dams, dramatically enhances the incidence of cleft palate formation in fetuses. Although both compounds are known teratogens, the combined treatment resulted in a synergistic effect on teratogenic outcome (167).
Studies by Yang et al. involving TCDD administration however provide data that support the opposite conclusion that retinol supplementation lessens xenobiotic hepatotoxicity. Yang et al. reported that pretreatment of mice with 2,500 IU retinol/kg body weight for one week before and throughout the period of TCDD exposure (lasting up to 42 days) attenuated, although did not fully eliminate, the severity of dioxin-induced hepatic injury after either administration of a single dose of TCDD (40 µg/kg body weight) or after repeated daily dosing at 0.1 µg/kg body weight for 42 days (11). However, in these studies, the rate of liver injury induced by TCDD administration was evaluated only semiquantitatively for relative liver weights and histological parameters, but not by liver function tests. Although this semiquantitative approach weakens the conclusions that can be reached from the study, the data do allow for the conclusion that the hepatic injury observed upon TCDD administration following retinoid pretreatment is diminished.
Other studies have suggested that the effects of retinoid-supplementation on xenobiotic-induced liver toxicity may depend on the specific xenobiotic being studied. High levels of dietary retinyl palmitate supplementation were reported to prevent hepatic injury in rats exposed to non-dioxin-like PCB153, but that supplementation was not found to be protective in rats exposed to dioxin-like PCB77 or a combination of both compounds. Histopathological examination of livers was convincing that high levels of dietary retinyl palmitate intake lessened the severity of hepatocellular necrosis and fatty changes induced by PCB77 alone or when given in combination with PCB153. It should be noted that supplementation with high levels of dietary retinyl palmitate diminished, but did not eliminate the number and volume of altered hepatic foci (107). Other studies in rats have shown that an increase of the retinoid content of a moderately supplemented experimental diet (from 0.6 to 6 retinol equivalents/g diet), after coplanar PCB77 administration, did not affect the rate of hepatic retinoid loss; however, this did increased renal accumulation of retinol and retinyl esters (168).
Conclusions and future direction
Overall, it is clear that retinoid-xenobiotic interactions are complex, multilevel and bilateral, as schematically illustrated in Figure 8. It is clear that much still needs to be learned regarding the molecular origins of these interactions. It remains to be definitively established what xenobiotic molecular targets, including nuclear receptors, regulatory proteins, enzymes, and transporters affect retinoid metabolism and signaling. Nor is it understood what contribution specific CYP isoforms make towards diminishing tissue levels of retinyl esters, retinol and ATRA/9CRA in vivo. A question also remains as to whether other CYP isoforms or oxygenases that can catalyze retinoid catabolism in vivo still remain to be identified? The answers to these fundamental questions will not only improve our understanding of the basic fundamental issues, but may also open the way for the development of practical interventions aimed at preventing the toxicity of xenobiotics.
Figure 8.

Graphical summary of retinoid-xenobiotic interaction.
Acknowledgements
The author gratefully acknowledges Dr. William S. Blaner from Columbia University in the City of New York for his critical reading and insightful review of the manuscript.
Footnotes
Conflicts of Interest: The author has no conflicts of interest to declare.
References
- 1.Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res 2005;28:249-68. [DOI] [PubMed] [Google Scholar]
- 2.Xie W, Evans RM. Orphan nuclear receptors: the exotics of xenobiotics. J Biol Chem 2001;276:37739-42. [DOI] [PubMed] [Google Scholar]
- 3.Omiecinski CJ, Vanden Heuvel JP, Perdew GH, et al. Xenobiotic metabolism, disposition, and regulation by receptors: from biochemical phenomenon to predictors of major toxicities. Toxicol Sci 2011;120 Suppl 1:S49-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woods CG, Heuvel JP, Rusyn I. Genomic profiling in nuclear receptor-mediated toxicity. Toxicol Pathol 2007;35:474-94. [DOI] [PubMed] [Google Scholar]
- 5.Aleksunes LM, Klaassen CD. Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPARalpha-, and Nrf2-null mice. Drug Metab Dispos 2012;40:1366-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barouki R, Aggerbeck M, Aggerbeck L, et al. The aryl hydrocarbon receptor system. Drug Metabol Drug Interact 2012;27:3-8. [DOI] [PubMed] [Google Scholar]
- 7.Song Z, Pollenz RS. Ligand-dependent and independent modulation of aryl hydrocarbon receptor localization, degradation, and gene regulation. Mol Pharmacol 2002;62:806-16. [DOI] [PubMed] [Google Scholar]
- 8.Labrecque MP, Prefontaine GG, Beischlag TV. The aryl hydrocarbon receptor nuclear translocator (ARNT) family of proteins: transcriptional modifiers with multi-functional protein interfaces. Curr Mol Med 2013;13:1047-65. [DOI] [PubMed] [Google Scholar]
- 9.Li S, Pei X, Zhang W, et al. Functional analysis of the dioxin response elements (DREs) of the murine CYP1A1 gene promoter: beyond the core DRE sequence. Int J Mol Sci 2014;15:6475-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol 1995;35:307-40. [DOI] [PubMed] [Google Scholar]
- 11.Yang YM, Huang DY, Liu GF, et al. Inhibitory effects of vitamin A on TCDD-induced cytochrome P-450 1A1 enzyme activity and expression. Toxicol Sci 2005;85:727-34. [DOI] [PubMed] [Google Scholar]
- 12.Ramadoss P, Marcus C, Perdew GH. Role of the aryl hydrocarbon receptor in drug metabolism. Expert Opin Drug Metab Toxicol 2005;1:9-21. [DOI] [PubMed] [Google Scholar]
- 13.Barouki R, Coumoul X, Fernandez-Salguero PM. The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett 2007;581:3608-15. [DOI] [PubMed] [Google Scholar]
- 14.Song J, Clagett-Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR, et al. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci U S A 2002;99:14694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henry EC, Welle SL, Gasiewicz TA. TCDD and a putative endogenous AhR ligand, ITE, elicit the same immediate changes in gene expression in mouse lung fibroblasts. Toxicol Sci 2010;114:90-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan KP, Wang B, Yang M, Boutros PC, Macaulay J, Xu H, et al. Aryl hydrocarbon receptor is a transcriptional activator of the human breast cancer resistance protein (BCRP/ABCG2). Mol Pharmacol 2010;78:175-85. [DOI] [PubMed] [Google Scholar]
- 17.Watson JD, Prokopec SD, Smith AB, Okey AB, Pohjanvirta R, Boutros PC. TCDD dysregulation of 13 AHR-target genes in rat liver. Toxicol Appl Pharmacol 2014;274:445-54. [DOI] [PubMed] [Google Scholar]
- 18.Yeager RL, Reisman SA, Aleksunes LM, et al. Introducing the “TCDD-inducible AhR-Nrf2 gene battery”. Toxicol Sci 2009;111:238-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tijet N, Boutros PC, Moffat ID, et al. Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries. Mol Pharmacol 2006;69:140-53. [DOI] [PubMed] [Google Scholar]
- 20.Celius T, Roblin S, Harper PA, et al. Aryl hydrocarbon receptor-dependent induction of flavin-containing monooxygenase mRNAs in mouse liver. Drug Metab Dispos 2008;36:2499-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kakizaki S, Karami S, Negishi M. Retinoic acids repress constitutive active receptor-mediated induction by 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene of the CYP2B10 gene in mouse primary hepatocytes. Drug Metab Dispos 2002;30:208-11. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Min G, Kemper B. Chromatin assembly enhances binding to the CYP2B1 phenobarbital-responsive unit (PBRU) of nuclear factor-1, which binds simultaneously with constitutive androstane receptor (CAR)/retinoid X receptor (RXR) and enhances CAR/RXR-mediated activation of the PBRU. J Biol Chem 2001;276:7559-67. [DOI] [PubMed] [Google Scholar]
- 23.Wahlang B, Falkner KC, Clair HB, et al. Human receptor activation by aroclor 1260, a polychlorinated biphenyl mixture. Toxicol Sci 2014;140:283-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luckert C, Ehlers A, Buhrke T, et al. Polycyclic aromatic hydrocarbons stimulate human CYP3A4 promoter activity via PXR. Toxicol Lett 2013;222:180-8. [DOI] [PubMed] [Google Scholar]
- 25.Xie W, Barwick JL, Simon CM, et al. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev 2000;14:3014-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blumberg B, Sabbagh W, Jr, Juguilon H, et al. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev 1998;12:3195-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf KK, Wood SG, Hunt JA, et al. Role of the nuclear receptor pregnane X receptor in acetaminophen hepatotoxicity. Drug Metab Dispos 2005;33:1827-36. [DOI] [PubMed] [Google Scholar]
- 28.Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci U S A 1997;94:5361-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel RD, Hollingshead BD, Omiecinski CJ, et al. Aryl-hydrocarbon receptor activation regulates constitutive androstane receptor levels in murine and human liver. Hepatology 2007;46:209-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaban Z, Soliman M, El-Shazly S, et al. AhR and PPARalpha: antagonistic effects on CYP2B and CYP3A, and additive inhibitory effects on CYP2C11. Xenobiotica 2005;35:51-68. [DOI] [PubMed] [Google Scholar]
- 31.Borland MG, Krishnan P, Lee C, et al. Modulation of aryl hydrocarbon receptor (AHR)-dependent signaling by peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) in keratinocytes. Carcinogenesis 2014;35:1602-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brtko J, Dvorak Z. Role of retinoids, rexinoids and thyroid hormone in the expression of cytochrome p450 enzymes. Curr Drug Metab 2011;12:71-88. [DOI] [PubMed] [Google Scholar]
- 33.Wan YJ, An D, Cai Y, et al. Hepatocyte-specific mutation establishes retinoid X receptor alpha as a heterodimeric integrator of multiple physiological processes in the liver. Mol Cell Biol 2000;20:4436-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai T, Wu Y, Leng AS, et al. RXRalpha-regulated liver SAMe and GSH levels influence susceptibility to alcohol-induced hepatotoxicity. Exp Mol Pathol 2003;75:194-200. [DOI] [PubMed] [Google Scholar]
- 35.Cai Y, Dai T, Ao Y, et al. Cytochrome P450 genes are differentially expressed in female and male hepatocyte retinoid X receptor alpha-deficient mice. Endocrinology 2003;144:2311-8. [DOI] [PubMed] [Google Scholar]
- 36.Oda Y, Nakajima M, Tsuneyama K, et al. Retinoid X receptor α in human liver is regulated by miR-34a. Biochem Pharmacol 2014;90:179-87. [DOI] [PubMed] [Google Scholar]
- 37.Noreault-Conti TL, Fellows A, Jacobs JM, et al. Arsenic decreases RXRalpha-dependent transcription of CYP3A and suppresses immune regulators in hepatocytes. Int Immunopharmacol 2012;12:651-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai Y, Konishi T, Han G, et al. The role of hepatocyte RXR alpha in xenobiotic-sensing nuclear receptor-mediated pathways. Eur J Pharm Sci 2002;15:89-96. [DOI] [PubMed] [Google Scholar]
- 39.Wu Y, Zhang X, Bardag-Gorce F, et al. Retinoid X receptor alpha regulates glutathione homeostasis and xenobiotic detoxification processes in mouse liver. Mol Pharmacol 2004;65:550-7. [DOI] [PubMed] [Google Scholar]
- 40.Dai G, Chou N, He L, et al. Retinoid X receptor alpha Regulates the expression of glutathione s-transferase genes and modulates acetaminophen-glutathione conjugation in mouse liver. Mol Pharmacol 2005;68:1590-6. [DOI] [PubMed] [Google Scholar]
- 41.Wang H, Liu K, Geng M, et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res 2013;73:3097-108. [DOI] [PubMed] [Google Scholar]
- 42.Howell SR, Shirley MA, Ulm EH. Effects of retinoid treatment of rats on hepatic microsomal metabolism and cytochromes P450. Correlation between retinoic acid receptor/retinoid x receptor selectivity and effects on metabolic enzymes. Drug Metab Dispos 1998;26:234-9. [PubMed] [Google Scholar]
- 43.Wang K, Mendy AJ, Dai G, et al. Retinoids activate the RXR/SXR-mediated pathway and induce the endogenous CYP3A4 activity in Huh7 human hepatoma cells. Toxicol Sci 2006;92:51-60. [DOI] [PubMed] [Google Scholar]
- 44.Chen S, Wang K, Wan YJ. Retinoids activate RXR/CAR-mediated pathway and induce CYP3A. Biochem Pharmacol 2010;79:270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pettersson F, Hanna N, Lagodich M, et al. Rexinoids modulate steroid and xenobiotic receptor activity by increasing its protein turnover in a calpain-dependent manner. J Biol Chem 2008;283:21945-52. [DOI] [PubMed] [Google Scholar]
- 46.Rühl R, Sczech R, Landes N, et al. Carotenoids and their metabolites are naturally occurring activators of gene expression via the pregnane X receptor. Eur J Nutr 2004;43:336-43. [DOI] [PubMed] [Google Scholar]
- 47.Gradelet S, Leclerc J, Siess MH, et al. beta-Apo-8'-carotenal, but not beta-carotene, is a strong inducer of liver cytochromes P4501A1 and 1A2 in rat. Xenobiotica 1996;26:909-19. [DOI] [PubMed] [Google Scholar]
- 48.Kawai M, Chen J, Cheung CY, et al. Transcript profiling of cytochrome P450 genes in HL-60 human leukemic cells: upregulation of CYP1B1 by all-trans-retinoic acid. Mol Cell Biochem 2003;248:57-65. [DOI] [PubMed] [Google Scholar]
- 49.Yamada H, Yamaguchi T, Oguri K. Suppression of the expression of the CYP2B1/2 gene by retinoic acids. Biochem Biophys Res Commun 2000;277:66-71. [DOI] [PubMed] [Google Scholar]
- 50.Saito K, Kobayashi K, Mizuno Y, et al. Constitutive androstane/active receptor is a target of retinoic acid receptor in humans. Biochem Pharmacol 2010;80:129-35. [DOI] [PubMed] [Google Scholar]
- 51.Wang XJ, Hayes JD, Henderson CJ, et al. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc Natl Acad Sci U S A 2007;104:19589-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inouye K, Mae T, Kondo S, et al. Inhibitory effects of vitamin A and vitamin K on rat cytochrome P4501A1-dependent monooxygenase activity. Biochem Biophys Res Commun 1999;262:565-9. [DOI] [PubMed] [Google Scholar]
- 53.Soprano DR, Gambone CJ, Sheikh SN, et al. The synthetic retinoid AGN 193109 but not retinoic acid elevates CYP1A1 levels in mouse embryos and Hepa-1c1c7 cells. Toxicol Appl Pharmacol 2001;174:153-9. [DOI] [PubMed] [Google Scholar]
- 54.Vecchini F, Lenoir-Viale MC, Cathelineau C, et al. Presence of a retinoid responsive element in the promoter region of the human cytochrome P4501A1 gene. Biochem Biophys Res Commun 1994;201:1205-12. [DOI] [PubMed] [Google Scholar]
- 55.Hoegberg P, Schmidt CK, Fletcher N, et al. Retinoid status and responsiveness to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking retinoid binding protein or retinoid receptor forms. Chem Biol Interact 2005;156:25-39. [DOI] [PubMed] [Google Scholar]
- 56.Ohno M, Ikenaka Y, Ishizuka M. All-trans retinoic acid inhibits the recruitment of ARNT to DNA, resulting in the decrease of CYP1A1 mRNA expression in HepG2 cells. Biochem Biophys Res Commun 2012;417:484-9. [DOI] [PubMed] [Google Scholar]
- 57.Fallone F, Villard PH, Seree E, et al. Retinoids repress Ah receptor CYP1A1 induction pathway through the SMRT corepressor. Biochem Biophys Res Commun 2004;322:551-6. [DOI] [PubMed] [Google Scholar]
- 58.Wanner R, Zhang J, Henz BM, et al. AP-2 gene expression and modulation by retinoic acid during keratinocyte differentiation. Biochem Biophys Res Commun 1996;223:666-9. [DOI] [PubMed] [Google Scholar]
- 59.Wanner R, Brommer S, Czarnetzki BM, et al. The differentiation-related upregulation of aryl hydrocarbon receptor transcript levels is suppressed by retinoic acid. Biochem Biophys Res Commun 1995;209:706-11. [DOI] [PubMed] [Google Scholar]
- 60.Wanner R, Panteleyev A, Henz BM, et al. Retinoic acid affects the expression rate of the differentiation-related genes aryl hydrocarbon receptor, ARNT and keratin 4 in proliferative keratinocytes only. Biochim Biophys Acta 1996;1317:105-11. [DOI] [PubMed] [Google Scholar]
- 61.Couroucli XI, Liang YW, Jiang W, et al. Attenuation of oxygen-induced abnormal lung maturation in rats by retinoic acid: possible role of cytochrome P4501A enzymes. J Pharmacol Exp Ther 2006;317:946-54. [DOI] [PubMed] [Google Scholar]
- 62.Qian L, Zolfaghari R, Ross AC. Liver-specific cytochrome P450 CYP2C22 is a direct target of retinoic acid and a retinoic acid-metabolizing enzyme in rat liver. J Lipid Res 2010;51:1781-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lai XS, Yang LP, Li XT, et al. Human CYP2C8: structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr Drug Metab 2009;10:1009-47. [DOI] [PubMed] [Google Scholar]
- 64.Lu Y, Bratton S, Heydel JM, et al. Effect of retinoids on UDP-glucuronosyltransferase 2B7 mRNA expression in Caco-2 cells. Drug Metab Pharmacokinet 2008;23:364-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Samokyszyn VM, Gall WE, Zawada G, et al. 4-hydroxyretinoic acid, a novel substrate for human liver microsomal UDP-glucuronosyltransferase(s) and recombinant UGT2B7. J Biol Chem 2000;275:6908-14. [DOI] [PubMed] [Google Scholar]
- 66.Xia C, Taylor JB, Spencer SR, Ketterer B. The human glutathione S-transferase P1-1 gene: modulation of expression by retinoic acid and insulin. Biochem J 1993;292:845-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xia C, Hu J, Ketterer B, Taylor JB. The organization of the human GSTP1-1 gene promoter and its response to retinoic acid and cellular redox status. Biochem J 1996;313:155-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beckmann JD, Palmatier R, Kliewer B. Retinoic acid inhibits hydrocortisone-stimulated expression of phenol sulfotransferase in bovine bronchial epithelial cells. J Cell Physiol 1996;166:281-7. [DOI] [PubMed] [Google Scholar]
- 69.Xu S, Sun AQ, Suchy FJ. A novel RARalpha/CAR-mediated mechanism for regulation of human organic solute transporter-beta gene expression. Am J Physiol Gastrointest Liver Physiol 2014;306:G154-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hessel S, Lampen A. All-trans retinoic acid enhances the transport of phase II metabolites of benzo[a]pyrene by inducing the Breast Cancer Resistance Protein expression in Caco-2 cells. Toxicol Lett 2010;197:151-5. [DOI] [PubMed] [Google Scholar]
- 71.Fletcher N, Hanberg A, Håkansson H. Hepatic vitamin a depletion is a sensitive marker of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure in four rodent species. Toxicol Sci 2001;62:166-75. [DOI] [PubMed] [Google Scholar]
- 72.Brouwer A, van den Berg KJ, Kukler A. Time and dose responses of the reduction in retinoid concentrations in C57BL/Rij and DBA/2 mice induced by 3,4,3',4'-tetrachlorobiphenyl. Toxicol Appl Pharmacol 1985;78:180-9. [DOI] [PubMed] [Google Scholar]
- 73.Roos R, Andersson PL, Halldin K, et al. Hepatic effects of a highly purified 2,2',3,4,4',5,5'-heptachlorbiphenyl (PCB 180) in male and female rats. Toxicology 2011;284:42-53. [DOI] [PubMed] [Google Scholar]
- 74.Schmidt CK, Hoegberg P, Fletcher N, et al. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the endogenous metabolism of all-trans-retinoic acid in the rat. Arch Toxicol 2003;77:371-83. [DOI] [PubMed] [Google Scholar]
- 75.Clugston RD, Jiang H, Lee MX, et al. Altered hepatic retinyl ester concentration and acyl composition in response to alcohol consumption. Biochim Biophys Acta 2013;1831:1276-86. [PubMed]
- 76.Kamata R, Shiraishi F, Nishikawa J, et al. Screening and detection of the in vitro agonistic activity of xenobiotics on the retinoic acid receptor. Toxicol In Vitro 2008;22:1050-61. [DOI] [PubMed] [Google Scholar]
- 77.Lemaire G, Balaguer P, Michel S, et al. Activation of retinoic acid receptor-dependent transcription by organochlorine pesticides. Toxicol Appl Pharmacol 2005;202:38-49. [DOI] [PubMed] [Google Scholar]
- 78.Schoff PK, Ankley GT. Effects of methoprene, its metabolites, and breakdown products on retinoid-activated pathways in transfected cell lines. Environ Toxicol Chem 2004;23:1305-10. [DOI] [PubMed] [Google Scholar]
- 79.Li J, Ma M, Wang Z. A two-hybrid yeast assay to quantify the effects of xenobiotics on retinoid X receptor-mediated gene expression. Toxicol Lett 2008;176:198-206. [DOI] [PubMed] [Google Scholar]
- 80.Sarath Josh MK, Pradeep S, Vijayalekshmi Amma KS, et al. Phthalates efficiently bind to human peroxisome proliferator activated receptor and retinoid X receptor α, β, γ subtypes: an in silico approach. J Appl Toxicol 2014;34:754-65. [DOI] [PubMed] [Google Scholar]
- 81.Beníšek M, Kubincová P, Bláha L, et al. The effects of PAHs and N-PAHs on retinoid signaling and Oct-4 expression in vitro. Toxicol Lett 2011;200:169-75. [DOI] [PubMed] [Google Scholar]
- 82.Chen Y, Reese DH. A screen for disruptors of the retinol (vitamin A) signaling pathway. Birth Defects Res B Dev Reprod Toxicol 2013;98:276-82. [DOI] [PubMed] [Google Scholar]
- 83.Aoki T, Takada T. Bisphenol A modulates germ cell differentiation and retinoic acid signaling in mouse ES cells. Reprod Toxicol 2012;34:463-70. [DOI] [PubMed] [Google Scholar]
- 84.Nishizawa H, Manabe N, Morita M, et al. Effects of in utero exposure to bisphenol A on expression of RARalpha and RXRalpha mRNAs in murine embryos. J Reprod Dev 2003;49:539-45. [DOI] [PubMed] [Google Scholar]
- 85.Nishizawa H, Morita M, Sugimoto M, et al. Effects of in utero exposure to bisphenol A on mRNA expression of arylhydrocarbon and retinoid receptors in murine embryos. J Reprod Dev 2005;51:315-24. [DOI] [PubMed] [Google Scholar]
- 86.Koda T, Morita M, Imai H. Retinoic acid inhibits uterotrophic activity of bisphenol A in adult ovariectomized rats. J Nutr Sci Vitaminol (Tokyo) 2007;53:432-6. [DOI] [PubMed] [Google Scholar]
- 87.Chen LC, Berberian I, Koch B, et al. Polychlorinated and polybrominated biphenyl congeners and retinoid levels in rat tissues: structure-activity relationships. Toxicol Appl Pharmacol 1992;114:47-55. [DOI] [PubMed] [Google Scholar]
- 88.Chen LC, Berberian I, Glauert HP, et al. Altered tissue levels of vitamin A by selected polycyclic aromatic hydrocarbons (PAHs). Polycyclic Aromatic Compounds 1994;4:173-82. [Google Scholar]
- 89.Leo MA, Lowe N, Lieber CS. Decreased hepatic vitamin A after drug administration in men and in rats. Am J Clin Nutr 1984;40:1131-6. [DOI] [PubMed] [Google Scholar]
- 90.Azaïs V, Rachman F, Gros S, et al. Cyclosporine administration decreases liver vitamin A stores in normal and vitamin A-deficient rats. Drug Nutr Interact 1987;5:81-8. [PubMed] [Google Scholar]
- 91.Schindler R, Fielenbach T, Rave G. Flupenthixol and cefotiam: effects on vitamin A metabolism in rats. Br J Nutr 2004;92:597-605. [DOI] [PubMed] [Google Scholar]
- 92.Safe SH. Polychlorinated biphenyls (PCBs): environmental impact, biochemical and toxic responses, and implications for risk assessment. Crit Rev Toxicol 1994;24:87-149. [DOI] [PubMed] [Google Scholar]
- 93.McKinney JD, Waller CL. Polychlorinated biphenyls as hormonally active structural analogues. Environ Health Perspect 1994;102:290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nishimura N, Yonemoto J, Miyabara Y, et al. Altered thyroxin and retinoid metabolic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin in aryl hydrocarbon receptor-null mice. Arch Toxicol 2005;79:260-7. [DOI] [PubMed] [Google Scholar]
- 95.Boverhof DR, Burgoon LD, Tashiro C, et al. Temporal and dose-dependent hepatic gene expression patterns in mice provide new insights into TCDD-Mediated hepatotoxicity. Toxicol Sci 2005;85:1048-63. [DOI] [PubMed] [Google Scholar]
- 96.Nilsson CB, Hoegberg P, Trossvik C, et al. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases serum and kidney retinoic acid levels and kidney retinol esterification in the rat. Toxicol Appl Pharmacol 2000;169:121-31. [DOI] [PubMed] [Google Scholar]
- 97.Fletcher N, Giese N, Schmidt C, et al. Altered retinoid metabolism in female Long-Evans and Han/Wistar rats following long-term 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treatment. Toxicol Sci 2005;86:264-72. [DOI] [PubMed] [Google Scholar]
- 98.Van Birgelen AP, Van der Kolk J, Fase KM, et al. Subchronic dose-response study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in female Sprague-Dawley rats. Toxicol Appl Pharmacol 1995;132:1-13. [DOI] [PubMed] [Google Scholar]
- 99.Yang YM, Huang DY, Liu GF, et al. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on vitamin A metabolism in mice. J Biochem Mol Toxicol 2005;19:327-35. [DOI] [PubMed] [Google Scholar]
- 100.Fattore E, Trossvik C, Hakansson H. Relative potency values derived from hepatic vitamin A reduction in male and female Sprague-Dawley rats following subchronic dietary exposure to individual polychlorinated dibenzo-p-dioxin and dibenzofuran congeners and a mixture thereof. Toxicol Appl Pharmacol 2000;165:184-94. [DOI] [PubMed] [Google Scholar]
- 101.Novák J, Benísek M, Hilscherová K. Disruption of retinoid transport, metabolism and signaling by environmental pollutants. Environ Int 2008;34:898-913. [DOI] [PubMed] [Google Scholar]
- 102.Chu I, Lecavalier P, Hakansson H, et al. Mixture effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and polychlorinated biphenyl congeners in rats. Chemosphere 2001;43:807-14. [DOI] [PubMed] [Google Scholar]
- 103.van der Plas SA, Lutkeschipholt I, Spenkelink B, et al. Effects of subchronic exposure to complex mixtures of dioxin-like and non-dioxin-like polyhalogenated aromatic compounds on thyroid hormone and vitamin A levels in female Sprague-Dawley rats. Toxicol Sci 2001;59:92-100. [DOI] [PubMed] [Google Scholar]
- 104.Al-Salman F, Plant N. Non-coplanar plychlorinated biphenyls (PCBs) are direct agonists for the human pregnane-X receptor and constitutive androstane receptor, and activate target gene expression in a tissue-specific manner. Toxicol Appl Pharmacol 2012;263:7-13. [DOI] [PubMed] [Google Scholar]
- 105.Han X, O’Connor JC, Donner EM, et al. Non-coplanar 2,2',3,3',4,4',5,5',6,6'-decachlorobiphenyl (PCB 209) did not induce cytochrome P450 enzyme activities in primary cultured rat hepatocytes, was not genotoxic, and did not exhibit endocrine-modulating activities. Toxicology 2009;255:177-86. [DOI] [PubMed] [Google Scholar]
- 106.Håkansson H, Manzoor E, Trossvik C, et al. Effect on tissue vitamin A levels in the rat following subchronic exposure to four individual PCB congeners (IUPAC 77, 118, 126, and 153). Chemosphere 1994;29:2309-13. [DOI] [PubMed] [Google Scholar]
- 107.Berberian I, Chen LC, Robinson FR, et al. Effect of dietary retinyl palmitate on the promotion of altered hepatic foci by 3,3',4,4'-tetrachlorobiphenyl and 2,2',4,4',5,5'-hexachlorobiphenyl in rats initiated with diethylnitrosamine. Carcinogenesis 1995;16:393-8. [DOI] [PubMed] [Google Scholar]
- 108.Morse DC, Brouwer A. Fetal, neonatal, and long-term alterations in hepatic retinoid levels following maternal polychlorinated biphenyl exposure in rats. Toxicol Appl Pharmacol 1995;131:175-82. [DOI] [PubMed] [Google Scholar]
- 109.Brouwer A, van den Berg KJ. Early and differential decrease in natural retinoid levels in C57BL/Rij and DBA/2 mice by 3,4,3',4'-tetrachlorobiphenyl. Toxicol Appl Pharmacol 1984;73:204-9. [DOI] [PubMed] [Google Scholar]
- 110.Murk AJ, van den Berg JH, Koeman JH, et al. The toxicity of tetrachlorobenzyltoluenes (Ugilec 141) and polychlorobiphenyls (Aroclor 1254 and PCB-77) compared in Ah-responsive and Ah-nonresponsive mice. Environ Pollut 1991;72:57-67. [DOI] [PubMed] [Google Scholar]
- 111.Azais V, Arand M, Rauch P, et al. A time-course investigation of vitamin A levels and drug metabolizing enzyme activities in rats following a single treatment with prototypic polychlorinated biphenyls and DDT. Toxicology 1987;44:341-54. [DOI] [PubMed] [Google Scholar]
- 112.Mercier M, Pascal G, Azais-Braesco V. Retinyl ester hydrolase and vitamin A status in rats treated with 3,3',4, 4'-tetrachlorobiphenyl. Biochim Biophys Acta 1990;1047:70-6. [DOI] [PubMed] [Google Scholar]
- 113.Fletcher N, Wahlstrom D, Lundberg R, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) alters the mRNA expression of critical genes associated with cholesterol metabolism, bile acid biosynthesis, and bile transport in rat liver: a microarray study. Toxicol Appl Pharmacol 2005;207:1-24. [DOI] [PubMed] [Google Scholar]
- 114.Nilsson CB, Hanberg A, Trossvik C, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin affects retinol esterification in rat hepatic stellate cells and kidney. Environ Toxicol Pharmacol 1996;2:17-23. [DOI] [PubMed] [Google Scholar]
- 115.Iida T, Todaka T, Hirakawa H, et al. Concentration and distribution of dioxins and related compounds in human tissues. Chemosphere 2007;67:S263-71. [DOI] [PubMed] [Google Scholar]
- 116.van Ede KI, Aylward LL, Andersson PL, et al. Tissue distribution of dioxin-like compounds: potential impacts on systemic relative potency estimates. Toxicol Lett 2013;220:294-302. [DOI] [PubMed] [Google Scholar]
- 117.Håkansson H, Hanberg A. The distribution of [14C]-2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and its effect on the vitamin A content in parenchymal and stellate cells of rat liver. J Nutr 1989;119:573-80. [DOI] [PubMed] [Google Scholar]
- 118.Riebniger D, Schrenk D. Nonresponsiveness to 2,3,7,8-tetrachlorodibenzo-p-dioxin of transforming growth factor beta1 and CYP 1A1 gene expression in rat liver fat-storing cells. Toxicol Appl Pharmacol 1998;152:251-60. [DOI] [PubMed] [Google Scholar]
- 119.Hanberg A, Kling L, Hakansson H. Effect of 2,3,7,8-tetrachlorodibenzo-P-dioxin (TCDD) on the hepatic stellate cell population in the rat. Chemosphere 1996;32:1225-33. [DOI] [PubMed] [Google Scholar]
- 120.Lu H, Cui W, Klaassen CD. Nrf2 protects against 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced oxidative injury and steatohepatitis. Toxicol Appl Pharmacol 2011;256:122-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pierre S, Chevallier A, Teixeira-Clerc F, et al. Aryl hydrocarbon receptor-dependent induction of liver fibrosis by dioxin. Toxicol Sci 2014;137:114-24. [DOI] [PubMed] [Google Scholar]
- 122.Azaïs-Braesco V, Hautekeete ML, Dodeman I, et al. Morphology of liver stellate cells and liver vitamin A content in 3,4,3',4'-tetrachlorobiphenyl-treated rats. J Hepatol 1997;27:545-53. [DOI] [PubMed] [Google Scholar]
- 123.Hoegberg P, Schmidt CK, Nau H, et al. 2,3,7,8-tetrachlorodibenzo-p-dioxin induces lecithin: retinol acyltransferase transcription in the rat kidney. Chem Biol Interact 2003;145:1-16. [DOI] [PubMed] [Google Scholar]
- 124.Håkansson H, Waern F, Ahlborg UG. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the lactating rat on maternal and neonatal vitamin A status. J Nutr 1987;117:580-6. [DOI] [PubMed] [Google Scholar]
- 125.Kransler KM, Tonucci DA, McGarrigle BP, et al. Gestational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin alters retinoid homeostasis in maternal and perinatal tissues of the Holtzman rat. Toxicol Appl Pharmacol 2007;224:29-38. [DOI] [PubMed] [Google Scholar]
- 126.Haraguchi K, Koga N, Kato Y. Comparative metabolism of polychlorinated biphenyls and tissue distribution of persistent metabolites in rats, hamsters, and Guinea pigs. Drug Metab Dispos 2005;33:373-80. [DOI] [PubMed] [Google Scholar]
- 127.Soprano DR, Blaner WS. Plasma retinol-binding protein. In: Sporn MB, Roberts AB, Goodman DS. editors. The Retinoids: Biology, Chemistry and Medicine. New York, NY: Raven Press, 1994:257-81. [Google Scholar]
- 128.Brouwer A, van den Berg KJ. Binding of a metabolite of 3,4,3',4'-tetrachlorobiphenyl to transthyretin reduces serum vitamin A transport by inhibiting the formation of the protein complex carrying both retinol and thyroxin. Toxicol Appl Pharmacol 1986;85:301-12. [DOI] [PubMed] [Google Scholar]
- 129.Isozaki M, Ito K, Masubuchi Y, et al. Plasma retinol binding protein for monitoring the acetaminophen-induced hepatotoxicity. Drug Metab Pharmacokinet 2002;17:540-5. [DOI] [PubMed] [Google Scholar]
- 130.Isozaki M, Masubuchi Y, Horie T. Evaluation of drug-induced hepatotoxicity by plasma retinol binding protein. In Vivo 2002;16:61-5. [PubMed] [Google Scholar]
- 131.Isozaki M, Masubuchi Y, Horie T. Retinol binding protein in plasma to evaluate the hepatotoxicity of rats treated with CCl4. Res Commun Mol Pathol Pharmacol 2001;110:303-10. [PubMed] [Google Scholar]
- 132.Kalin JR, Wells MJ, Hill DL. Effects of phenobarbital, 3-methylcholanthrene, and retinoid pretreatment on disposition of orally administered retinoids in mice. Drug Metab Dispos 1984;12:63-7. [PubMed] [Google Scholar]
- 133.Chen PJ, Padgett WT, Moore T, et al. Three conazoles increase hepatic microsomal retinoic acid metabolism and decrease mouse hepatic retinoic acid levels in vivo. Toxicol Appl Pharmacol 2009;234:143-55. [DOI] [PubMed] [Google Scholar]
- 134.Kelley SK, Nilsson CB, Green MH, et al. Mobilization of vitamin A stores in rats after administration of 2,3, 7,8-tetrachlorodibenzo-p-dioxin: a kinetic analysis. Toxicol Sci 2000;55:478-84. [DOI] [PubMed] [Google Scholar]
- 135.Jacobs H, Dennefeld C, Feret B, et al. Retinoic acid drives aryl hydrocarbon receptor expression and is instrumental to dioxin-induced toxicity during palate development. Environ Health Perspect 2011;119:1590-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Murphy KA, Villano CM, Dorn R, et al. Interaction between the aryl hydrocarbon receptor and retinoic acid pathways increases matrix metalloproteinase-1 expression in keratinocytes. J Biol Chem 2004;279:25284-93. [DOI] [PubMed] [Google Scholar]
- 137.Shmarakov IO, Borschovetska VL, Marchenko MM, et al. Retinoids modulate thioacetamide-induced acute hepatotoxicity. Toxicol Sci 2014;139:284-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Andreola F, Fernandez-Salguero PM, Chiantore MV, et al. Aryl hydrocarbon receptor knockout mice (AHR-/-) exhibit liver retinoid accumulation and reduced retinoic acid metabolism. Cancer Res 1997;57:2835-8. [PubMed] [Google Scholar]
- 139.Zolfaghari R, Ross AC. Lecithin:retinol acyltransferase from mouse and rat liver. CDNA cloning and liver-specific regulation by dietary vitamin a and retinoic acid. J Lipid Res 2000;41:2024-34. [PubMed] [Google Scholar]
- 140.Weston WM, Nugent P, Greene RM. Inhibition of retinoic-acid-induced gene expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem Biophys Res Commun 1995;207:690-4. [DOI] [PubMed] [Google Scholar]
- 141.Amacher DE. The effects of cytochrome P450 induction by xenobiotics on endobiotic metabolism in pre-clinical safety studies. Toxicol Mech Methods 2010;20:159-66. [DOI] [PubMed] [Google Scholar]
- 142.Elsherbiny ME, Brocks DR. The ability of polycyclic aromatic hydrocarbons to alter physiological factors underlying drug disposition. Drug Metab Rev 2011;43:457-75. [DOI] [PubMed] [Google Scholar]
- 143.Ross AC, Cifelli CJ, Zolfaghari R, et al. Multiple cytochrome P-450 genes are concomitantly regulated by vitamin A under steady-state conditions and by retinoic acid during hepatic first-pass metabolism. Physiol Genomics 2011;43:57-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kedishvili NY. Enzymology of retinoic acid biosynthesis and degradation. J Lipid Res 2013;54:1744-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tay S, Dickmann L, Dixit V, Isoherranen N. A comparison of the roles of peroxisome proliferator-activated receptor and retinoic acid receptor on CYP26 regulation. Mol Pharmacol 2010;77:218-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Taimi M, Helvig C, Wisniewski J, et al. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J Biol Chem 2004;279:77-85. [DOI] [PubMed] [Google Scholar]
- 147.Thatcher JE, Zelter A, Isoherranen N. The relative importance of CYP26A1 in hepatic clearance of all-trans retinoic acid. Biochem Pharmacol 2010;80:903-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen H, Howald WN, Juchau MR. Biosynthesis of all-trans-retinoic acid from all-trans-retinol: catalysis of all-trans-retinol oxidation by human P-450 cytochromes. Drug Metab Dispos 2000;28:315-22. [PubMed] [Google Scholar]
- 149.Choudhary D, Jansson I, Stoilov I, et al. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab Dispos 2004;32:840-7. [DOI] [PubMed] [Google Scholar]
- 150.Tomita S, Okuyama E, Ohnishi T, et al. Characteristic properties of a retinoic acid synthetic cytochrome P-450 purified from liver microsomes of 3-methylcholanthrene-induced rats. Biochim Biophys Acta 1996;1290:273-81. [DOI] [PubMed] [Google Scholar]
- 151.Zhang QY, Dunbar D, Kaminsky L. Human cytochrome P-450 metabolism of retinals to retinoic acids. Drug Metab Dispos 2000;28:292-7. [PubMed] [Google Scholar]
- 152.Nelson CH, Buttrick BR, Isoherranen N. Therapeutic potential of the inhibition of the retinoic acid hydroxylases CYP26A1 and CYP26B1 by xenobiotics. Curr Top Med Chem 2013;13:1402-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Marill J, Cresteil T, Lanotte M, et al. Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol Pharmacol 2000;58:1341-8. [DOI] [PubMed] [Google Scholar]
- 154.Roberts ES, Vaz AD, Coon MJ. Role of isozymes of rabbit microsomal cytochrome P-450 in the metabolism of retinoic acid, retinol, and retinal. Mol Pharmacol 1992;41:427-33. [PubMed] [Google Scholar]
- 155.Andreola F, Hayhurst GP, Luo G, et al. Mouse liver CYP2C39 is a novel retinoic acid 4-hydroxylase. Its down-regulation offers a molecular basis for liver retinoid accumulation and fibrosis in aryl hydrocarbon receptor-null mice. J Biol Chem 2004;279:3434-8. [DOI] [PubMed] [Google Scholar]
- 156.Nadin L, Murray M. Participation of CYP2C8 in retinoic acid 4-hydroxylation in human hepatic microsomes. Biochem Pharmacol 1999;58:1201-8. [DOI] [PubMed] [Google Scholar]
- 157.Spear PA, Garcin H, Narbonne JF. Increased retinoic acid metabolism following 3,3',4,4',5,5'-hexabromobiphenyl injection. Can J Physiol Pharmacol 1988;66:1181-6. [DOI] [PubMed] [Google Scholar]
- 158.McSorley LC, Daly AK. Identification of human cytochrome P450 isoforms that contribute to all-trans-retinoic acid 4-hydroxylation. Biochem Pharmacol 2000;60:517-26. [DOI] [PubMed] [Google Scholar]
- 159.Wang T, Ma X, Krausz KW, et al. Role of pregnane X receptor in control of all-trans retinoic acid (ATRA) metabolism and its potential contribution to ATRA resistance. J Pharmacol Exp Ther 2008;324:674-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sass JO, Forster A, Bock KW, et al. Glucuronidation and isomerization of all-trans- and 13-cis-retinoic acid by liver microsomes of phenobarbital- or 3-methylcholanthrene-treated rats. Biochem Pharmacol 1994;47:485-92. [DOI] [PubMed] [Google Scholar]
- 161.Rosengren RJ, Sauer JM, Hooser SB, et al. The interactions between retinol and five different hepatotoxicants in the Swiss Webster mouse. Fundam Appl Toxicol 1995;25:281-92. [DOI] [PubMed] [Google Scholar]
- 162.Sauer JM, Hooser SB, Badger DA, et al. Alterations in chemically induced tissue injury related to all-trans-retinol pretreatment in rodents. Drug Metab Rev 1995;27:299-323. [DOI] [PubMed] [Google Scholar]
- 163.Pumford NR, Roberts DW, Benson RW, et al. Immunochemical quantitation of 3-(cystein-S-yl)acetaminophen protein adducts in subcellular liver fractions following a hepatotoxic dose of acetaminophen. Biochem Pharmacol 1990;40:573-9. [DOI] [PubMed] [Google Scholar]
- 164.Bray BJ, Goodin MG, Inder RE, et al. The effect of retinol on hepatic and renal drug-metabolising enzymes. Food Chem Toxicol 2001;39:1-9. [DOI] [PubMed] [Google Scholar]
- 165.Bray BJ, Rosengren RJ. Retinol potentiates acetaminophen-induced hepatotoxicity in the mouse: mechanistic studies. Toxicol Appl Pharmacol 2001;173:129-36. [DOI] [PubMed] [Google Scholar]
- 166.elSisi AE, Hall P, Sim WL, et al. Characterization of vitamin A potentiation of carbon tetrachloride-induced liver injury. Toxicol Appl Pharmacol 1993;119:280-8. [DOI] [PubMed] [Google Scholar]
- 167.Birnbaum LS, Harris MW, Stocking LM, et al. Retinoic acid and 2,3,7,8-tetrachlorodibenzo-p-dioxin selectively enhance teratogenesis in C57BL/6N mice. Toxicol Appl Pharmacol 1989;98:487-500. [DOI] [PubMed] [Google Scholar]
- 168.Pélissier MA, Siess MH, Lhuissier M, et al. Effect of prototypic polychlorinated biphenyls on hepatic and renal vitamin contents and on drug-metabolizing enzymes in rats fed diets containing low or high levels of retinyl palmitate. Food Chem Toxicol 1992;30:723-9. [DOI] [PubMed] [Google Scholar]