

Abstract
Exposure to genotoxic agents, such as ionizing radiation (IR) produces DNA damage leading to DNA double-strand breaks (DSBs); IR toxicity is augmented when the DNA repair is impaired. We reported that radiosensitization by a PARP inhibitor (PARPi) was highly prominent in prostate cancer (PCa) cells expressing the TMPRSS2-ERG gene fusion protein. Here, we show that TMPRSS2-ERG blocks non-homologous end-joining (NHEJ) DNA repair by inhibiting DNA-PKcs. VCaP cells, which harbor TMPRSS2-ERG and PC3 cells that stably express it displayed γH2AX and 53BP1 foci constitutively, indicating persistent DNA damage that was absent if TMPRSS2-ERG was depleted by siRNA in VCaP cells. The extent of DNA damage was enhanced and associated with TMPRSS2-ERG’s ability to inhibit DNA-PKcs function, as indicated by its own phosphorylation (Thr2609, Ser2056) and that of its substrate, Ser1778-53BP1. DNA-PKcs deficiency caused by TMPRSS2-ERG destabilized critical NHEJ components on chromatin. Thus, XRCC4 was not recruited to chromatin, with retention of other NHEJ core factors being reduced. DNA-PKcs autophosphorylation was restored to the level of parental cells when TMPRSS2-ERG was depleted by siRNA. Following IR, TMPRSS2-ERG-expressing PC3 cells had elevated Rad51 foci and homologous recombination (HR) activity, indicating that HR compensated for defective NHEJ in these cells, hence addressing why TMPRSS2-ERG alone did not lead to radiosensitization. However, the presence of TMPRSS2-ERG, by inhibiting NHEJ DNA repair, enhanced PARPi-mediated radiosensitization. IR in combination with PARPi resulted in enhanced DNA damage in TMPRSS2-ERG-expressing cells. Thus, by inhibiting NHEJ, TMPRSS2-ERG provides a synthetic lethal interaction with PARPi in PCa patients expressing TMPRSS2-ERG.
Keywords: TMPRSS2-ERG, DNA-PKcs, XRCC4, PARP, DNA repair
Introduction
The occurrence of gene fusion by chromosomal rearrangement is well established in hematological tumors, a discovery that changed the way cancer is understood today (1). In epithelial tumors, however, gene fusions have only recently been ascertained, yet they are encountered in ~ 50% of prostate-specific antigen (PSA)-screened human prostate tumors (2). The most prevalent prostate cancer (PCa) gene fusion joins the 5′ untranslated region of an androgen-regulated gene, TMPRSS2, with members of the E-twenty six (ETS) transcription factor family (ERG or ETV1) through frequent chromosomal rearrangements (3). Such ERG gene fusions occur during the initiation of PCa progression, which can then lead to the transition from high-grade prostatic intraepithelial neoplasia lesions to invasive carcinoma (4, 5). The TMPRSS2-ERG rearrangement can occur either by interstitial deletion (2, 6, 7) or by chromosomal translocation (8) as both fusion protein partners are located on chromosome 21q just 3 Mb apart. Recent studies reported that androgen stimulation facilitates chromosomal proximity of the TMPRSS2 and ERG genomic loci in several cell lines (9, 10), which can then drive the formation of TMPRSS2-ERG rearrangements at low frequency (9, 11).
As ETS gene fusions can lead to DNA double-strand breaks (DSBs) in PCa (12, 13), therapeutic strategies that target the DNA repair pathway as well as the ETS gene fusions can be effective in patients with tumors harboring such fusions. The ETS gene fusion status may thus predict a patient’s response to therapy either indirectly, such as through the effects of anti-androgens, or directly, through novel agents that target the function of ETS gene fusions.
Three pathways are required for repair of DNA DSBs: classical non-homologous end joining (c-NHEJ), homologous recombination (HR), and alternative NHEJ (5). Ionizing radiation (IR)-induced DNA damage is predominantly repaired by c-NHEJ, which is rapid and active throughout the cell cycle. However, HR and alternative NHEJ, that are slower, could also be important for repairing IR-induced DNA DSBs (14–16). The c-NHEJ DNA repair proceeds through four sequential steps: (i) recognition of DNA DSBs and association of c-NHEJ components at DSBs, (ii) stabilization of DNA ends by tethering, (iii) processing of DNA ends, and (iv) ligation of DNA ends (17, 18). The initial step of c-NHEJ involves DSB recognition by the Ku70/80 heterodimer, which is followed by DNA-dependent protein kinase catalytic subunit (DNA-PKcs) recruitment (19). Broken DNA ends placed in close proximity by DNA-PKcs are then processed by various factors, such as the nuclease Artemis, polynucleotide kinase, and the aprataxin and PNK-like factor, before finally being ligated by the XLF–XRCC4–ligase IV complex (20). Once recruited to the DNA, DNA-PKcs undergoes trans- and auto-phosphorylation at several sites, which induces its enzymatic activity, ultimately contributing to its dissociation from DNA. DNA-PKcs phosphorylation at Thr2609 is essential for its dissociation from chromatin and recruitment of downstream c-NHEJ components (21). The phosphorylation can be mediated either by ataxia telangiectasia mutated kinase (ATM) or by trans-autophosphorylation (22).
In this study, we define a unique molecular mechanism that elucidates how the TMPRSS2-ERG fusion protein inhibits DNA-PKcs, the critical kinase involved in c-NHEJ DNA repair. We show for the first time that TMPRSS2-ERG perturbs c-NHEJ DNA repair by inhibiting DNA-PKcs auto/trans-phosphorylation, blocking recruitment of XRCC4 to the chromatin, and diminishing the retention of the other core c-NHEJ components. Therefore, in TMPRSS2-ERG-expressing cells, the XRCC4-DNA-PKCs-dependent c-NHEJ is disrupted. As a consequence, PARPi radiosensitizes cells expressing the TMPRSS2-ERG gene fusion protein that represents most PCa patients.
Materials and Methods
Cell culture and treatments
Human PC3 and VCaP PCa cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and cultured as described previously (13). PC3 cells were authenticated by the Cell Line Authentication Services (Genetica DNA Laboratories) by comparative analysis with database profiling, where our cell line was compared with the known reference profile from ATCC. VCaP cells were bought within six months prior to performing the experiments.
The TMPRSS2-ERG fusion III (the most common) isoform was transfected using lipofectamine 2000 (Life Technologies), followed by selection in 1 mg/ml G418 (Invitrogen). For siERG-mediated knock-down, 50 ng of the siERG pool (Dharmacon) was delivered using lipofectamine 2000. IR was administered using a cesium-137 irradiator (JL Shepherd Associates, San Fernando, CA), at a dose rate of 146 cGy/min, as described (13). Stock solutions of the PARP inhibitor rucaparib (provided by Pfizer), and DNA-PKcs inhibitor [(NU7441; Selleck Chemicals (Houston, TX)] were made in DMSO (Sigma Aldrich). Rucaparib (2.5 µM) or NU7441 (500 nM) was administered simultaneously with radiation, as described (13).
Confocal immunostaining for DNA damage markers
Cells were plated on coverslips in 35-mm culture dishes. After treatment, cells were fixed with 2.0% paraformaldehyde for 20 min at room temperature, washed three times for 5 min with phosphate-buffered saline (PBS), permeabilized with 0.2% Triton X-100 in PBS for 10 min, and blocked in 3% fetal bovine serum in PBS containing 0.1% Triton X-100 for 1 h. The coverslips were then immunostained using γH2AX (Millipore), 53BP1 (Abcam), ERG (Epitomics), phospho-Ser25-53BP1, Ser1778-53BP1, PARP (Cell Signaling Technology); phospho-Thr2609-DNA-PKcs, phospho-Ser2056-DNA-PKcs, DNA Ligase IV (Abcam), Rad51, Ku70 (Santa Cruz Biotechnology), XRCC4 (Serotec), V5 (Life Technologies), XLF (Bethyl Laboratories) β-actin (Sigma-Aldrich) primary antibodies, followed by a fluorescently conjugated (Invitrogen) secondary antibody. Mounting and staining of the nuclei were performed using Vectashield containing 4,6-diamidino-2-phenylindole (DAPI; Vector Laboratories). Images were collected using an HCX Plan Apo 63×/1.4N.A. oil immersion objective lens on a Leica TCS-SP2 confocal microscope (Leica Microsystems). The number of cells counted, representing different fields was > 70. The number of foci in each nucleus was counted manually. The average number of foci was divided by number of nuclei to determine foci/nuclei.
Homologous Recombination (HR) assay
HR was measured as described (23–26). Briefly, 1 µg p-DR-GFP (HR substrate) and 500 ng of I-SceI expressing plasmid were co-transfected in parental and TMPRSS2-ERG-expressing PC3 cells. After 48 h cells were harvested and the GFP-positive cells were detected by BD FACS Calibur flow cytometry. Functional GFP was generated after I-SceI cutting and subsequent repair by HR. The GFP expressing plasmid was used as transfection control and the pCDNA plasmid as I-SceI carrier control.
Comet assay
Neutral Comet assay was performed in PC3 cells, with and without TMPRSS2-ERG expression, using the Comet Assay kit (Trevigen) according to manufacturer’s instructions. Comets were analyzed for olive moment using CometScore software (TriTek) as described earlier (13, 23).
Chromatin recruitment assay
Irradiated or untreated cells were fractionated and then subjected to a chromatin recruitment assay as described (13, 27). Two consecutive extractions were carried out for obtaining insoluble pellet. Cells were washed twice in ice-cold PBS and harvested by centrifugation then resuspended for 20 min on ice in 200 µl extraction buffer 1 (50 mM HEPES pH 7.5, 1 mM EDTA, 150 mM NaCl, 0.1% Triton X-100) supplemented with protease (Roche-Diagnostics) and phosphatase inhibitors (Sigma-Aldrich). The pellet was collected after centrifugation at 14,000g for 3.5 min, then incubated at room temperature in a shaker for 30 min after addition of 200 µl extraction buffer 2 (50 mM HEPES pH 7.5, 1 mM EDTA and 150 mM NaCl) supplemented with 200 µg/ml RNAse A (Sigma-Aldrich). The pellet was collected again after centrifugation at 14,000g for 3.5 min, then suspended in PBS containing 1% SDS. The samples were heated for 10 min and sonicated for 10 s before separation by SDS–PAGE and immunoblotted with the above indicated antibodies. Protein levels were quantified by ImageJ (NIH).
Flow cytometry
Cell cycle distribution was determined by flow cytometry as indicated previously (26) for cells treated with 4 Gy IR, with or without rucaparib for 24 and 48 h. After treatment, cells were collected and incubated in a solution containing propidium iodide (PI) (50 µg/ml), RNase A (0.1 mg/ml), Triton X (0.05%) and analyzed on a BD FACS Calibur flow cytometer (Beckton Dickinson). The raw data obtained was analyzed by CellQuest (Version 5.2.1) software. The results were normalized to control cells.
Statistical analysis
Statistical comparisons between groups were conducted using a 2-tailed student’s t-test or two-way ANOVA in Prism (Version 4.0c, GraphPad). Standard deviation (SD) was calculated from experiments conducted in triplicate and is indicated by error bars on the figures. All experiments were repeated three times, independently. The statistical significance was set to a level < 0.05.
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
Results
The TMPRSS2-ERG fusion gene induces DNA damage
The TMPRSS2-ERG fusion gene is prevalent in PCa (28) and its expression is associated with constitutive DNA damage before any treatment (12, 13). To address how TMPRSS2-ERG contributes to DNA damage, we first examined the constitutive and IR-induced DNA damage in VCaP cells, which harbor TMPRSS2-ERG, compared to derivative cells in which ERG expression was depleted by siRNA-mediated knockdown (Fig. 1). Untreated VCaP cells displayed constitutively γH2AX and 53BP1 foci, two prominent DNA damage markers, thus indicating a basal level of DNA damage. At 30 and 60 min after IR, VCaP but not siRNA-expressing derivative cells, showed an elevated number of γH2AX and 53BP1 IR-induced foci (IRIFs), indicating accumulation of DNA damage (Fig. 1A and B). These data suggest that in VCaP cells, which endogenously express the fusion gene, the resolution of DNA damage foci is delayed compared to the cells in which TMPRSS2-ERG is depleted, indicating that IR-induced DNA damage repair in these cells is impaired and occurs with slower kinetics. The significant increase (P < 0.001) in γH2AX and 53BP1 IRIF number at 60 min following IR (Fig. 1C) indicates that TMPRSS2-ERG expression results in constitutive DNA damage, which is augmented by IR.
Figure 1. The TMPRSS2/ERG fusion gene induces DNA damage in VCaP cells.

Confocal immunostaining for γH2AX (A) and 53BP1 (B) at the indicated times following 10 Gy ionizing radiation (IR) in VCaP parental and siERG-expressing derivative cells. C, Quantification of the number of IR-induced foci (IRIFs) in VCaP parental and derivative cells with ERG depleted by siERG. Left top panel represents western blot for levels of ERG in control (Ct) and siERG-treated VCaP cells. Typically, > 70 cells, representing different fields, were scored for IRIFs. β-actin was used as a loading control. Error bars represent SD (n=3). P < 0.0001 was calculated by two-way ANOVA.
As the VCaP cells in which the fusion gene was depleted were unable to proliferate proficiently because of their dependency on TMPRSS2-ERG (29), we expressed it stably in PC3 cells, which, unlike VCaP cells, do not express it endogenously. Constitutive DNA damage was prominent in cells expressing TMPRSS2-ERG, but not in parental PC3 cells. Thus, PC3 cells expressing TMPRSS2-ERG displayed constitutive γH2AX and 53BP1 foci, which increased following IR treatment (Fig. 2A and 2B). In parental PC3 cells most of the γH2AX IRIFs were resolved by 6 h after IR; in contrast, cells expressing TMPRSS2-ERG showed more persistent γH2AX IRIFs at 3 and 6 h, indicating that IR-induced DNA damage repair was impaired (Fig. 2A). Nevertheless, the number of γH2AX IRIFs was reduced significantly (P=0.0002) by 6 h compared to that found at 1 h (Fig. 2C), suggesting that the DNA repair was not completely inhibited in the fusion-expressing cells, but it rather proceeded with a slower kinetics. Expression of ERG was determined by immnunoblot in cells that do or do not express it, including following siERG-mediated silencing (Fig. 2D). 53BP1 IRIFs also increased significantly (P<0.0001) following IR in TMPRSS2-ERG-expressing cells at all-time points compared to parental PC3 cells (Fig. 2B and 2C).
Figure 2. TMPRSS2-ERG enhances DNA damage and decreases the DNA repair kinetics.

Confocal immunostaining for (A) γH2AX and (B) 53BP1 foci following IR (10 Gy) treatment in PC3 parental and derivative cells expressing the TMPRSS2-ERG fusion gene. C, Quantification of the number of γH2AX, and 53BP1 foci at the indicated times. P < 0.0001 was calculated by two-way ANOVA. Error bars represent SD (n=3). D, TMPRSS2-ERG-V5 tag expression was detected with antibody against the V5-tag and the V5-TMPRSS2-ERG expression specificity examined by its siERG-mediated down-regulation.
Rad51 is a key component in HR repair of IR-induced DNA damage that has been reported to compensate for diminished c-NHEJ (30). Examination of Rad51 revealed a significant (P=0.001) increase in the number of IRIFs in TMPRSS2-ERG-expressing PC3 cells compared to parental cells as early as 3 h following IR, and which remained elevated at 6 h (P=0.001), at a time when such foci were substantially diminished in parental cells (Fig. 3A and 3B). Importantly, using an HR reporter (DR-GFP) described previously (23–26) an increased HR activity was observed (P < 0.01) in TMPRSS2-ERG-expressing PC3 cells as compared to parental cells (Fig. 3C). This functional assay further supports the concept that PC3 fusion III-expressing cells compensate for the diminished NHEJ repair by enhancing HR activity.
Figure 3. Homologous recombination is enhanced in TMPRSS2-ERG-expressing cells.

A, Rad51 foci at the indicated times following IR (10 Gy), administered alone or in combination, concomitant with PARPi, in PC3 parental and TMPRSS2-ERG-expressing cells. B, Quantification of the number of Rad51 foci at the indicated times following IR treatment in PC3 parental and derivative cells expressing the TMPRSS2-ERG fusion gene. Typically, > 70 cells were scored for IRIFs. P < 0.0001 was calculated by two-way ANOVA. C, HR activation in PC3 cells with and without TMPRSS2-ERG expression transiently co-transfected with the DR-GFP reporter and the I-Sce-I expression construct. The GFP-positive cells were detected by flow cytometry. D, Olive moment of Comet assay performed in PC3 parental and TMPRSS2-ERG-expressing cells following IR (10 Gy), alone and in combination with PARPi (2.5 µM). P values were calculated with the Student’s t-test. Error bars in (B–D) represent SD (n=3).
We previously reported that rucaparib, a potent poly (ADP-ribose) polymerase (PARP) inhibitor (PARPi) was most effective in radiosensitizing PCa cells that harbor TMPRSS2-ERG (13). Treatment with PARPi and radiation led to diminished Rad51 IRIFs indicating diminished HR repair (Fig. 3A). Next we used the neutral Comet single cell gel electrophoresis assay to measure DSBs under neutral conditions, as it detects DSB- and not single-stranded DNA damage. There was significant (P<0.001) increase in DNA damage at 1 and 24 h following this combination treatment, as determined by olive moment in cells expressing TMPRSS2-ERG compared to parental PC3 cells (Fig. 3D). Together, these data indicate enhanced DNA damage and diminished DNA repair when IR is administered in combination with PARPi in TMPRSS2-ERG-expressing cells.
Presence of TMPRSS2-ERG inhibits radiation-induced cell cycle arrest
The DNA damage response following exposure to IR involves activation of cell cycle checkpoints. IR prominently promotes G2-M arrest (31), which can be sustained longer in radiosensitive cells (17, 32). At 24 h following IR, PC3 parental cells underwent robust G2-M arrest, with PARPi enhancing the IR-induced arrest ~ 1.5-fold (Fig. 4A and Supplementary Table S1). IR-induced cell cycle arrest was attenuated by 48 h in PC3 cells, whereas cells undergoing a combination treatment of PARPi with IR still displayed an enhanced and persistent arrest in G2-M (Fig. 4B and Supplementary Table S1). In contrast, TMPRSS2-ERG-positive PC3 cells displayed a markedly diminished cell cycle arrest after IR, with only a 1.2-fold increase in G2-M compared to the 2.9-fold increase in parental PC3 cells, indicating that radiation has a reduced effect on these cells. IR in combination with PARPi increased the proportion of fusion-positive PC3 cells undergoing G2-M arrest at 24 h that was maintained at 48 h after treatment. The proportion of cells in S-phase at 24 h following the IR and PARPi combination treatment was 2-fold greater in TMPRSS2-ERG compared to parental PC3 cells. These data could partially explain the increased radiosensitivity of TMPRSS2-ERG-positive cells with PARPi treatment (13), as cell cycle arrest due to checkpoint activation after IR was reduced in the presence of TMPRSS2-ERG compared to PC3 parental cells, as recently reported (33), yet PARP inhibition compensated for this reduction.
Figure 4. TMPRSS2-ERG expression prevents cell cycle arrest.
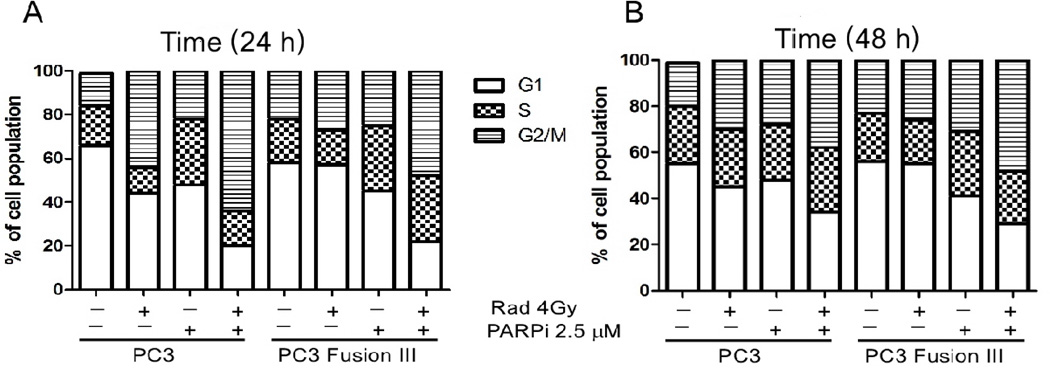
Cell cycle analysis of parental and TMPRSS2-ERG-expressing PC3 cells at 24 h (A) and 48 h (B) following IR (10 Gy) administered alone or in combination with PARPi (2.5 µM).
Defective recruitment of XRCC4 to chromatin and retention of core c-NHEJ factors
IR-induced DNA damage is predominantly repaired by rapid, error-prone c-NHEJ (34). The initiation step of c-NHEJ involves Ku70/Ku80 heterodimers binding to the free DSB ends, which then act as a scaffolding protein to recruit other essential DNA repair components, such as DNA-PKcs, XRCC4, XLF, and Ligase IV (35). To determine the impact of TMPRSS2-ERG on the c-NHEJ pathway, chromatin recruitment of c-NHEJ factors was examined in PC3 parental and derivative cells expressing TMPRSS2-ERG. Strikingly, XRCC4 was not recruited in TMPRSS2-ERG-expressing cells following IR. In contrast, XRCC4 was recruited abundantly to the chromatin of parental PC3 cells (Fig. 5A). This difference could not be accounted for by differences in XRCC4 levels, which were comparable in the two cell lines (Fig. 5B). Levels of Ku70, XLF, and Ligase IV that were recruited to the chromatin and which increased after IR were also diminished in TMPRSS2-ERG-expressing compared to parental PC3 cells (Fig. 5A, C). There was no significant difference in the recruitment and retention of ATM, a critical DNA damage sensor, and of its activated form, Ser1981-ATM (Fig. 5A), suggesting that TMPRSS2-ERG does not affect ATM expression or function, indicating that the DNA damage response was not perturbed in TMPRSS2-ERG-expressing cells (Fig. 5A). Surprisingly, the V5-tagged TMPRSS2-ERG fusion protein was constitutively bound to chromatin. The levels of PARP, a chromatin-bound protein known to be involved in the DNA damage response, were comparable between the two cell lines (Fig. 5A and 5C). These results indicate that TMPRSS2-ERG is present on the chromatin and perturbs DNA-PKcs function and XRCC4 recruitment. Together, these data suggest that the repair of the DNA damage in cells expressing TMPRSS2-ERG is mediated by a repair pathway independent of XRCC4/DNA-PKcs.
Figure 5. TMPRSS2-ERG impairs the recruitment and retention of NHEJ components.

A, Chromatin recruitment of indicated proteins at 0, 30, and 60 min after IR (10 Gy), examined with the corresponding primary antibodies; cell fractionation was performed as described in Materials and Methods. B, XRCC4, Ligase IV, and XLF levels in PC3 and TMPRSS2-ERG-expressing cells at the indicated times following IR. β-actin was used as a loading control. C, Chromatin recruitment of indicated proteins at 0, 30, and 60 min following IR (10 Gy). The data are representative of three independent experiments.
TMPRSS2-ERG inhibits DNA-PKcs
The most deleterious form of IR-induced DNA damage are DSBs. These DSBs are predominantly repaired by c-NHEJ, for which DNA-PKcs is the principal kinase required (34). The best characterized enzymatic substrate of DNA-PKcs is DNA-PKcs itself, with the phosphorylation sites being present in several clusters (18). Phosphorylation of DNA-PKcs in the ABCDE cluster on Thr2609 plays an important role in enabling the dissociation of DNA-PKcs from chromatin (36). Following IR, the chromatin-bound cellular fraction from parental cells contained DNA-PKcs phosphorylated on Thr2609 as early as 30 min, whereas TMPRSS2-ERG-expressing PC3 showed delayed and reduced DNA-PKcs phosphorylation (Fig. 5C). This finding is consistent with the higher retention, and, therefore more abundant DNA-PKcs presence on the chromatin in PC3 cells expressing TMPRSS2-ERG.
To directly examine the role of DNA-PKcs, we examined its activation in VCaP cells, which express TMPRSS2-ERG endogenously. Indeed, VCaP cells had minimal, if any Thr2609 foci at 30 and 60 min following IR (Fig. 6A) compared to a high number of such foci in derivative cells in which TMPRSS2-ERG expression was depleted by siRNA. PC3 cells with stable expression of TMPRSS2-ERG also had abrogated Thr2609 DNA-PKcs phosphorylation both before and following IR (Fig. 6B). Similarly, Ser2056 DNA-PKcs phosphorylation in the PQR cluster was absent in TMPRSS2-ERG-expressing, but not in parental cells at 30 and 60 min following IR (Supplementary Fig. S1). These results indicate that by interacting with DNA-PKcs TMPRSS2-ERG does indeed inhibit DNA-PKcs Thr2609 and Ser2056 auto-phosphorylation required for its function. Finally, we and others have shown that Ser1778 is a 53BP1 C-terminal phosphorylation target of DNA-PKcs (37, 38) that contributes significantly to DNA repair after IR (39). Ser1778 53BP1 foci were also absent in PC3 cells expressing TMPRSS2-ERG (Fig. 6C), indicating defective DNA repair, similar to results obtained when PC3 cells were pretreated with the specific DNA-PKcs inhibitor NU7441 (Fig. 6C). Taken together, these findings establish conclusively that DNA-PKcs activity is blocked in cells expressing TMPRSS2-ERG.
Figure 6. TMPRSS2-ERG inhibits DNA-PKcs auto-phosphorylation and activity.

Confocal immunostaining for phospho-DNA-PKcs Thr2609 at the indicated times following IR in VCaP (A) and PC3 cells (B), with or without expression of the TMPRSS2-ERG fusion. C, Ser1778-53BP1 phosphorylation as a DNA-PKcs target was examined by confocal immunostaining following IR in PC3 parental and TMPRSS2-ERG-expressing derivative cells without (left) or following pre-treatment with NU7441 (right). Right side panels, represent quantification of the respective foci on the left. Error bars represent SD (n=3).
TMPRSS2-ERG reduces MDC1 IRIFs
In addition to Ser1778, 53BP1 is also phosphorylated following IR-induced DNA damage on Ser25/29. This modification, which is known to depend predominantly on ATM and less on DNA-PKcs (37), has been shown to be critical for an efficient DNA damage response (40). However, we found no detectable difference in the number of Ser25-53BP1 IRIFs between TMPRSS2-ERG-expressing and parental PC3 cells (Supplementary Fig. S2A). MDC1, a pivotal mediator of the DNA damage response (25), has been proposed to be an upstream regulator of 53BP1. Both 53BP1 and MDC1 are downstream targets of the ATM-mediated DNA repair pathway, with ATM-mediated Ser25/29-53BP1 phosphorylation being regulated via MDC1 (41). At 60 min after IR, the number of Ser964-MDC1 IRIFs was reduced in TMPRSS2-ERG-expressing, but not parental PC3 cells (Supplementary Fig. S2B). These results indicate that in TMPRSS2-ERG-expressing cells, ATM-regulated Ser25/29 phosphorylation of 53BP1 is not solely mediated via MDC-1 because diminished MDC1 IRIFs did not affect 53BP1 phosphorylation on Ser25/29.
Discussion
The TMPRSS2 gene fusion to the oncogenic ETS family members ERG and ETV1 is common in PCa and has significant implications for understanding PCa tumorigenesis and developing novel approaches for diagnosis and therapy. These rearrangements are present in the majority of patients with PCa, being reported in up to 60% of PCa incident cases (27). A previous report has shown that the TMPRSS2-ERG fusion protein interacts with DNA-PKcs (12), suggesting that TMPRSS2-ERG is involved in c-NHEJ. Our data demonstrate for the first time that TMPRSS2-ERG perturbs c-NHEJ by inhibiting DNA-PKcs phosphorylation and XRCC4 recruitment.
Expression of TMPRSS2-ERG leads to constitutive DNA damage (12, 13). We found that VCaP cells that express TMPRSS2-ERG endogenously and PC3 cells that express it ectopically, displayed γH2AX and 53BP1 foci constitutively. The abundance of these foci was further elevated following IR as diminished or delayed DNA repair kinetics led to accumulation or/and persistence of DNA damage. This observation suggests that DNA-PKcs-dependent c-NHEJ is defective in these cells. Depletion of TMPRSS2-ERG in VCaP cells restored the DNA repair kinetics and reduced the DNA damage to a level comparable to that found in parental PC3 cells, indicating that TMPRSS2-ERG triggers/facilitates DNA damage and/or prevents its effective repair. Strikingly, in TMPRSS2-ERG-expressing, but not parental cells, the recruitment of XRCC4 to chromatin following IR was completely abrogated. Moreover, the retention of Ku70, XLF, and ligase IV was diminished in the presence of TMPRSS2-ERG. As a previous report suggested that DNA-PKcs binds to the ERG fusion gene product (12), this interaction might hinder the recruitment or retention of other c-NHEJ factors. We show that DNA-PKcs activity is suppressed in the presence of TMPRSS2-ERG, as indicated by the absence of foci of Thr2609 and Ser2056 DNA-PKcs and a critical DNA-PKcs substrate, Ser1778-53BP1 (37), as well as limited DNA-PKcs auto-phosphorylation. The higher levels of chromatin-associated DNA-PKCs in the presence of TMPRSS2-ERG suggests that its dissociation from DSBs was impaired, consistent with its defective autophosphorylation. siRNA-mediated depletion of TMPRSS2-ERG fully restored the DNA-PKcs activity in these cells. These data are consistent with the interpretation that the presence of TMPRSS2-ERG on the chromatin interferes with c-NHEJ. A recent report suggests that Ligase IV contributes to DNA-PKcs autophosphorylation and that end joining occurs by early formation of a supra-molecular entity composed of the DNA-PKcs, XLF, and Ligase IV complexes on DNA ends (42). Our finding of decreased chromatin retention of Ligase IV and XLF suggests that TMPRSS2-ERG is perturbing the formation of the end-joining complex and as a consequence, it is inhibiting c-NHEJ DNA repair. Interestingly, PC3 cells express another fusion gene, ETV4 (43), yet that has no effect on IR-induced DNA damage and its repair. Therefore, the effect on DNA repair is specific to the TMPRSS2-ERG fusion, which has been shown to interact with DNA-PKcs via its Y373 residue (12).
Prior studies have suggested that the choice of DNA damage repair pathway, HR vs. c-NHEJ, depends on DNA-PKcs autophosphorylation status (30). When the phosphorylation of DNA-PKcs at the ABCDE cluster (e.g. Thr2609) is defective, HR is also inhibited due to the restricted access to DNA ends. However, subsequent defective phosphorylation of the PQR cluster (e.g. Ser2056) increases HR due to partial rescue from the end-blocking effect by DNA-PKcs at DSBs (30), thus suggesting that cells deficient in phosphorylation of DNA-PKcs at both the Thr2609 and Ser2056 sites have augmented HR. Rad51 is a major component of HR repair following IR, which has been suggested to compensate for diminished c-NHEJ (30). Indeed, our data indicate that in TMPRSS2-ERG-expressing, but not parental cells, elevated Rad51 foci are present as early as 3 h following IR, indicating earlier or accelerated occurrence of HR, corroborated by a functional HR reporter assay.
However the Rad51 foci were diminished in TMPRSS2-ERG-expressing cells upon PARP inhibition and the persistent DNA damage enhanced, strongly suggesting that PARP is involved in repairing IR-induced DNA damage in these cells. As reported, PARPi binds to PARP on single-stranded DNA breaks, which cannot be repaired and become DSBs following DNA replication (16). As the TMPRSS2-ERG-expressing cells are extremely sensitive to PARPi co-administered with IR (13), there is an enhanced likelihood of involvement of PARP in the DNA repair process following IR-induced DNA damage.
Interestingly, we reported that there was no significant difference in radiosensitivity between cells expressing TMPRSS2-ERG compared to isogenic derivatives that did not express it, whether it was down-regulated (in VCaP that express it) or overexpressed (in PC3 that do not express it) (13), suggesting an adaptive cellular response to chronic expression of TMPRSS2-ERG. Here we extend these observations by showing that there was no difference in DNA damage initiation as indicated by activation of ATM and Comet assays. Clinical data indicating that TMPRSS2-ERG is not prognostic for following PCa radiotherapy (44) support this conclusion. A recent study found that ERG expression conferred a small enhancement in radio-sensitization (45). These differences may be caused by the different constructs (ERG vs. TMPRSS2-ERG) or the experimental conditions used in the two studies.
Replication stress driven by oncogenes or other growth-stimulating factors in tumor cells can produce collapsed replication forks and other DNA structures that lead to additional DNA damage that needs to be repaired (46) Thus, tumor cells expressing TMPRSS2-ERG may also have higher endogenous levels of DNA damage, with IR leading to additional lesions requiring repair. Under proficient DNA repair conditions, regardless of TMPRSS2-ERG expression, cells may be able to complete DNA repair without saturating the DNA repair ability. However, if DNA repair is prevented by PARP inhibition, an additional DNA lesion burden in cells with TMPRSS2-ERG may saturate the DNA repair capacity and sensitize these cells more relative to cells that do not express TMPRSS2-ERG. This possibility may explain why PARP inhibition is more effective in cells expressing TMPRSS2-ERG (13, 47). The diminished ability to repair the DNA damage by c-NHEJ in cells chronically expressing TMPRSS2-ERG may lead to an adaptive response through a compensatory increase in HR; therefore, the overall DNA repair capacity might not be noticeably disrupted in these cells, unless PARP is inhibited.
In contrast to DNA-PKcs, we found that ATM regulation was not affected in TMPRSS2-ERG-expressing cells because the amounts of recruited total and Ser1981-phosphorylated forms of ATM after IR were comparable to those in PC3 parental cells. Moreover, there was no detectable difference between these cells in phosphorylation of Ser25/29-53BP1, an ATM target (37). A recent report has shown that ERG directly represses the expression of the checkpoint kinase 1 (CHK1), a key DNA damage response cell cycle regulator that is essential for the maintenance of genome integrity. This study found that ERG expression correlates with CHK1 downregulation in human patients and that CHK1 downregulation sensitized PCa cells to DNA-damage (etoposide) but not docetaxel-based treatment (33). This finding provides support for the cell cycle differences we observed in the TMPRSS2-ERG-expressing cells. However, phosphorylation on Ser964 of the mediator protein MDC1 was reduced in TMPRSS2-ERG-expressing cells, indicating that some ATM targets may be affected.
In summary, our studies provide new insights into the molecular understanding of IR-induced DNA repair deregulation by TMPRSS2-ERG, which interferes with the assembly of c-NHEJ factors at DSBs on the chromatin. By inhibiting c-NHEJ via defective recruitment of XRCC4 and impaired DNA-PKcs phosphorylation, TMPRSS2-ERG rearrangement may reveal a “synthetic lethal” interaction with HR, blocking repair of lesions at collapsed DNA replication forks induced by PARPi. As PARP1 and Ku70/80 compete for repair of DSBs by distinct DNA repair pathways, it is possible that inhibition of c-NHEJ may activate PARP1-mediated DNA repair. PARP1 has been suggested to participate in a back-up, alternative NHEJ pathway that is more active in c-NHEJ-defective cells and in some instances has been shown to be predominant over HR (48). The role of alternative NHEJ, which has a specific requirement for PARP1 and a resection factor (e.g. CtIP), could be examined in the future in the response of TMPRSS2-ERG-expressing cells to IR. Recently, it has been shown that a hormone-DNA repair signaling circuit defines the response to genotoxic insults in PCa, thus implicating androgen receptor signaling, the main therapeutic target in PCa (49, 50). Collectively these findings help in our understanding the DNA repair mechanism following IR-induced DNA damage in cells expressing TMPRSS2-ERG, clinically significant as this gene fusion is present in most patients with PCa.
Supplementary Material
Acknowledgements
We would like to thank Dr. Alan Kraker (Pfizer) for rucaparib and our colleagues, Drs. Judy Drazba and John Peterson (Imaging Core) for technical assistance, Drs. Janet Houghton and Tapati Mazumdar for flow cytometry. Dr. Cassandra Talerico (Cleveland Clinic) provided substantive editing and comments.
Grant Support: This study was supported by the National Institutes of Health (NCI) grants CA127264 to A. Almasan and CA154625 to J. Zhang.
Footnotes
Conflict of interest: The authors disclose no potential conflicts of interest
References
- 1.Rowley JD. Molecular cytogenetics: Rosetta stone for understanding cancer--twenty-ninth G.H.A. Clowes memorial award lecture. Cancer Res. 1990;50:3816–3825. [PubMed] [Google Scholar]
- 2.Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, Setlur S, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006;66:8337–8341. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 3.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 4.Perner S, Mosquera JM, Demichelis F, Hofer MD, Paris PL, Simko J, et al. TMPRSS2-ERG fusion prostate cancer: an early molecular event associated with invasion. Am J Surg Pathol. 2007;31:882–888. doi: 10.1097/01.pas.0000213424.38503.aa. [DOI] [PubMed] [Google Scholar]
- 5.Park K, Dalton JT, Narayanan R, Barbieri CE, Hancock ML, Bostwick DG, et al. TMPRSS2:ERG gene fusion predicts subsequent detection of prostate cancer in patients with high-grade postatic intraepithelial noplasia. J Clinical Oncol: 2014;32:206–211. doi: 10.1200/JCO.2013.49.8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iljin K, Wolf M, Edgren H, Gupta S, Kilpinen S, Skotheim RI, et al. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer Res. 2006;66:10242–10246. doi: 10.1158/0008-5472.CAN-06-1986. [DOI] [PubMed] [Google Scholar]
- 7.Yoshimoto M, Joshua AM, Chilton-Macneill S, Bayani J, Selvarajah S, Evans AJ, et al. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia. 2006;8:465–469. doi: 10.1593/neo.06283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teixeira MR. Chromosome mechanisms giving rise to the TMPRSS2-ERG fusion oncogene in prostate cancer and HGPIN lesions. Am J Surg Pathol. 2008;32:642–644. doi: 10.1097/PAS.0b013e31815b6056. [DOI] [PubMed] [Google Scholar]
- 9.Mani RS, Tomlins SA, Callahan K, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–678. doi: 10.1016/j.ccr.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chatterjee P, Choudhary GS, Sharma A, Singh K, Heston WD, Ciezki J, et al. PARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cells. PLoS One. 2013;8:e60408. doi: 10.1371/journal.pone.0060408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray S, Almasan A. Apoptosis induction in prostate cancer cells and xenografts by combined treatment with Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand and CPT-11. Cancer Res. 2003;63:4713–4723. [PubMed] [Google Scholar]
- 15.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 16.Groth P, Orta ML, Elvers I, Majumder MM, Lagerqvist A, Helleday T. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res. 2012;40:6585–6594. doi: 10.1093/nar/gks315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin OA, Ivashkevich A, Choo S, Woodbine L, Jeggo PA, Martin RF, et al. Statistical analysis of kinetics, distribution and co-localisation of DNA repair foci in irradiated cells: cell cycle effect and implications for prediction of radiosensitivity. DNA Repair. 2013;12:844–855. doi: 10.1016/j.dnarep.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Davis AJ, Chen DJ. DNA double strand break repair via non-homologous end-joining. Transl Cancer Res. 2013;2:130–143. doi: 10.3978/j.issn.2218-676X.2013.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 20.Lieber MR, Wilson TE. SnapShot: Nonhomologous DNA end joining (NHEJ) Cell. 2010;142:496-.e1. doi: 10.1016/j.cell.2010.07.035. [DOI] [PubMed] [Google Scholar]
- 21.Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, et al. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem. 2011;286:32638–32650. doi: 10.1074/jbc.M111.272641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neal JA, Sugiman-Marangos S, Vandervere-Carozza P, Wagner M, Turchi J, Lees-Miller SP, et al. Unraveling the complexities of DNA-PK autophosphorylation. Mol Cell Biol. 2014;34:2162–2175. doi: 10.1128/MCB.01554-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng Z, Zhang J. A dual role of BRCA1 in two distinct homologous recombination mediated repair in response to replication arrest. Nucleic Acids Res. 2012;40:726–738. doi: 10.1093/nar/gkr748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Ma Z, Vanderwaal RP, Feng Z, Gonzalez-Suarez I, Wang S, et al. The mTOR inhibitor rapamycin suppresses DNA double-strand break repair. Radiat Res. 2011;175:214–224. doi: 10.1667/rr2323.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Ma Z, Treszezamsky A, Powell SN. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat Struct Mol Biol. 2005;12:902–909. doi: 10.1038/nsmb991. [DOI] [PubMed] [Google Scholar]
- 26.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drouet J, Delteil C, Lefrancois J, Concannon P, Salles B, Calsou P. DNA-dependent protein kinase and XRCC4-DNA ligase IV mobilization in the cell in response to DNA double strand breaks. J Biol Chem. 2005;280:7060–7069. doi: 10.1074/jbc.M410746200. [DOI] [PubMed] [Google Scholar]
- 28.Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. J Clinical Oncol. 2011;29:3659–3668. doi: 10.1200/JCO.2011.35.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Cai Y, Yu W, Ren C, Spencer DM, Ittmann M. Pleiotropic biological activities of alternatively spliced TMPRSS2/ERG fusion gene transcripts. Cancer Res. 2008;68:8516–8524. doi: 10.1158/0008-5472.CAN-08-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol. 2005;25:10842–10852. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crosby ME, Jacobberger J, Gupta D, Macklis RM, Almasan A. E2F4 regulates a stable G2 arrest response to genotoxic stress in prostate carcinoma. Oncogene. 2007;26:1897–1909. doi: 10.1038/sj.onc.1209998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krempler A, Deckbar D, Jeggo PA, Lobrich M. An imperfect G2M checkpoint contributes to chromosome instability following irradiation of S and G2 phase cells. Cell Cycle. 2007;6:1682–1686. doi: 10.4161/cc.6.14.4480. [DOI] [PubMed] [Google Scholar]
- 33.Lunardi A, Varmeh S, Chen M, Taulli R, Guarnerio J, Ala U, et al. Suppression of CHK1 by ETS family members promotes DNA damage response by-pass and tumorigenesis. Cancer Dscov. 2015 doi: 10.1158/2159-8290.CD-13-1050. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair. 2006;5:1042–1048. doi: 10.1016/j.dnarep.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 35.Neal JA, Dang V, Douglas P, Wold MS, Lees-Miller SP, Meek K. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol Cell Biol. 2011;31:1719–1733. doi: 10.1128/MCB.01298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, et al. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol. 2007;177:219–229. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harding SM, Bristow RG. Discordance between phosphorylation and recruitment of 53BP1 in response to DNA double-strand breaks. Cell Cycle. 2012;11:1432–1444. doi: 10.4161/cc.19824. [DOI] [PubMed] [Google Scholar]
- 38.Chatterjee P, Plesca D, Mazumder S, Boutros J, Yannone SM, Almasan A. Defective chromatin recruitment and retention of NHEJ core components in human tumor cells expressing a Cyclin E fragment. Nucleic Acids Res. 2013;41:10157–10169. doi: 10.1093/nar/gkt812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JH, Cheong HM, Kang MY, Kim SY, Kang Y. Ser1778 of 53BP1 Plays a Role in DNA Double-strand Break Repairs. Korean J Physiol Pharmacol. 2009;13:343–348. doi: 10.4196/kjpp.2009.13.5.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Munoz IM, Jowsey PA, Toth R, Rouse J. Phospho-epitope binding by the BRCT domains of hPTIP controls multiple aspects of the cellular response to DNA damage. Nucleic Acids Res. 2007;35:5312–5322. doi: 10.1093/nar/gkm493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minter-Dykhouse K, Ward I, Huen MSY, Chen J, Lou ZK. Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. J Cell Biol. 2008;181:727–735. doi: 10.1083/jcb.200801083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cottarel J, Frit P, Bombarde O, Salles B, Négrel A, Bernard S, et al. A noncatalytic function of the ligation complex during nonhomologous end joining. J Cell Biol. 2013;200:173–186. doi: 10.1083/jcb.201203128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pellecchia A, Pescucci C, De Lorenzo E, Luceri C, Passaro N, Sica M, et al. Overexpression of ETV4 is oncogenic in prostate cells through promotion of both cell proliferation and epithelial to mesenchymal transition. Oncogenesis. 2012;1:e20. doi: 10.1038/oncsis.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dal Pra A, Lalonde E, Sykes J, Warde F, Ishkanian A, Meng A, et al. TMPRSS2-ERG status is not prognostic following prostate cancer radiotherapy: implications for fusion status and DSB repair. Clin Cancer Res. 2013;19:5202–5209. doi: 10.1158/1078-0432.CCR-13-1049. [DOI] [PubMed] [Google Scholar]
- 45.Han S, Brenner JC, Sabolch A, Jackson W, Speers C, Wilder-Romans K, et al. Targeted radiosensitization of ETS fusion-positive prostate cancer through PARP1 inhibition. Neoplasia. 2013;15:1207–1217. doi: 10.1593/neo.131604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartkova J, Horejsi Z, Koed K, Krämer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen TJ. Enhancing radiosensitivity: targeting the DNA repair pathways. Cancer Biol Ther. 2009;8:665–670. doi: 10.4161/cbt.8.8.8304. [DOI] [PubMed] [Google Scholar]
- 48.Iliakis G. Backup pathways of NHEJ in cells of higher eukaryotes: cell cycle dependence. Radiother Oncol. 2009;92:310–315. doi: 10.1016/j.radonc.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 49.Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–1271. doi: 10.1158/2159-8290.CD-13-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3:1245–1253. doi: 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.