
Figure 7. Central illustration.
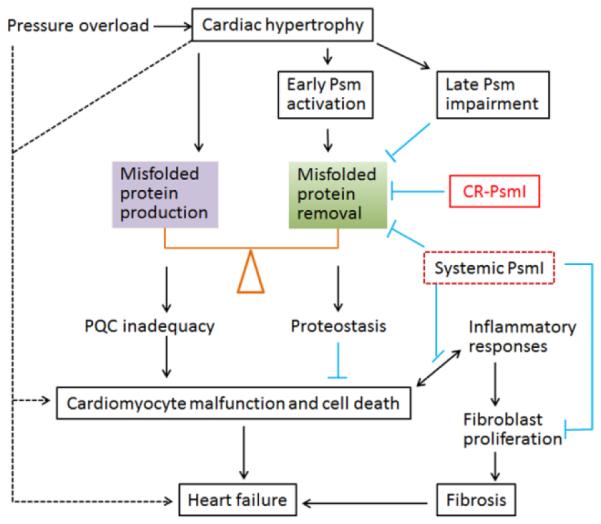
Maintaining cardiac proteostasis requires timely removal of misfolded proteins whereas increased production of misfolded proteins tilts the balance toward PQC inadequacy. In response to acute systolic overload, the heart undergoes hypertrophy, which inevitably increases protein synthesis and by-production of misfolded proteins. To counter the increased proteotoxic stress during hypertrophy, cardiomyocytes adaptively mobilizes proteasomal reserve and thereby elevates proteasome (Psm) activities and UPS performance; however, when the stress sustains, proteasome function and UPS performance are impaired by unknown factors, which diminishes the capability of cardiomyocytes to maintain proteostasis and suppresses cell survival signaling, leading to cardiomyocyte malfunction and cell death. Cardiomyocyte death, especially in the form of necrosis, triggers inflammatory responses which could further fuel cardiomyocyte death and stimulate proliferation of interstitial cells (e.g., fibroblasts), leading to fibrosis. Both loss of cardiomyocytes and fibrosis are main pathogenic factors of heart failure. CR-PsmI prevents the initial adaptive proteasomal activation and hastens UPS impairment, thereby facilitating heart failure during systolic overload. Systemic PsmI inhibits UPS-mediated proteolysis in both cardiomyocytes and non-cardiomyocyte compartments. The non-cardiac PsmI could potentially attenuate inflammatory responses and the proliferation of fibroblasts, thereby being anti-fibrotic; hence, systemic PsmI might be beneficial at the certain stage of systolic overload. The dashed lines denote other potential pathways that are directly tested by the present study.