

Abstract
In sepsis, the severity-dependent decrease of von Willebrand factor (VWF)–inactivating protease, a disintegrin and metalloproteinase with thrombospondin motifs 13 (ADAMTS13), results in platelet aggregation and consumption, leading to sepsis-associated thrombotic microangiopathy (TMA) and organ failure. Previous reports assessing its functional deficiency have pinpointed involvement of autoantibodies or mutations to propagate thrombotic thrombocytopenic purpura (TTP). However, mechanisms of acquired ADAMTS13 deficiency during host response remain unclear. To enhance understanding of ADAMTS13 deficiency in sepsis, we evaluated changes in expression of mRNA coding ADAMTS13 during septic conditions using primary cellular sources of the protease. We hypothesized that proinflammatory cytokines and constituents of serum from septic patients affect the transcriptional level of ADAMTS13 in vitro, and previously recommended therapeutic agents as adjunctive therapy for sepsis interact therewith. Cultured hepatic stellate cells (HSCs), endothelial cells (HMEC) and human precision-cut liver slices as an ex vivo model were stimulated with sepsis prototypic cytokines, bacterial endotoxin and pooled serum obtained from septic patients. Stimulation resulted in a significant decrease in ADAMTS13 mRNA between 10% and 80% of basal transcriptional rates. Costimulation of selenite or recombinant activated protein C (APC) with serum prevented ADAMTS13 decrease in HSCs and increased ADAMTS13 transcripts in HMEC. In archived clinical samples, the activity of ADAMTS13 in septic patients treated with APC (n = 5) increased with an accompanying decrease in VWF propeptide as surrogate for improved endothelial function. In conclusion, proinflammatory conditions of sepsis repress mRNA coding ADAMTS13 and the ameliorating effect by selenite and APC may support the concept for identification of beneficial mechanisms triggered by these drugs at a molecular level.
INTRODUCTION
Despite therapy with appropriate antibiotics and intensive supportive care, sepsis carries a persistently high morbidity and mortality (1–4). Sepsis is a progressive injurious process aggravating from a systemic inflammatory response to infection (5). The continuum of sepsis pathophysiology involves a complex integrated response involving immune cell activation, inflammatory mediators and the coagulation system. Central to these responses are alterations in endothelial cell function and a shift to a proinflammatory and procoagulant surface, which amplifies the initial signal intensity (6,7). Alterations in microcirculation and microvascular permeability precede early tissue injury and organ failure (8). However, pathogenic mechanisms and messengers involved in processes resulting in remote organ injury require further investigation.
During sepsis, von Willebrand factor (VWF), a multimeric acute phase protein, is abundantly secreted into plasma by activated endothelial cells and platelets (9,10). VWF in its native, ultralarge isoform (ulVWF) is only detectable in conditions of an activated endothelium (11) and functions as a determinant of platelet activation and aggregation (12,13). ADAMTS13 is a plasma protease primarily synthesized and secreted from hepatic stellate cells (HSCs) and is the only enzyme reported to inactivate ulVWF, thereby reducing its thrombogenicity (14,15). Congenital or immunologically induced deficiency of ADAMTS13 has been identified to result in thrombotic thrombocytopenic purpura (TTP) (16). In sepsis, the term thrombotic microangiopathy (TMA) was introduced to characterize thrombotic occlusion of arterioles less than 100 μm diameter independent of the underlying pathophysiology (17,18). Hence, thrombocytopenia is a risk factor for mortality in critically ill patients (19) and is also an independent predictor of multiple organ failure and death (20–22).
Further evidence suggests ADAMTS13 deficiency in patients with systemic inflammation and severe sepsis (9,23–25). This deficiency is associated with an increase in ulVWF and is postulated to cause TMA in patients (9,23,26). The impact of ADAMTS13 deficiency in thrombocytopenia and organ failure is supported by the observation that plasma exchange reversed organ dysfunction in thrombocytopenia-associated multiple organ failure in children (27) and renal deposition of thrombi enriched from activated platelets and VWF in a porcine sepsis model (28). Also, an inverse relationship between severity of inflammation and ADAMTS13 activity is described in clinical studies (29,30). However, the actual cause for ADAMTS13 deficiency is controversial, ranging from its consumption (31), secretion defects (32) or proteolytic degradation by enzymes circulating in plasma of critically ill patients (33,34). Furthermore, an unbalanced expression of ADAMTS13 and VWF was reported in mouse endotoxemia (35) and in the cecal ligation and puncture model of sepsis (36). However, VWF secretion was found to function as the major determinant of sepsis-induced mortality because the complete absence of its regulating protease had only a minor impact in the murine sepsis model.
To gain insight into mechanisms of ADAMTS13 deficiency in systemic inflammation and sepsis, we investigated the effects of proinflammatory cytokines, endotoxins and constituents of serum obtained from septic patients on ADAMTS13 expression on a transcriptional level. Furthermore, we evaluated the contribution of selenite and APC as adjuvant sepsis therapeutics in regulating inflammation-induced ADAMTS13 changes in transcription. Finally, we compared data obtained from in vitro experiments to an observational study cohort for consistency of our findings.
MATERIALS AND METHODS
Institutional ethical approval by the local ethics committee of the medical faculty of Friedrich-Schiller University Hospital in Jena (A-1827-07/06 and A-1840-08/06) and written informed consent of each patient were obtained.
Cell Culture Experiments
Using cell culture models, an immortalized hepatic stellate cell line (LX-2) (37,38) and a human microvascular endothelial cell line (HMEC) (39), both of human origin, were cultured under standard conditions (LX-2: 1% heat inactivated fetal calf serum [FCS] in Dulbecco’s Modified Eagle Medium (DMEM) media, 5% CO2; HMEC: 10% heat inactivated FCS in DMEM media, 5% CO2). Cells (1 × 106) were seeded in media supplemented with pooled human AB-serum (1%), equilibrated for 2 h and stimulated for 6 or 24 h with human recombinant cytokines (tumor necrosis factor-α [TNFα], interferon γ [IFNγ], interleukin-1β [IL-1β], IL-2 or IL-6; reconstituted in saline supplemented with 0.1% human albumin) to final concentration of 100 ng/mL, with endotoxin (200 ng/mL E. coli, serotype B4; saline supplemented with 0.1% AB-serum), and a mixture of cytokines (TNFα, IFNγ and IL-1β [10 ng/mL] each combined with 100 ng/mL of endotoxin as described previously) (40). The stimulation period was limited to a maximum of 24 h because ADAMTS13 mRNA transcripts remain stable during this period, therefore providing a threshold to identify significant changes in ADAMTS13 transcripts. Longer stimulation times may provoke mRNA changes due to artifacts resulting from cellular stress induced by starvation and accumulation of cell debris and not the intended effect observed by the inflammatory stimuli. A concentration of 5% pooled serum from septic patients (n = 12) was used for stimulation where indicated. Serum samples were obtained within 24 h after the patients met the criteria for sepsis or severe sepsis (5). Pivotal parameters of the serum pool were determined as follows (normal values are given in brackets): creatinine 121 μmol/L [44–106], blood urea nitrogen 37.2 mg/dL [2.6–6.4], γ-glutamyltransferase 80 U/L [6–28], glutamatoxalacetate-transaminase 209 U/L [<18], lactate dehydrogenase 463 U/L [135–223], ammonia 128 μg/dL [19–94], creatine-phosphokinase 640 U/L [<174], albumin 2.6 g/dL [3,5–5.5], glucose 6.9 mmol/L [3.9–5.6], IL-6 2282.75 pg/mL, TNFα below detection limit (10 pg/mL).
Cells were also coincubated with one of the following substances: noradrenalin (NA), protein C–activated (Eli Lilly, Indianapolis, IN, USA), arginine vasopressin (AVP) or desmopressin (DDAVP), sodium selenite (biosyn Arzneimittel GmbH, Fellbach, Germany) and tolvaptan (Sigma-Aldrich, St. Louis, MO, USA).
Human High Precision–Cut Liver Slices (HPLS)
As an established ex vivo model, HPLS from patients who underwent liver resection spatially distant to metastases controlled by macroscopic inspection by two independent and experienced scientists were obtained. Specimens were precision-cut into 200-μm slices (5 mm diameter) with a Krumdieck slicer (Alabama Research and Development, Munford, AL, USA). Slices were then incubated into primary cultures for 24 h before stimulation as described previously (41).
RNA Preparation and Determination of Transcript Level
RNA was isolated with RNeasy kit (Qiagen, Hilden, Germany), reverse transcribed (Bio-Rad, Berkeley, CA, USA) and ADAMTS13 transcripts were quantified using an iCycler. Data were normalized to unstimulated controls with unvaried transcripts (hypoxanthine-guanine phosphoribosyltransferase [HPRT])/disintegrin and metalloproteinase domain-containing protein 9 [ADAM9] for HSCs–HPRT/porphobilinogen deaminase [PBGD] for HMEC) and ratios analyzed according to Pfaffl (42). ADAM9 (stability value [M] < 0.340), GUSB (M < 0.305), PBGD (M < 0.303) and HPRT (M < 0.554) were most suitable for data normalization.
Characterization of Septic Patients and Determination of Laboratory Parameters
Blood was collected in sodium citrate anticoagulant. ADAMTS13 activity was determined using a von Willebrand factor fluorescent resonance energy transfer 73 (VWF-FRETS73) substrate as previously described (43,44). von Willebrand factor antigen (VWF:Ag), von Willebrand collagen binding activity (VWF:CB), von Willebrand ristocetin cofactor assay (VWF:RCo), VWF propeptides were determined as described previously (9,30,45).
Statistics
Statistical analyses were performed using SPSS 22 (SPSS [IBM, Armonk, NY, USA]). The Student t test or Mann–Whitney test was used to calculate statistical significance of data sets (P < 0.05). Data are shown as means ± standard deviation of at least four independent experiments. Based on normalized expression data, a log2-threshold value of +1.0 or −1.0 signifying a two-fold variation was used to define transcripts differentially expressed.
RESULTS
Proinflammatory Cytokines and Serum from Septic Patients Suppresses ADAMTS13 Transcription in Primary Cellular Sources
TNFα, IL-1β and IL-6 decreased the normalized amount of transcript encoding ADAMTS13 in HSCs, which was found differentially regulated by two fold across both time points (Figure 1A). However, IFNγ and IL-2 displayed moderate effects within the cutoff value, while endotoxin failed to significantly regulate ADAMTS13 transcription in HSCs, but reached minimum values around 60% of basal value in HMEC. Interestingly, the mixture of prototypic mediators (cytokines and endotoxin) did not impair the transcriptional rate significantly. Pooled serum from patients with sepsis significantly reduced the ADAMTS13 transcription rate to 10% compared with an unstimulated control group. In endothelial cells, the effect of TNFα, IL-2, IL-6 and the mixture of cytokines in suppressing ADAMTS13 transcription was more pronounced. However, serum from septic patients reduced ADAMTS13 transcription rate to a minor extent (Figure 1B).
Figure 1.

ADAMTS13 expression in inflammatory conditions. (A) ADAMTS13 transcript is decreased significantly in HSCs after stimulation with TNFα (100 ng/mL), IFNγ (100 ng/mL), IL-1β (100 ng/mL), IL-2 (100 ng/mL), IL-6 (100 ng/mL) and pooled serum from septic patients (pooled SepSe) for 6 h or 24 h. Expression of ADAMTS13 based on Pfaffl method and normalized to reference transcripts (ADAM9 and HPRT). (B) ADAMTS13 transcript is significantly decreased in HMEC after stimulation with TNFα (100 ng/mL), IFNγ (100 ng/mL), IL-2 (100 ng/mL), IL-6 (100 ng/mL), cytokine mix (CM) and pooled SepSe for 6 h or 24 h. Expression of ADAMTS13 is normalized to reference genes (PBGD and HPRT ). Bars represent mean values of log2-ratios ± SEM for at least four independent experiments.
Incubation with Adjuvant Septic Therapeutics Regulates ADAMTS13 Transcription
We assessed the role of drugs used in patients with septic shock that promote vasoconstriction (norepinephrine, AVP) (46), of a stabilized analogue of AVP triggering the release of VWF from endothelial cells (DDAVP) (47), selenite supplementation with known antioxidative properties (48) and APC which is reported to regulate severe inflammatory response (49). In HSCs, all tested compounds displayed nonsignificant effects on ADAMTS13 transcription at 6 h (Figure 2A). However, after 24 h, the amount of mRNA encoding ADAMTS13 was significantly diminished, up to 26% of basal value. Incubation with selenite reduced the expression rate to 50% (24 h). In endothelial cells, ADAMTS13 transcription was only affected by AVP and APC at the late observation point while incubation with selenite increased ADAMTS13 transcription rate after 24 h (Figure 2B). To elucidate the signaling mechanism involved in DDAVP reduction of ADAMTS13 transcripts, we coincubated endothelial cells with DDAVP and a V2-receptor antagonist (tolvaptan) for 24 h (Figure 5). The choice of endothelial cells was selected because ADAMTS13 transcription was significantly diminished in endothelial cells and at 24 h. We observed that by inhibiting the V2 receptor, ADAMTS13 suppression was completely prohibited while there was no significant change of ADAMTS13 in the presence of the pharmacological agent alone (Figure 3).
Figure 2.
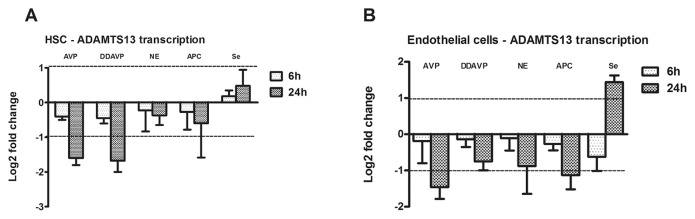
ADAMTS13 expression after stimulation with agents relevant for sepsis management. (A) ADAMTS13 transcript is decreased significantly in HSCs after stimulation with AVP and DDAVP (10 ng/mL), and activated protein C (APC, 10 ng/mL) for 6 h or 24 h. Expression of ADAMTS13 based Pfaffl method normalized to reference transcripts (ADAM9 and HPRT ). (B) ADAMTS13 transcript in HMEC is altered after stimulation with AVP and DDAVP (10 ng/mL), with NE (10 ng/mL), APC (10 ng/mL) and selenite (1 ng/mL) for 6 h or 24 h. Expression of ADAMTS13 based on Pfaffl method and normalized to reference transcripts (PBGD and HPRT). Bars represent mean values of log2-ratios ± SEM for at least four independent experiments.
Figure 5.

DDAVP reduction of ADAMTS13 is mediated via the V2-receptor signaling. Using a V2-selective receptor antagonist (tolvaptan, 50 nmol/L) prevented DDAVP-induced reduction of ADAMTS13 transcripts in endothelial cells after 24 h of incubation.
Figure 3.

Decrease of ADAMTS13 expression level in human ex vivo model. HPLS were stimulated with TNFα (100 ng/mL), AVP (10 ng/mL) and DDAVP (10 ng/mL), LPS (200 ng/mL), IFNγ (100 ng/mL), IL-1β (100 ng/mL), IL-2 (100 ng/mL), IL-6 (100 ng/mL), a mix of cytokines (CM), endotoxin, pooled serum from septic patients (SepSe) and pooled serum from healthy individuals (control) for 6 h. Expression of ADAMTS13 based on Pfaffl method and normalized to ADAM9 and HPRT. Expression of ADAM12 as a marker for inflammatory response. Bars represent mean values of log2-ratios ± SEM for at least four independent experiments.
Proinflammatory Conditions Affect ADAMTS13 Transcription in a Human Ex Vivo Liver Model
To investigate the effects of proinflammatory stimulation on ADAMTS13 transcription in a human ex vivo liver model, we evaluated the expression value of disintegrin and metalloproteinase domain-containing protein 12 (ADAM12) as a marker specific for the activation state of HSCs. We observed an increase of ADAM12 expression values at 6 h after stimulation with TNFα, endotoxin, IL-1β, IL-2 and cytokine mixture (see Figure 3). IFNγ and IL-6 displayed moderate effects that were completely absent following incubation with AVP and DDAVP. Furthermore, stimulation with serum obtained from patients with sepsis also exhibited an increase in ADAM12 expression compared with un-stimulated controls. Additionally, in this model, proinflammatory stimulation resulted in a prominently decreased expression of ADAMTS13 by DDAVP and IL-1β (see Figure 3). Stimulation with serum from septic patients resulted in a decreased expression value of ADAMTS13 as compared with unstimulated controls.
Selenite and APC Prevent ADAMTS13 Suppression during Proinflammatory Conditions
Because APC and selenite regulate ADAMTS13 expression, we elucidated the effects of serum obtained from patients with sepsis in the presence of both therapeutic agents. In HSCs, APC completely abrogated the depressive effects of serum constituents on ADAMTS13 expression while selenite stabilized ADAMTS13 expression rate without attaining the twofold threshold value of differential expression (Figure 4A). In endothelial cells, APC and selenite induced an increase in ADAMTS13 expression after 24 h (Figure 4B). To elucidate the mechanism of action of APC-mediated rescue of ADAMTS13, we used a specific blocking antibody targeting the endothelial protein C receptor (EPCR). It was evident that blocking the EPCR prevented APC-mediated rescue of ADAMTS13 transcription in the presence of septic stimuli (Figure 4C). To elucidate a potential synergism between APC and selenite, we used a combination of both APC and selenite to determine the transcription rate of ADAMTS13 in the presence of septic stimuli. We observed a minimal additive effect when both drugs were used (Figure 4D). Use of a specific inhibitor confirmed V2-receptor mediated signaling as essential for DDAVP-induced ADAMTS13 depression (Figure 5).
Figure 4.

Decrease of ADAMTS13 expression is abrogated by selenite and APC. (A) ADAMTS13 transcripts decreased in HSCs after stimulation with serum from septic patients (SepSe). Costimulation with APC (SepSe + APC) and selenite (SepSe + selenite) for 6 h or 24 h nullified ADAMTS13 transcript reduction as normalized to ADAM9 and HPRT. (B) The decrease of ADAMTS13 transcripts in HMEC is nullified after costimulation with APC (SepSe + APC) and selenite (SepSe + selenite) for 6 h or 24 h. (C) APC-mediated rescue of ADAMTS13 is triggered via the endothelial protein C receptor (EPCR) in endothelial cells. (D) Coadministration of APC and selenite increased ADAMTS13 mRNA transcription rate slightly. Bars represent mean values of log2-ratios ± SEM for at least four independent experiments.
APC Administration Increased ADAMTS13 Activity in Patients with Sepsis
To determine if ADAMTS13 levels and related parameters are affected after adjunctive therapy with APC, we used archival samples from a small characterized cohort of septic patients who received APC over time (Table 1). ADAMTS13 activity was diminished at the day of enrollment (range 0.19–0.53 U/mL) without a significant difference as compared with the placebo group. However, ADAMTS13 levels increased during the study period in the APC treatment group (Figure 6A). VWF:Ag ranged between 3.6 and 6.9 U/mL at d 1 with a broad spectrum in patients without adjunctive therapy and an intermittent decrease at d 2 and d 3 in APC-treated patients (Figure 6B). To gain insights into associations with thrombotic microangiopathy, we evaluated collagen-binding affinity (CB). VWF:CB decreased in the treatment group ranging between 146% to 232% at the day of enrollment. However, only a moderate variation was found in patients without APC therapy (Figure 6C). As a marker of endothelial activation, VWF propeptide levels were found enhanced in both groups at d 1 (APC group: 3.4 versus non-APC: 5.9 U/mL, P > 0.05), which decreased over time, however, in the APC-treated group to a greater extent with minor variations and with significant differences from d 4 of treatment with respect to d 1 (Figure 6D).
Table 1.
Patient characteristics at the day of enrollment.
| Adjunctive APC-treatmenta | No adjunctive APC-treatmentb | |
|---|---|---|
| Age, years | 47 (40.0/66.5)c | 64.5 (58.25/74.25)c |
| Number of patients | 5 | 4 |
| Sex distribution, male | 60% | 75% |
| Scores, points | ||
| APACHE II | 25 (23.5/28.5)c | 25 (19.75/34.75)c |
| SAPS | 50 (49/59.5)c | 62 (47.75/67.75)c |
| ISTH | 3 (3/3)c | 3 (3/3)c |
| SOFA | 9 (8/11.5)c | 11 (9.25/12.75)c |
| Pathogen | ||
| Gram-positive | 3 | 4 |
| Gram-negative | 2 | 0 |
| Focus | ||
| Lung | 2 | 2 |
| Abdomen | 2 | 3 |
| Urogenital | 2 | 1 |
| Various | 1 | 0 |
| Laboratory findings | ||
| Procalcitonin, ng/mL | 20.57 (4.79/33.09)c | 1.18 (0.37/29.66)c |
| Leukocytes, Gpt/L | 18.5 (4.4/37.25)c | 27.2 (4.25/40.75)c |
| Outcome | ||
| Length of stay at ICU, days | 8 (7/24)c | 13.5 (4/40.25)c |
| Length of stay at hospital, days | 34 (22/60)c | 19 (4.25/65.25)c |
| Mortality rate, % | 0 | 50 |
Patients who received APC (n = 5).
Patients with placebo (n = 4).
Data given as median (25th/75th percentile).
Figure 6.

Laboratory findings in patients with adjunctive therapy. ADAMTS13 activity and related parameters were determined in patients with severe sepsis with APC treatment (n = 5) and without APC (n = 4) therapy over time. (A) ADAMTS13 activity was diminished at the day of enrollment (range 0.19 to 0.53 u/mL) without significance between subgroups but increased during the course of APC treatment. (B) VWF:Ag ranged between 3.6 to 6.9 U/mL at d 1 with a broad spreading tendency in patients without adjunctive therapy and intermittently decrease in APC-treated patients. (C) VWF:CB (collagen binding) decreased in the APC group (range: 146% to 232 %) at the day of enrollment while a moderate variation was observed in the non-APC group. (D) VWF propeptide levels were found enhanced in both groups at d 1 (APC group: 3.4 versus non-APC: 5.9 U/mL, not significant [n.s.]) which decreased over time. However, there was a significant difference from treatment d 4 with respect to d 1. Asterisks indicate significant differences during the patients’ course in comparison to day of enrollment (*P < 0.05).
DISCUSSION
In this study, we demonstrate a mechanism of ADAMTS13 suppression originating from a decrease in mRNA transcription encoding ADAMTS13. This suppression of transcription was induced by proinflammatory cytokines, which are abundantly secreted in the circulation of septic patients due to an overwhelming immune response during infection (3,4). HSCs and endothelial cells are primary sources of ADAMTS13, but it is unclear to what extent they regulate ADAMTS13 activity. Therefore, we explored the effect of proinflammatory stimulation in HSCs and endothelial cells as the primary cellular sources of ADAMTS13 synthesis and secretion in circulation. The transcriptional suppression of ADAMTS13 mRNA by proinflammatory cytokines may explain the observed loss of proteolytic activity because ADAMTS13 is secreted directly from the Golgi to the cell exterior without storage (50). In addition, secreted ADAMTS13 is inactivated by plasma proteases such as elastase, plasmin or thrombin (29,34) abundantly present in the circulation of septic patients. A significant reduction in ADAMTS13 mRNA in this study is consistent with data obtained from primary HSCs in rats and endothelial cells in humans (32). ADAMTS13 activity is reduced markedly during apoptosis of HSC following treatment with dimethylnitrosamine in rats, but not in animals treated with tetra-chloromethane (CCl4) or thioacetamide, where tissue injury is rather characterized by hepatic stellate cell activation and fibrogenesis (51). Transactivation of rat HSC, triggered by cultivation on a plastic surface after isolation or in vivo by administration of CCl4, only controlled the concordant activity of plasma-secreted ADAMTS13 activity marginally (52). We also demonstrate the regulation of ADAMTS13 transcription by vasoactive agents (DDAVP and AVP). At 6 h and 24 h post-stimulation, HSCs and endothelial cells expressed less ADAMTS13 mRNA. However, ADAMTS13 mRNA deficiency should not significantly influence proteolysis of circulating or endothelium-bound VWF because an increase in shear stress during administration of the vasoconstrictors results in the exposure of selective epitopes of VWF, thereby enhancing proteolysis by residual ADAMTS13 as a compensatory event (53). In our study, ADAMTS13 transcript levels and proteolytic activity are downregulated by vasopressin in vitro. Because it is reported that DDAVP action is mediated via the V2 receptor, we determined the role of the V2 receptor using a potent V2-receptor antagonist. From our study, we identified that inhibition of the V2 receptor prevented ADAMTS13 mRNA suppression, suggesting a potential role of the V2 receptor in mediating ADAMTS13 mRNA down-regulation. However, the predefined two-fold threshold value for definition of differentially expressed genes was not attained in HPLS. This may lead to dilution of HSC-specific transcripts in a mixed tissue sample with the reference transcripts simultaneously expressed in hepatocytes. Nevertheless, the variation of ADAM12 upregulation as a marker and ADAMTS13 downregulation in our study supports our proposed mechanism. Given that DDAVP is mediated via the arginine vasopressin receptor 2 (V2 receptor) (54) and suppressed ADAMTS13 mRNA, it could be speculated that a V2-receptor subtype might be present and functionally active in HSCs. Because increased VWF:Ag levels following DDAVP administration are paralleled by a mild decrease of plasma ADAMTS13 activity in human volunteers (55,56), our current observations provide first evidence that the decline might be caused by transcriptional repression.
There is evidence of splice variants of ADAMTS13 in a hepatoma cell line resulting in synthesis of an intracellularly accumulated protease (57). To the best of our knowledge, there was no systematic report on splice variants of ADAMTS13 in human hepatic tumor tissue. Very recent reports (58) found a maintained balance of platelet activation status in patients with cirrhosis and cancer compared with age-related controls.
We also demonstrate that the administration of human recombinant APC resulted in preserved basal transcriptional rate of ADAMTS13 in vitro as well as an increased plasma activity. APC treatment was accompanied by an improved clinical condition of patients, as reflected by enhanced levels of VWF-related parameters and of endothelial dysfunction such as VWF propeptide (59,60). Following APC administration, the mechanism resulting in a decrease in collagen-binding activity, which is the measure of decreased VWF-adhesive activity, is unclear. It might be speculated that an increase in activity of ADAMTS13 by the presence of APC should have induced this phenomenon. The interaction between APC and human HSCs showed an expression of EPCR and an abrogation of thrombin-induced TNFα expression (61). As shown by our blocking experiments, the mechanism of action is strongly associated to the activity of EPCR-receptor. It might be speculated by which pathway the protective effect of APC is triggered along intracellular signaling events, resulting in a rather antiinflammatory condition. Otherwise, events of oxidative stress might be involved in the regulation of ADAMTS13 transcription, while the antioxidative properties of selenite are capable of interrupting the unfavorable effect with respect to ADAMTS13. There is mounting evidence that the prevention of oxidative damage and the attenuation of inflammatory response by selenite is reflected by either a decrease of the transcriptional rate of endothelial adhesion molecules (62) and cytokine-induced cellular activation (63) or the restoration of antioxidative selenoenzymes (64). Strikingly, costimulation with APC and selenite exert at least an additive effect on rescue of ADAMTS13 transcription. The mechanisms involved warrant further investigation. Furthermore, a direct interaction between ADAMTS13 activity and inflammation is provided by the evidence that ADAMTS13 deficiency results in increased leukocyte rolling and adhesion, as well as enhanced extravasation of neutrophils, where both processes are dependent on the presence of ulVWF and platelets (65). In accordance with these observations, our findings suggest that ADAMTS13 deficiency during systemic inflammation and infection accelerate inflammatory responses by slowing down leukocytes and facilitating their extravasation, which functions intrinsically as a protective mechanism, but becomes destructive due to an overwhelming systemic response which might be limited by APC.
In septic patients, the role of selenite supplementation to modulate reactive oxygen species generation, lower endogenous antioxidative capacity and improve clinical outcome is controversial (66–68). Here, we demonstrate that selenite abrogates the inhibitory effect of serum constituents from septic patients on cultivated HSCs and endothelial cells. It could be speculated that this mechanism involves genes regulating cell cycle, apoptosis and intermediary metabolism (69).
In this study, we obtained a data set with respect to ADAMTS13 regulation from surgical waste material, which is not representative for healthy, naïve tissue, but clearly reflects the comorbid conditions which are decisive for a patient cohort which is susceptible for generalized infection compared with healthy controls. For evaluation of the response generated to inflammatory stimuli, we determined the expression of ADAM9, which is a well-established and validated approach. ADAM9 was decreased in the presence of inflammatory stimuli. The use of HPLS with preserved tissue architecture was to overcome limitations of cell culture experiments. This model is an alternative to mouse models where VWF plasma concentration during the complete absence of ADAMTS13 activity is the major determinant of sepsis-induced mortality, which is unrepresentative of the clinical situation in humans (36). As observed in the mouse endotoxemia model, LPS administration affects both VWF and ADAMTS13 transcription in an opposite manner in hepatic and renal tissue accompanied by variations in plasma concentration (35).
CONCLUSION
In conclusion, we provide consistent data from in vitro experiments and an observational patient cohort implicating a possible benefit of selenite and APC in mediating ADAMTS13 regulation during sepsis. We also report that the dysbalance ratio between VWF and ADAMTS13 might be governed by treatment of endothelial dysfunction with APC. Proinflammatory cytokines highly abundant in plasma during sepsis, endotoxin and serum from septic patients suppress ADAMTS13 transcripts in primary production sources. These observations may reflect the decline of proteolytic activity of ADAMTS13 in sepsis and sepsis-associated TMA. Moreover, selenite and APC are able to overcome ADAMTS13 suppression on RNA-level in vitro. Suppression of ADAMTS13 also was overcome in patients treated with APC alone. Despite the fact that neither selenite nor APC is currently recommended by the international guidelines for treatment of severe sepsis or septic shock (70), we provide evidence of a direct manipulation of ADAMTS13 transcript level by use of pharmacological agents in sepsis, which may help to characterize and further evaluate the relevance of this therapeutic agents in the management of sepsis. The beneficial action of APC is nearly completely EPC-receptor driven. Furthermore, the interaction of APC with the receptor is also significant for abrogation of effects triggered by proinflammatory cytokines as present in our serum obtained from septic patients.
ACKNOWLEDGMENTS
The authors thank Gordon Philipp Otto, Sina Coldewey, Brigitte Specht, Barbara Schmidt and Edith Walther for their technical support and helpful comments. The authors acknowledge Utz Settmacher for providing surgical waste material and Dieter Muller for assistance in handling HPLS. This study was supported by grants from the German Research Foundation to RA Claus (DFG CL 173/4-1); German Federal Ministry of Education and Research within the Center for Sepsis Control and Care (grant 01 EO 1002, Project D1.9; PhD fellowship to ML Ekaney); and by the United States National Institutes of Health (DK56621 to SL Friedman).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Ekaney ML, et al. (2015) Preserved expression of mRNA coding von Willebrand factor–cleaving protease ADAMTS13 by selenite and activated protein C. Mol. Med. 21:355–63.
REFERENCES
- 1.Angus DC, Pereira CA, Silva E. Epidemiology of severe sepsis around the world. Endocr Metab Immune Disord Drug Targets. 2006;6:207–12. doi: 10.2174/187153006777442332. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity. 2014;40:463–75. doi: 10.1016/j.immuni.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–51. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- 5.Bone RC, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. 1992. Chest. 2009;136:e28. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 6.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 7.Aird WC. Endothelium as a therapeutic target in sepsis. Curr Drug Targets. 2007;8:501–7. doi: 10.2174/138945007780362782. [DOI] [PubMed] [Google Scholar]
- 8.Czabanka M, Peter C, Martin E, Walther A. Microcirculatory endothelial dysfunction during endotoxemia—insights into pathophysiology, pathologic mechanisms and clinical relevance. Curr Vasc Pharmacol. 2007;5:266–75. doi: 10.2174/157016107782023389. [DOI] [PubMed] [Google Scholar]
- 9.Bockmeyer CL, et al. Inflammation-associated ADAMTS13 deficiency promotes formation of ultra-large von Willebrand factor. Haematol. 2008;93:137–140. doi: 10.3324/haematol.11677. [DOI] [PubMed] [Google Scholar]
- 10.Kayal S, Jais JP, Aguini N, Chaudiere J, Labrousse J. Elevated circulating E-selectin, intercellular adhesion molecule 1, and von Willebrand factor in patients with severe infection. Am J Respir Crit Care Med. 1998;157:776–84. doi: 10.1164/ajrccm.157.3.9705034. [DOI] [PubMed] [Google Scholar]
- 11.Ruggeri ZM. von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost. 2003;1:1335–42. doi: 10.1046/j.1538-7836.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- 12.Arya M, et al. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood. 2002;99:3971–7. doi: 10.1182/blood-2001-11-0060. [DOI] [PubMed] [Google Scholar]
- 13.Andrews RK, Berndt MC. Platelet physiology and thrombosis. Thromb Res. 2004;114:447–53. doi: 10.1016/j.thromres.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 14.Uemura M, et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005;106:922–4. doi: 10.1182/blood-2005-01-0152. [DOI] [PubMed] [Google Scholar]
- 15.Levy GG, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–94. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Z, Nguyen TC, Guchhait P, Dong JF. von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura. Sem Thromb Hemost. 2010;36:71–81. doi: 10.1055/s-0030-1248726. [DOI] [PubMed] [Google Scholar]
- 17.Symmers WS. Thrombotic microangiopathic haemolytic anaemia (thrombotic microangiopathy) Br Med J. 1952;2:897–903. doi: 10.1136/bmj.2.4790.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, et al. Sepsis-induced disseminated intravascular coagulation with features of thrombotic thrombocytopenic purpura: a fatal fulminant syndrome. Clin Appl Thromb Hemost. 2010;17:251–3. doi: 10.1177/1076029609357739. [DOI] [PubMed] [Google Scholar]
- 19.Kinasewitz GT, Zein JG, Lee GL, Nazir SA, Taylor FB., Jr Prognostic value of a simple evolving disseminated intravascular coagulation score in patients with severe sepsis. Crit Care Med. 2005;33:2214–21. doi: 10.1097/01.ccm.0000181296.53204.de. [DOI] [PubMed] [Google Scholar]
- 20.Chertow GM, et al. Mortality after acute renal failure: models for prognostic stratification and risk adjustment. Kidney Int. 2006;70:1120–6. doi: 10.1038/sj.ki.5001579. [DOI] [PubMed] [Google Scholar]
- 21.Vincent JL, Yagushi A, Pradier O. Platelet function in sepsis. Crit Care Med. 2002;30:S313–7. doi: 10.1097/00003246-200205001-00022. [DOI] [PubMed] [Google Scholar]
- 22.Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J Thromb Haemost. 2006;4:1186–93. doi: 10.1111/j.1538-7836.2006.01949.x. [DOI] [PubMed] [Google Scholar]
- 23.Kremer Hovinga JA, et al. ADAMTS-13, von Willebrand factor and related parameters in severe sepsis and septic shock. J Thromb Haemost. 2007;5:2284–90. doi: 10.1111/j.1538-7836.2007.02743.x. [DOI] [PubMed] [Google Scholar]
- 24.Larkin D, et al. Severe Plasmodium falciparum malaria is associated with circulating ultra-large von Willebrand multimers and ADAMTS13 inhibition. PLoS Pathog. 2009;5:e1000349. doi: 10.1371/journal.ppat.1000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peigne V, et al. The prognostic value of ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats, member 13) deficiency in septic shock patients involves interleukin-6 and is not dependent on disseminated intravascular coagulation. Crit Care. 2013;17:R273. doi: 10.1186/cc13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claus RA, Bockmeyer CL, Sossdorf M, Losche W. The balance between von-Willebrand factor and its cleaving protease ADAMTS13: bio-marker in systemic inflammation and development of organ failure? Curr Mol Med. 2010;10:236–48. doi: 10.2174/156652410790963367. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen TC, et al. Intensive plasma exchange increases ADAMTS-13 activity and reverses organ dysfunction in children with thrombocytopenia-associated multiple organ failure. Crit Care Med. 2008;36:2878–87. doi: 10.1097/ccm.0b013e318186aa49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bockmeyer CL, et al. ADAMTS13 activity is decreased in a septic porcine model. Significance for glomerular thrombus deposition. Thromb Haemost. 2011;105:145–53. doi: 10.1160/TH10-03-0153. [DOI] [PubMed] [Google Scholar]
- 29.Ono T, et al. Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood. 2006;107:528–34. doi: 10.1182/blood-2005-03-1087. [DOI] [PubMed] [Google Scholar]
- 30.Claus RA, et al. Variations in the ratio between von Willebrand factor and its cleaving protease during systemic inflammation and association with severity and prognosis of organ failure. Thromb Haemost. 2009;101:239–47. [PubMed] [Google Scholar]
- 31.Mannucci PM, et al. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood. 2001;98:2730–5. doi: 10.1182/blood.v98.9.2730. [DOI] [PubMed] [Google Scholar]
- 32.Cao WJ, Niiya M, Zheng XW, Shang DZ, Zheng XL. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J Thromb Haemost. 2008;6:1233–5. doi: 10.1111/j.1538-7836.2008.02989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104:100–6. doi: 10.1182/blood-2004-01-0107. [DOI] [PubMed] [Google Scholar]
- 34.Crawley JT, et al. Proteolytic inactivation of ADAMTS13 by thrombin and plasmin. Blood. 2005;105:1085–93. doi: 10.1182/blood-2004-03-1101. [DOI] [PubMed] [Google Scholar]
- 35.Mimuro J, et al. Unbalanced expression of ADAMTS13 and von Willebrand factor in mouse endotoxinemia. Thromb Res. 2008;122:91–7. doi: 10.1016/j.thromres.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 36.Lerolle N, et al. von Willebrand factor is a major determinant of ADAMTS-13 decrease during mouse sepsis induced by cecum ligation and puncture. J Thromb Haemost. 2009;7:843–50. doi: 10.1111/j.1538-7836.2009.03313.x. [DOI] [PubMed] [Google Scholar]
- 37.Xu L, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–51. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiskirchen R, et al. Genetic characteristics of the human hepatic stellate cell line LX-2. PloS One. 2013;8:e75692. doi: 10.1371/journal.pone.0075692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ades EW, et al. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–90. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 40.Ceppi ED, Smith FS, Titheradge MA. Effect of multiple cytokines plus bacterial endotoxin on glucose and nitric oxide production by cultured hepatocytes. Biochem J. 1996;317(Pt 2):503–7. doi: 10.1042/bj3170503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuhn UD, Rost M, Muller D. Para-nitro-phenol glucuronidation and sulfation in rat and human liver slices. Exp Toxicol Pathol. 2001;53:81–7. doi: 10.1078/0940-2993-00153. [DOI] [PubMed] [Google Scholar]
- 42.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kokame K, Nobe Y, Kokubo Y, Okayama A, Miyata T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005;129:93–100. doi: 10.1111/j.1365-2141.2005.05420.x. [DOI] [PubMed] [Google Scholar]
- 44.Claus RA, Bockmeyer CL, Sossdorf M, Losche W, Hilberg T. Physical stress as a model to study variations in ADAMTS-13 activity, von Wille-brand factor level and platelet activation. J Thromb Haemost. 2006;4:902–5. doi: 10.1111/j.1538-7836.2006.01837.x. [DOI] [PubMed] [Google Scholar]
- 45.Reuken PA, et al. Imbalance of von Wille-brand factor and its cleaving protease ADAMTS13 during systemic inflammation superimposed on advanced cirrhosis. Liver Int. 2014;35:37–45. doi: 10.1111/liv.12657. [DOI] [PubMed] [Google Scholar]
- 46.Bai X, et al. Early versus delayed administration of norepinephrine in patients with septic shock. Crit Care. 2014;18:532. doi: 10.1186/s13054-014-0532-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pecori Giraldi F, et al. von Willebrand factor and fibrinolytic parameters during the desmopressin test in patients with Cushing’s disease. Br J Clin Pharmacol. 2011;71:132–6. doi: 10.1111/j.1365-2125.2010.03812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dziaman T, et al. Selenium supplementation reduced oxidative DNA damage in adnexectomized BRCA1 mutations carriers. Cancer Epidemiol Biomarkers Prev. 2009;18:2923–8. doi: 10.1158/1055-9965.EPI-09-0529. [DOI] [PubMed] [Google Scholar]
- 49.Christiaans SC, Wagener BM, Esmon CT, Pittet JF. Protein C and acute inflammation: a clinical and biological perspective. Am J Physiol Lung Cell Mol Physiol. 2013;305:L455–66. doi: 10.1152/ajplung.00093.2013. [DOI] [PubMed] [Google Scholar]
- 50.Turner NA, Nolasco L, Ruggeri ZM, Moake JL. Endothelial cell ADAMTS-13 and VWF: production, release, and VWF string cleavage. Blood. 2009;114:5102–11. doi: 10.1182/blood-2009-07-231597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kume Y, et al. Hepatic stellate cell damage may lead to decreased plasma ADAMTS13 activity in rats. FEBS Lett. 2007;581:1631–4. doi: 10.1016/j.febslet.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 52.Niiya M, et al. Increased ADAMTS-13 proteolytic activity in rat hepatic stellate cells upon activation in vitro and in vivo. J Thromb Haemost. 2006;4:1063–70. doi: 10.1111/j.1538-7836.2006.01893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grillberger R, et al. A novel flow-based assay reveals discrepancies in ADAMTS-13 inhibitor assessment as compared with a conventional clinical static assay. J Thromb Haemost. 2014;12:1523–32. doi: 10.1111/jth.12653. [DOI] [PubMed] [Google Scholar]
- 54.Juul KV, Bichet DG, Nielsen S, Norgaard JP. The physiological and pathophysiological functions of renal and extrarenal vasopressin V2 receptors. Am J Physiol renal Physiol. 2014;306:F931–40. doi: 10.1152/ajprenal.00604.2013. [DOI] [PubMed] [Google Scholar]
- 55.Reiter RA, Knobl P, Varadi K, Turecek PL. Changes in von Willebrand factor-cleaving protease (ADAMTS13) activity after infusion of desmopressin. Blood. 2003;101:946–8. doi: 10.1182/blood-2002-03-0814. [DOI] [PubMed] [Google Scholar]
- 56.Reiter RA, Varadi K, Turecek PL, Jilma B, Knobl P. Changes in ADAMTS13 (von-Willebrand-factor-cleaving protease) activity after induced release of von Willebrand factor during acute systemic inflammation. Thromb Haemost. 2005;93:554–8. doi: 10.1160/TH04-08-0467. [DOI] [PubMed] [Google Scholar]
- 57.Shomron N, et al. A splice variant of ADAMTS13 is expressed in human hepatic stellate cells and cancerous tissues. Thromb Haemost. 2010;104:531–5. doi: 10.1160/TH09-12-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alkozai EM, et al. No evidence for increased platelet activation in patients with hepatitis B- or C-related cirrhosis and hepatocellular carcinoma. Thromb Res. 2015;135:292–7. doi: 10.1016/j.thromres.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 59.Fukushima H, et al. Ratio of von Willebrand factor propeptide to ADAMTS13 is associated with severity of sepsis. Shock. 2013;39:409–14. doi: 10.1097/SHK.0b013e3182908ea7. [DOI] [PubMed] [Google Scholar]
- 60.Page AV, Liles WC. Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence. 2013;4:507–16. doi: 10.4161/viru.24530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamaguchi M, et al. Decreased protein C activation in patients with fulminant hepatic failure. Scand J Gastroenterol. 2006;41:331–7. doi: 10.1080/00365520500287574. [DOI] [PubMed] [Google Scholar]
- 62.Zhang F, et al. Inhibition of TNF-alpha induced ICAM-1, VCAM-1 and E-selectin expression by selenium. Atheroscler. 2002;161:381–6. doi: 10.1016/s0021-9150(01)00672-4. [DOI] [PubMed] [Google Scholar]
- 63.Tolando R, Jovanovic A, Brigelius-Flohe R, Ursini F, Maiorino M. Reactive oxygen species and proinflammatory cytokine signaling in endothelial cells: effect of selenium supplementation. Free Radic Biol Med. 2000;28:979–86. doi: 10.1016/s0891-5849(00)00183-0. [DOI] [PubMed] [Google Scholar]
- 64.Steinbrenner H, Bilgic E, Alili L, Sies H, Brenneisen P. Selenoprotein P protects endothelial cells from oxidative damage by stimulation of glutathione peroxidase expression and activity. Free Radic Res. 2006;40:936–43. doi: 10.1080/10715760600806248. [DOI] [PubMed] [Google Scholar]
- 65.Chauhan AK, et al. ADAMTS13: a new link between thrombosis and inflammation. J Exp Med. 2008;205:2065–74. doi: 10.1084/jem.20080130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heyland DK, Dhaliwal R, Suchner U, Berger MM. Antioxidant nutrients: a systematic review of trace elements and vitamins in the critically ill patient. Intensive Care Med. 2005;31:327–37. doi: 10.1007/s00134-004-2522-z. [DOI] [PubMed] [Google Scholar]
- 67.Sakr Y, et al. Adjuvant selenium supplementation in the form of sodium selenite in postoperative critically ill patients with severe sepsis. Crit Care. 2014;18:R68. doi: 10.1186/cc13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forceville X. Effects of high doses of selenium, as sodium selenite, in septic shock patients a placebo-controlled, randomized, double-blind, multi-center phase II study—selenium and sepsis. J Trace Elem Med Biol. 2007;21(Suppl 1):62–5. doi: 10.1016/j.jtemb.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 69.Bosse AC, et al. Impact of selenite and selenate on differentially expressed genes in rat liver examined by microarray analysis. Biosci Rep. 2010;30:293–306. doi: 10.1042/BSR20090089. [DOI] [PubMed] [Google Scholar]
- 70.Dellinger RP, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013;41:580–637. doi: 10.1097/CCM.0b013e31827e83af. [DOI] [PubMed] [Google Scholar]