

Abstract

We report herein the design and synthesis of a series of potent and selective GPR119 agonists. Our objective was to develop a GPR119 agonist with properties that were suitable for fixed-dose combination with a DPP4 inhibitor. Starting from a phenoxy analogue (1), medicinal chemistry efforts directed toward reducing half-life and increasing solubility led to the synthesis of a series of benzyloxy analogues. Compound 28 was chosen for further profiling because of its favorable physicochemical properties and excellent GPR119 potency across species. This compound exhibited a clean off-target profile in counterscreens and good in vivo efficacy in mouse oGTT.
Keywords: Type II diabetes, GPR119 agonists, chiral cis-cyclopropanes, benzyloxy analogues, oGTT
Diabetes mellitus is a serious and chronic medical condition that is rapidly growing throughout the world. Generally, diagnosis is determined by measurement of fasting plasma glucose and recently an international recommendation has been made for the use of hemoglobin A1C (HbA1C) as a time-integrated marker of glucose levels.1 Approximately, 90–95% of diabetics have type 2 diabetes (T2D), and the pathogenesis of this disease involves hepatic glucose overproduction, insulin resistance, and dysfunction of insulin producing β-cells.2 T2D is initially managed by regular exercise and dietary modifications; then, as it progresses, the disease is managed by oral medications or injectables such as insulin and GLP-1 agonists. Standard medications focus on glycemic control, either through monotherapies or combination therapies.3 In spite of the large number of available medications, there is still an unmet medical need for orally effective new treatments for diabetes that will offer better safety profiles and reduced adverse effects.
GPR119 is a G protein-coupled receptor whose expression in humans, mice, and rats is highly restricted to pancreatic islets and specific intestinal regions.4−6 GPR119 agonists are proposed to regulate glucose homeostasis through stimulating glucose-dependent insulin release both directly by enhancing pancreatic-cell function and indirectly by the release of incretin hormones from the small intestine.7−9 Theoretically, the selectivity of expression, coupled with an ability to potentiate insulin secretion only when glucose levels are elevated, makes GPR119 a very attractive target for the development of new medications for T2D. Indeed, the industry has advanced multiple GPR119 agonists, for example, GSK-1292263, PSN-821, and MBX-2982, into the Phase II clinical trials.8 However, despite tremendous efforts by academia and industry, the theoretical benefits that GPR119 agonists can offer have thus far been a challenge to demonstrate clinically.10,11
We were interested in developing a fixed-dose combination (FDC) of a GPR119 agonist and a DPP4 inhibitor in the hope that such a combination would provide greater effects than either agent alone.12 Previously we reported the design and synthesis of a series of GPR119 agonists with a cis-cyclopropane as the core structure.13,14 Herein we report our continued efforts in this area.
Compound 1, our previous preclinical candidate, had excellent potency, selectivity, and in vivo efficacy,14 but this compound had a long half-life (t1/2) in rat (Figure 1), and the clinically observed human half-life after single dose administration was significantly longer than desired,15 which rendered 1 unsuitable for FDC with sitagliptin (qd). Furthermore, this compound exhibited poor solubility in biorelevant media, including fasted-state simulated intestinal fluid (FaSSIF).16−18 The low solubility resulted in a high calculated dose number, an increase in absorption risk and anticipated need for enhanced formulation complexity.19−22 Since formulation complexity would increase FDC development challenges, it was desirable to identify compounds with improved physicochemical properties.23 Therefore, the lead optimization objective was to reduce the projected human half-life and increase solubility, while simultaneously maintaining other favorable properties.
Figure 1.

Previous preclinical candidate. (a)EC50 data: mean ± SD (% control at max dose). (b)Calculated from Cl 2.9 mL/min/kg, Vd 3.4 L/kg, and fu 0.2%. (c)HPLC LogD at pH 7.
The plasma half-life is determined by two pharmacokinetic parameters, intrinsic clearance (Clint), and unbound volume of distribution (Vdu).24 Half-life can be shortened either by increasing Clint or by decreasing Vdu of a compound. In most cases, increasing Clint also leads to an increased dose projection and potentially higher levels of metabolites, thus decreasing Vdu is a preferable strategy to reduce half-life. Vdu can be modified by adjusting the lipophilicity or introducing basic and acidic functional groups.25 For compounds in the same class, reducing lipophilicity usually leads to decreased Vdu. From phenoxy analogue 1 to benzyloxy analogue 2, a significant drop in lipophilicity was predicted (cLogP 3.8 vs 2.2), and measured LogD dropped by 1.2. Indeed, lower unbound volume (81 vs 1725 L/kg) and shorter rat t1/2 (1.8 vs 14.7 h) were observed in compound 2, where GPR119 potency was almost maintained (human EC50 = 8.7 vs 3.3 nM). Similarly, from phenoxy analogue 3 to benzyloxy analogue 4, unbound volume was decreased from 242 to 62 L/kg, and t1/2 was reduced from 6.5 to 1.5 h (Figure 2). Although Clint changed in both cases, it was the magnitude of Vdu reduction that caused the ratio of Vdu to Clint to drop by 8-fold from 1 to 2 and 4-fold from 3 to 4, which was approximately reflected in the reduction in t1/2, respectively. In addition, from phenoxy analogues to benzyloxy analogues (1, 3 vs 2, 4), the decreased cLogP and LogD value correlated with an increase in solubility. For example, compound 4 demonstrated improved FaSSIF solubility over 3 (127 vs 61 μM). Finally, to our delight, compound 4 also displayed higher GPR119 potency (0.8 vs 1.7 nM).
Figure 2.

Comparison of benzyloxy and phenoxy analogues. (a)EC50 data: mean ± SD (% control at max dose). (b)From Cl 9.0 mL/min/kg, Vd 1.3 L/kg, and fu 1.6%. (c)HPLC LogD at pH 7. (d)fu of the corresponding methylsulfone analogue (2%) used; Cl 13.5 mL/min/kg, Vd 4.8 L/kg. (e)From Cl 13.5 mL/min/kg, Vd 4.8 L/kg, and fu 5.8%.
In recent years the concept of lipophilic ligand efficiency (LLE), which combines both potency and lipophilicity, has been shown to be very useful in measuring the progress of the lead optimization process.26 In this regard, the LLE was significantly increased from phenoxy analogues 1 and 3 to benzyloxy analogues 2 and 4: LLE was 4.7 vs 5.9 for compounds 1 and 2, and 3.8 vs 5.0 for compounds 3 and 4.27
Encouraged by these initial results, further structure–activity relationship (SAR) efforts were directed toward finding a compound with an even more favorable physicochemical profile. Our optimization efforts kept the central cyclopropyl piperidine core intact and focused on surveying a variety of functional groups at both ends, including the favorable piperidine right-hand side “capping” groups previously reported for GPR119 agonists.12 As illustrated in Scheme 1, the synthesis of the central cyclopropyl piperidine piece, the chiral synthon 9, started with the commercially available piperidine aldehyde 5. Corey–Fuchs type reaction was used to convert aldehyde 5 to propargyl alcohol 7 by a two-step sequence via dibromo-olefin intermediate 6.28 Reduction of the alkyne in 7 with Lindlar’s catalyst gave cis-alkene 8, which was subject to Charette’s enantioselective cyclopropanation reaction.29 Thus, using the dioxaborolane ligand derived from (S,S)-N,N,N′,N′-tetramethyltartaric acid diamide as a chiral auxiliary, the desired chiral synthon (R,R)-9 was obtained as a white solid (>98% de and >98% ee) after recrystallization from heptane.30
Scheme 1. Synthesis of Chiral Synthon (R,R)-9.

Reagents and conditions: (a) CBr4, PPh3; (b) nBuLi, THF, then paraformaldehyde; (c) H2, quinoline, Lindlar catalyst, EtOAc; (d) Charette cyclopropanation, Et2Zn/CH2I2, dichloromethane, −20–0 °C.
With key intermediate 9 in hand, the benzyloxy analogues can be efficiently assembled (Scheme 2).30 In the presence of a base such as NaH or NaHMDS, SN2 type reaction between 9 and an appropriate benzyl halide gave benzyl ether 10, which upon deprotection of the Boc group provided piperidine intermediate 11. The synthesis was divergent at this point depending on the type of the piperidine capping groups. If the capping group is a pyrimidine, an SNAr reaction was performed (2, 18, 19, 23–31, 33, and 34). To introduce the 1-methylcyclopropyloxycarbonyl capping group in 22, intermediate 11 was reacted with the preformed succinimide 12.31 In the case of the oxadiazole capping group (4, 20, and 21), intermediate 11 was converted to cyano substituted piperidine 13 by treatment with cyanogen bromide; then the cyano group was converted to the isopropyl oxadiazole by zinc chloride mediated reaction with N-hydroxyisopropylimidamide, followed by acid mediated cyclization.32
Scheme 2. Synthesis of Benzyloxy Analogues.

Reagents and conditions: (a) Base, THF; (b) TFA, dichloromethane; (c) Het-X, Cs2CO3, DMF; (d) Et3N, dichloromethane; (e) Br-CN, K2CO3, CHCl3, reflux; (f) iPr–C(=NH)NHOH, ZnCl2, THF, reflux.
The synthetic sequence for acetamide analogue 32 is shown in Scheme 3. After three straightforward manipulations, Boc deprotection, pyrimidine installation, and benzyl ether formation, aryl bromide 16 was transformed to aryl acetate 17 through an efficient Negishi type coupling.33 Finally, tert-butyl ester hydrolysis and amide formation completed the synthesis of 32.
Scheme 3. Synthesis of Acetamide Analogue 32.

Reagents and conditions: (a) 4 N HCl in dioxane; (b) 2-chloro-5-methoxymethylpyrimidine, Cs2CO3, DMSO, 60 °C, 76%; (c) NaH, 4-bromo-1-(bromomethyl)-2-fluorobenzene, DMF, 96%.; (d) BrZnCH2COOtBu, Pd2(dba)3, X-Phos, THF, 60 °C, 98%; (e) TFA, dichloromethane, 100%; (f) morpholine, HOBT, EDC, diisopropylethylamine, dichloromethane, 69%.
The in vitro agonist activity for human and mouse receptors was evaluated by two measures: the potency measure as expressed in EC50 values in a cAMP assay and the magnitude of agonist activity measure as expressed in % control at the maximum dose compared to an internal agonist control.34 The data is summarized in Table 1. In general, these benzyloxy analogues possessed good to excellent potency and a full agonist response if the appropriate piperidine capping groups were chosen on the right, such as 5-substitued pyrimidine, isopropyl oxadiazole, and 1-methylcyclopropyloxycarbonyl, and favorable substituents were incorporated on the left, such as methylsulfone and tetrazole.12−14 Several trends were observed. While hydroxyethylsulfone somewhat increased potency (18 vs 2), ethylsulfone somewhat decreased potency (31 vs 28). Replacing 2-pyridyl with 2-fluorophenyl group resulted in significant improvements in FaSSIF solubility (27 vs 19), which could not be predicted by the cLogP and measured LogD values. The 5-position of the pyrimidine capping group was compatible with many functional groups of which methoxy and methoxymethyl (MOM) were effective at improving solubility (23, 25, and 28). Finally, all the benzyloxy analogues in Table 1 exhibited relatively short t1/2s in rat compared to the phenoxy analogues and had predicted human half-lives in the range of 2 to 38 h.35
Table 1. SAR Efforts and Identification of Compound 28.
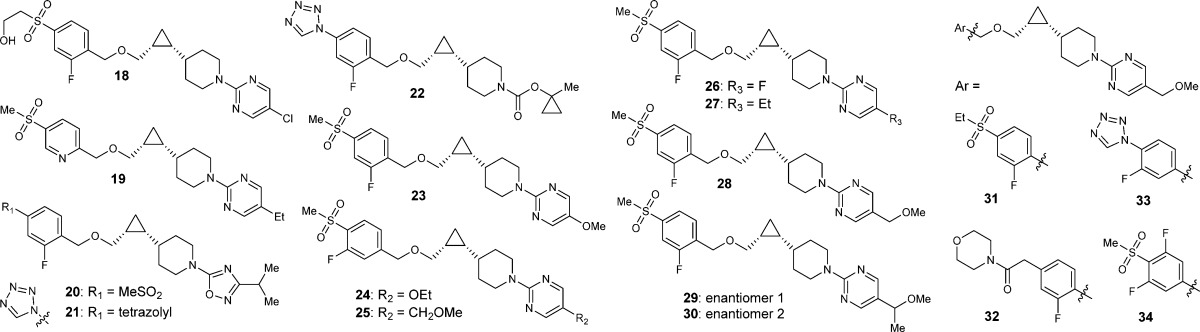
| cmpd | hGPR119a EC50 (nM) | mGPR119a EC50 (nM) | FaSSIFb (μM) | cLogP | HPLC LogD (pH 7) | rat t1/2 (h) | pred. human t1/2 (h)d |
|---|---|---|---|---|---|---|---|
| 2 | 8.7 ± 3.4 (53%) | 3.6 ± 0.1 (56%) | nd | 2.2 | 3.7 | 1.8 | nd |
| 18 | 2.3 ± 0.3 (68%) | 1.7 ± 0.4 (121%) | 83 | 2.9 | 4.8 | 0.9 | 5–7 |
| 19 | 1.5 ± 0.1 (83%) | 2.5 ± 2.1 (153%) | 7 | 2.5 | 3.6 | 1.3 | 8–9 |
| 20 | 4.2 ± 1.0 (87%) | 9.9 ± 5.5 (76%) | 144 | 3.6 | 3.8 | 2.2 | 9–14 |
| 21 | 1.7 ± 0.1 (129%) | 4.8 ± 0.1 (105%) | 103 | 4.1 | 3.9 | 1.4 | 5–9 |
| 22 | 4.8 ± 1.8 (121%) | 15 ± 0.3 (108%) | 127 | 3.6 | 3.8 | 1.0 | 6–7 |
| 23 | 4.5 ± 2.9 (79%) | 1.4 ± 1.2 (85%) | 151 | 3.1 | 4.0 | 0.6 | 4–6 |
| 24 | 1.7 ± 1.1 (75%) | 2.3 ± 0.6 (96%) | 101 | 3.7 | 4.2 | 0.8 | 5–7 |
| 25 | 3.3 ± 1.9 (94%) | 5.2 ± 2.1 (87%) | 155 | 2.4 | 3.5 | 1.2c | 8–9 |
| 26 | 2.4 ± 2.1 (85%) | 4.5 ± 5.6 (131%) | 92 | 2.8 | 4.2 | 1.5 | 12–13 |
| 27 | 2.5 ± 1.9 (98%) | 5.5 ± 2.7 (115%) | 140 | 3.6 | 4.3 | 1.0c | 13 |
| 28 | 2.1 ± 1.3 (88%) | 1.9 ± 0.8 (97%) | 161 | 2.4 | 3.5 | 1.0 | 26 |
| 29 | 3.1 ± 1.2 (87%) | 8.9 ± 9.5 (109%) | 143 | 2.7 | 3.9 | 1.4 | 26 |
| 30 | 4.6 ± 5.0 (95%) | 3.3 ± 1.6 (70%) | 137 | 2.7 | 3.9 | 1.0c | 21 |
| 31 | 7.1 ± 1.4 (58%) | 12.5 ± 0.8 (87%) | 129 | 2.9 | 3.8 | 2.1 | 21 |
| 32 | 3.2 ± 0.6 (104%) | 3.7 ± 0.8 (99%) | 172 | 3.0 | 3.4 | 0.4 | 2 |
| 33 | 1.1 ± 0.3 (84%) | 3.8 ± 1.0 (75%) | 133 | 2.9 | 3.7 | 3.0c | 38 |
| 34 | 4.5 ± 1.9 (80%) | 5.8 ± 3.3 (67%) | 115 | 2.6 | 3.6 | 0.5 | 4 |
Human and mouse GPR119 EC50 data expressed as mean ± SD (n ≥ 2 independent experiments). The percentage in parentheses, % control at max dose: magnitude of cAMP stimulation expressed in % compared to an internal agonist control; the control was defined to have 100% cAMP stimulation.
Kinetic solubility in fasted-state simulated intestinal fluid (FaSSIF) at pH 6.5; in parentheses are cLogP values.
Effective t1/2 calculated from MRT × 0.693 (for compounds exhibiting biphasic PK).
Predicted solely based on rat PK. Where there is only one number (compounds 27–34), allometry was not used since rat Clint was much higher than human. Also, a predicted human half-life in the range of 8–48 h was considered acceptable for further profiling and could potentially support once daily dosing.
Compound 28, with FaSSIF solubility of 161 μM, cLogP of 2.4, and LLE of 6.3, was identified as a potent GPR119 agonist with suitable physicochemical properties for a fixed-dose-combination and therefore was further profiled. An off-target screen of this compound was performed against a panel of 168 receptors, ion channels, and enzymes, and no off-target activities were found with IC50s < 10 μM. In a cardiac ion channel functional blockade activity panel,3628 exhibited mild inhibition of hERG with a calculated IC50 of 13 μM.37 There were no effects on IKs or INa up to the highest tested concentration of 30 μM. Compound 28 was also selective over CYP 3A4 (IC50 = 40 μM) and hPXR (EC50 > 30 μM, 39% Act).38
The promising in vitro profile of 28 was followed by characterization of this compound in vivo. In a mouse oGTT39 and PK/PD dose–response study, compound 28 showed a dose-dependent lowering of blood glucose excursion, as represented by the excursion AUC from 0–120 min (Figure 3). Based upon this data, the MEDmax (minimal efficacious dose for maximal efficacy) was 3 mpk. The blood concentrations of 28 at 90 min post-dosing increased approximately linearly at 0.3, 1, 3, and 10 mpk (18 ± 5, 86 ± 8, 302 ± 57, and 622 ± 160 nM, respectively).
Figure 3.
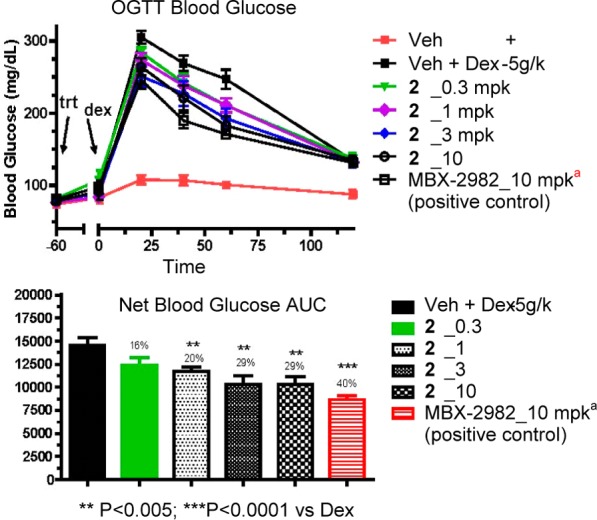
oGTT and PK/PD dose response of compound 28 in lean mice. (a)Positive control MBX-298240 was more efficacious than 28 probably because it had better mouse PK; the two compounds had comparable mouse potency.
The pharmacokinetic profile of 28 was evaluated in rat, dog, and monkey (Table 2). Overall, this compound possessed good PK properties in dogs (t1/2 17 h, F 60%), but lower oral bioavailability in rats and monkeys, likely due to more extensive metabolism in these species relative to dogs (Clint was 1104, 319, 60, and 42 mL/min/kg in rat, monkey, dog, and human liver microsomes, respectively).41 Compared to lead compound 1, 28 had a much lower Vdu (1725 vs 54 L/kg) in rat, which contributed heavily to t1/2 reduction (rat t1/2 14.7 vs 1.0 h). Based upon a plasma trough target of 549 nM (from the MEDmax in mouse and a blood/plasma ratio of 0.55), the projected human half-life of 28 was ∼15 h and estimated human QD dose was ∼300 mg.42
Table 2. Plasma Pharmacokinetic Profile of Compound 28a.
| species | Plasma Cl (mL/min/kg) | Vd (L/kg) | t1/2 (h) | F (%) |
|---|---|---|---|---|
| ratb | 41 | 2.2 | 1.0 | 27 |
| dogc | 6.0 | 6.5 | 17 | 60 |
| monkeyd | 16 | 2.0 | 2.3 | 5 |
PPB for 28 (fu): r 4.1%, d 5.5%, rh 3.2, h 4.9%.
Rat Wistar Han: Clint = 1000 mL/min/kg; Vdu = 54 L/kg.
Dog Beagle.
Rhesus monkey.
The pharmaceutical properties of 28 were also assessed. The crystalline free base of this compound was obtained from recrystallization with water, and it was characterized as its solid state form. The solubility of this form was evaluated in IV and PO vehicles that were acceptable for safety studies. After equilibration in vehicles and biorelevant media, compound 28 demonstrated solubility of 129 mg/mL in PEG200, 325 mg/mL in PEG400, 56 μg/mL in FaSSIF, and 0.87 mg/mL in simulated gastric fluid (SGF). Finally, compound 28 falls nicely within the druglike property space: MW 464, cLogP 2.4, HPLC LogD 3.5, LLE 6.3, and PFI 5.5.43
In summary, we have designed and synthesized a series of GPR119 agonists with properties that may be suitable for fixed-dose combination. A major focus of the optimization effort was to reduce the predicted t1/2 in humans and improve solubility of the phenoxy lead. Half-life was reduced by modifying the unbound volume of distribution (Vdu) through reducing lipophilicity, and solubility was improved by incorporating solubilizing groups such as MeO and MOM moieties. These efforts led to the identification of compound 28 that possessed excellent potency, selectivity, and improved physicochemical properties. Further optimization to improve overall PK profile and reduce projected human dose will be reported in due course.
Acknowledgments
We thank Dr. Jeffrey J. Hale for insightful suggestions.
Supporting Information Available
Synthetic procedures and characterization data of selected compounds, conditions for the biological assays, and protocol for pharmacokinetic and pharmacodynamic studies. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00207.
The authors declare no competing financial interest.
Supplementary Material
References
- International Expert Committee Report on the Role of the A1C Assay in the Diagnosis of Diabetes. Diabetes Care 2009, 32, 1327–1334. 10.2337/dc09-9033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skyler J. S. Pathogenesis of type 2 diabetes mellitus. J. Med. Chem. 2004, 47, 4113–4117. 10.1021/jm0306273. [DOI] [PubMed] [Google Scholar]
- Goldberg R. B.; Holman R.; Drucker D. J. Management of type 2 diabetes. N. Engl. J. Med. 2008, 358, 293–297. 10.1056/NEJMclde0708469. [DOI] [PubMed] [Google Scholar]
- Soga T.; Ohishi T.; Matsui T.; Saito T.; Matsumoto M.; Takasaki J.; Matsumoto S.; Kamohara M.; Hiyama H.; Yoshida S.; Momose K.; Ueda Y.; Matsushime H.; Kobori M.; Furuichi K. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G protein-coupled receptor. Biochem. Biophys. Res. Commun. 2005, 326, 744–751. 10.1016/j.bbrc.2004.11.120. [DOI] [PubMed] [Google Scholar]
- Chu Z.; Carroll C.; Alfonso J.; Gutierrez V.; He H.; Lucman A.; Pedraza M.; Mondala H.; Gao H.; Bagnol D.; Chen R.; Jones R. M.; Behan D. P.; Leonard J. A role for intestinal endocrine cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucagon-like peptide-1 and glucose-dependent insulinotropic peptide release. Endocrinology 2008, 149, 2038–2047. 10.1210/en.2007-0966. [DOI] [PubMed] [Google Scholar]
- Sakamoto Y.; Inoue H.; Kawakami S.; Miyawaki K.; Miyamoto T.; Mizuta K.; Itakura M. Expression and distribution of Gpr119 in the pancreatic islets of mice and rats: Predominant localization in pancreatic polypeptide-secreting PP-cells. Biochem. Biophys. Res. Commun. 2006, 351, 474–480. 10.1016/j.bbrc.2006.10.076. [DOI] [PubMed] [Google Scholar]
- Gao J.; Tian L.; Weng G.; Bhagroo N. V.; Sorenson R. L.; O’Brien T. D.; Luo J.; Guo Z. Stimulating beta Cell Replication and Improving Islet Graft Function by GPR119 Agonists. Transplant Int. 2011, 24, 1124–1134. 10.1111/j.1432-2277.2011.01332.x. [DOI] [PubMed] [Google Scholar]
- Ohishi T.; Yoshida S. The Therapeutic Potential of GPR119. Agonists for Type 2 Diabetes. Expert Opin. Invest. Drugs 2012, 21, 321–328. 10.1517/13543784.2012.657797. [DOI] [PubMed] [Google Scholar]
- Dhayal S.; Morgan N. G. The Significance of GPR119 Agonists as a Future Treatment for Type 2 Diabetes. Drug News Perspect. 2010, 23, 418–424. 10.1358/dnp.2010.23.7.1468395. [DOI] [PubMed] [Google Scholar]
- Polli J. W.; Hussey E.; Bush M.; Generaux G.; Smith G.; Collins D.; McMullen S.; Turner N.; Nunez D. J. Evaluation of drug interactions of GSK1292263 (a GPR119 agonist) with statins: from in vitro data to clinical study design. Xenobiotica 2013, 43, 498–508. 10.3109/00498254.2012.739719. [DOI] [PubMed] [Google Scholar]
- Katz L. B.; Gambale J. J.; Rothenberg P. L.; Vanapalli S. R.; Vaccaro N.; Xi L.; Sarich T. C.; Stein P. P. Effects of JNJ-38431055, a Novel GPR119 Receptor Agonist, in Randomized, Double-Blind, Placebo-Controlled Studies in Subjects with Type 2 Diabetes. Diabetes, Obes. Metab. 2012, 14, 709–716. 10.1111/j.1463-1326.2012.01587.x. [DOI] [PubMed] [Google Scholar]
- Shah U. GPR119 agonists: A promising new approach for the treatment of type 2 diabetes and related metabolic disorders. Curr. Opin. Drug Discovery Dev. 2009, 12, 519–532. [PubMed] [Google Scholar]
- Wood H. B.; Szewczyk J. W.; Huang Y.; Adams A. D.. Substituted cyclopropyl compounds, compositions containing such compounds and methods of treatment. WO 2009129036 A1, 2009.
- Szewczyk J. W.; Acton J.; Adams A. D.; Chicchi G.; Freeman S.; Howard A. D.; Huang Y.; Li C.; Mosely R.; Murphy E.; Samuel R.; Santini C.; Yang M.; Zhang Y.; Zhao K.; Wood H. B. Design of potent and selective GPR119 agonists for type II diabetes. Bioorg. Med. Chem. Lett. 2011, 21, 2665–2669. 10.1016/j.bmcl.2010.12.086. [DOI] [PubMed] [Google Scholar]
- Unpublished results.
- For the composition of the FaSSIF, see Table 13 inDressman J. B.; Amidon G. L.; Reppas C.; Shah V. P. Dissolution Testing as a Prognostic Tool for Oral Drug Absorption: Immediate Release Dosage Forms. Pharm. Res. 1998, 15, 11–22. 10.1023/A:1011984216775. [DOI] [PubMed] [Google Scholar]
- For an introduction to biorelevant media, such as fasted state simulated intestinal fluid (FaSSIF), see:Galia E.; Nicolaides E.; Horter D.; Lobenberg R.; Reppas C.; Dressman J. B. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998, 15, 698–705. 10.1023/A:1011910801212. [DOI] [PubMed] [Google Scholar]
- FaSSIF solubility was used as one of the key parameters during the optimization of this lead series since it was demonstrated that dissolution testing in biorelevant media such as FaSSIF mimics the in vivo situation more closely, which provides better in vitro/in vivo correlation; see:Dressman J. B.; Reppas C. In vitro–in vivo correlations for lipophilic, poorly water-soluble drugs. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), S73–S80. 10.1016/S0928-0987(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Oh D. M.; Curl R.; Amidon G. Estimating the fraction dose absorbed from suspensions of poorly soluble compounds in humans: a mathematical model. Pharm. Res. 1993, 10, 264–270. 10.1023/A:1018947113238. [DOI] [PubMed] [Google Scholar]
- Martinez M.; Amidon G. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J. Clin. Pharmacol. 2002, 42, 620–643. 10.1177/00970002042006005. [DOI] [PubMed] [Google Scholar]
- Rohrs B. R.Biopharmaceutics Modeling and the Role of Dose and Formulation on Oral Exposure. In Optimizing the “Drug-Like” Properties of Leads in Drug Discovery; Borchardt R. T., Hageman M. J., Stevens J. L., Kerns E. H., Thakker D. R., Eds; Springer: New York, 2006; Vol. IV, pp 151– 166. [Google Scholar]
- Wuelfing W. P.; Daublain P.; Kesisoglou F.; Templeton A.; McGregor C. Preclinical dose number and its application in understanding drug absorption risk and formulation design for preclinical species. Mol. Pharmaceutics 2015, 12, 1031–1039. 10.1021/mp500504q. [DOI] [PubMed] [Google Scholar]
- Desai D.; Wang J.; Wen H.; Li X.; Timmins P. Formulation design, challenges, and development considerations for fixed dose combination (FDC) of oral solid dosage forms. Pharm. Dev. Technol. 2013, 18, 1265–1276. 10.3109/10837450.2012.660699. [DOI] [PubMed] [Google Scholar]
- Benet L. Z.; Zia-Amirhosseini P. Basic Principles of Pharmacokinetics. Toxicol. Pathol. 1995, 23, 115–123. 10.1177/019262339502300203. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. Volume of Distribution in Drug Design. J. Med. Chem. 2015, 10.1021/acs.jmedchem.5b00201. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Calculated from human GPR119 EC50 and cLogP values according to the equation: LLE = pEC50 – cLogP.
- Corey E. J.; Fuchs P. L. A synthetic method for formyl to ethynyl conversion. Tetrahedron Lett. 1972, 13, 3769–3772. 10.1016/S0040-4039(01)94157-7. [DOI] [Google Scholar]
- Charette A. B.; Juteau H.; Lebel H.; Molinaro C. Enantioselective Cyclopropanation of Allylic Alcohols with Dioxaborolane Ligands: Scope and Synthetic Applications. J. Am. Chem. Soc. 1998, 120, 11943–11952. 10.1021/ja982055v. [DOI] [Google Scholar]
- For detailed procedure, see Supporting Information.
- Zhu H. Y.; Cooper A. B.; Desai J. A.; Wang J.-S.; Rane D. F.; Doll R. J.; Njoroge F. G.; Girijavallabhan V. M.. Novel farnesyl protein transferase inhibitors as antitumor agents. WO 2005014577 A1, 2005.
- Jones R. M.; Semple G.; Xiong Y.; Shin Y.-J.; Ren A. S.; Calderon I.; Choi J. S. K.; Fioravanti B.; Lehmann J.; Bruce M. A.. Trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prophylaxis and treatment of disorders related thereto. WO 2005007647 A1, 2005.
- Sham H. L.; Konradi A. W.; Hom R. K.; Probst G. D.; Bowers S.; Truong A.; Neitz R. J.; Sealy J.; Toth G.. Preparation of N-(thiophen-3-yl)acetamide derivatives as inhibitors of JNK N-terminal kinase. WO 2010091310 A1, 2010.
- For assay conditions, see Supporting Information.
- The half-lives in humans were predicted based upon allometric scaling from rats and/or in vitro Clint data in human vs rat liver microsomes.
- Measured by PatchXpress systems. For a review on ion channels, see:Aidley D. J., Stanfield P. R., Eds. Ion Channels: Molecules in Action; Cambridge University Press: Cambridge, U.K., 1996. [Google Scholar]
- There were no findings in CV guinea pig studies at 64× over target clinical exposure (unpublished results).
- Pregnane X receptor (PXR) is a nuclear receptor known to regulate expression of drug-metabolizing enzymes including cytochrome P450 (CYP450), see:Chu V.; et al. In vitro and in vivo induction of cytochrome P450: A survey of the current practices and recommendations: A Pharmaceutical Research and Manufacturers of America perspective. Drug Metab. Dispos. 2009, 37, 1339–1354. 10.1124/dmd.109.027029. [DOI] [PubMed] [Google Scholar]
- Andrikopoulos S.; Blair A. R.; Deluca N.; Fam B. C.; Proietto J. Evaluating the glucose tolerance test in mice. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1323–1332. 10.1152/ajpendo.90617.2008. [DOI] [PubMed] [Google Scholar]
- Roberts B.; Gregoire F. M.; Karpf D. B.; Clemens E.; Lavan B.; Johnson F.; Mcwherter C. A.; Martin R.; Wilson M.. MBX-2982, a Novel Oral GPR119 Agonist for the Treatment of Type 2 Diabetes: Results of Single & Multiple Dose Studies, American Diabetes Association, 69th Meetings, New Orleans, 2009. [Google Scholar]
- Met ID studies suggested that the two ether linkages, methyl ether on the right and cyclopropylmethyl ether in the middle, were metabolic hotspots in 28.
- Using cross species microsomal stability data and allometry from dogs, the predicted human plasma clearance was 3 mL/min/kg. The Vd in humans (mean of 3.8 L/kg) was calculated using the values in the preclinical species adjusted for plasma protein binding differences. Bioavailability in humans was estimated to be 60% based upon dog PK data.
- Leeson P. D.; Young R. J. Molecular Property Design: Does Everyone Get It?. ACS Med. Chem. Lett. 2015, 6, 722–725. 10.1021/acsmedchemlett.5b00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.