

Abstract
History is replete with emergent pandemic infections that have decimated the human population. Given the shear mass of humans that now crowd the earth, there is every reason to suspect history will repeat itself. We describe three RNA viruses that have recently emerged in the human population to mediate severe neurological disease. These new diseases are results of new mutations in the infectious agents or new exposure pathways to the agents or both. To appreciate their pathogenesis, we summarize the essential virology and immune response to each agent. Infection is described in the context of known host defenses. Once the viruses evade immune defenses and enter central nervous system (CNS) cells, they rapidly co‐opt host RNA processing to a cataclysmic extent. It is not clear why the brain is particularly susceptible to RNA viruses; but perhaps because of its tremendous dependence on RNA processing for physiological functioning, classical mechanisms of host defense (eg, interferon disruption of viral replication) are diminished or not available. Effectiveness of immunity, immunization and pharmacological therapies is reviewed to contextualize the scope of the public health challenge. Unfortunately, vaccines that confer protection from systemic disease do not necessarily confer protection for the brain after exposure through unconventional routes.
Keywords: avian influenza A virus, human parechovirus, infection, neuropathology, Rift Valley fever virus, virus
Introduction
There are no “new world” Native Americans who can recall smallpox because those indigenous civilizations were destroyed by novel Eurasian pathogens such as smallpox, measles and influenza. No one reading this article has any personal memory of the “Spanish Flu,” yet without it the course of human history might have been quite different. A few readers may remember a much lesser epidemic of polio, but even here it stretches our imagination to think that in the 1950s more people knew a scientist named Jonas Salk and the polio field trials than knew the full name of the President of the United States 90.
With memory of these devastating pandemics behind us, it would be fair to say that most citizens of the modern, developed world have grown complacent about the potential of infectious agents to lay low our civilization. Perhaps the panic in the early years of the AIDS epidemic, the fear over the brief SARS outbreak or the more recent Ebola outbreak in West Africa might foretell the type of hysteria society could face with the next plague, but developed nations have mostly dodged those bullets. Examining the AIDS epidemic in the developed world shows it primarily caused panic in a defined segment of society that was particularly susceptible to infection. The virus was only modestly successful in spreading through sexual contact and it killed slowly. Even today there is no effective HIV vaccine, but we were fortunate in developing combination drug therapy that has significantly abated morbidity and mortality and diminished spread of the virus at the same time. Nevertheless, the denizens of sub‐Saharan Africa have a much different perspective on the scourge of AIDS, and if HIV did not have to stand in line behind a number of other lethal infectious diseases, it might have had an even greater cultural and historical impact.
The goal of this brief review is to focus on three emerging infectious agents that have a particular propensity to damage the brain. It is probably not a coincidence that all three are RNA viruses. The human brain has evolved with a highly complex diversification of gene expression. Recent studies have suggested that aberrant RNA processing may underlie many neurodegenerative diseases 4. Indeed our entire understanding of brain functioning and physiology must be reexamined in the context of the role of RNA metabolism. This review presents the proverbial tip of the iceberg by highlighting how once RNA viruses evade immune defenses and enter central nervous system (CNS) cells, they are able to rapidly co‐opt the efficient host brain RNA processing to an unprecedented and cataclysmic extent.
To appreciate this common theme of the susceptibility of the brain to RNA virus infection, each of the three examples is described in a standard framework. The purpose of the review is not to comprehensively review the pathogenesis of the three emerging neurological diseases, as that will require further study. Rather our intent is to acquaint the reader with the threats and encourage broad collaboration with other specialists to gain insight into how we can combat emergence of these and similar infectious agents. The essential virology of each agent is presented first in order to appreciate the implications of molecular replicative strategies on the host/pathogen duel. Next, the immunological response to infection is described in the context of known host defenses and means of evasion by the pathogen. After setting the molecular and immunological stage, a brief description of the natural history of infection follows. The effectiveness of immunity, immunization and pharmacological therapies is then described to contextualize the medical problem. Finally, a pathological description of the disease and animal models is provided to highlight the severity of the disease processes and scope of the challenge.
Influenza A virus
Virology
Influenza A virus (IAV) is a member of the Orthomyxoviridae family consisting of enveloped viruses with single‐stranded, negative‐sense, segmented RNA genomes. The family has six genera (Influenza A, B, C, Thogotovirus, Isavirus and Quaranjavirus) that do not exchange genetic segments (reassortment) between genera. Based upon comparative sequence analysis of hemagglutinin (HA) genes, influenza A, B and C viruses were derived from a common ancestor diverging approximately 2000, 4000 and 8000 years ago, respectively 109. Therefore, even on a human historical scale they are relatively “new” agents. While both influenza A and B viruses are currently important human pathogens, the following discussion will be limited to IAV, a major cause of morbidity and mortality in man.
Since its first isolation in 1933, the molecular and physical ultrastructure of IAV has been thoroughly elucidated, offering numerous targets to disrupt viral pathogenesis 67. Historically, subtyping of IAV strains was accomplished serologically utilizing the immune system's recognition of two of the main surface proteins: HA and neuraminidase (NA). Current molecular analysis has shown that there are 18 different HA and 11 different NA subtypes that can occur in any combination (ie, 198 possible different subtypes of IAV) but tend to be selective to specific host species.
The molecular steps of IAV replication are known in exquisite detail 67 (Figure 1) and will not be comprehensively reviewed here other than to note key features that pertain to pathogenesis as an emerging threat to the CNS. Embedded in the surface envelope of IAV, HA binds to neuraminic acids (sialic acids) on the surface of host cell membranes. Sialic acids are universally expressed on the extracellular portions of a wide variety of cell membrane proteins from many animal species; however, subtle differences in HA amino acid sequence determine its capacity to be activated by different cellular proteases and to bind preferentially to different terminal sugar linkages. Cleavage of HA is critical to permitting effective binding and thus to overall virulence. IAVs with HAs requiring tissue or cellular‐specific extracellular proteases that are restricted to limited kinds of cells or tissue types such as trypsin‐like proteases for cleavage activation of the surface receptor are of limited virulence (low pathogenic strains), while IAVs with HAs that can be cleaved by ubiquitous proteases (eg, furin‐like proteases) recognizing polybasic residues are readily activated and consequently more capable of virulence with spread to any body compartment (highly pathogenic strains) 65.
Figure 1.
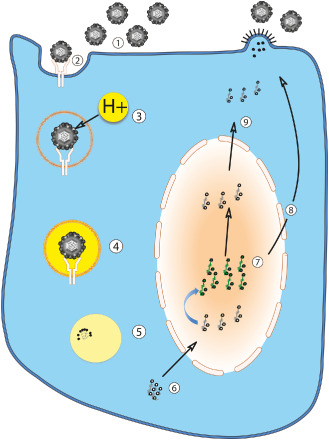
Diagram of influenza A virus infection of the cell. Free extracellular influenza A viruses (1) containing hemagglutinin on their envelopes bind sialylated glycoprotein receptors on the host cell surface (2). The virus enters the host cell by receptor‐mediated endocytosis (2,3). The resulting endosome becomes acidified by proton transport (3), allowing fusion of the host and viral membranes (4). Blocking acidification of the virion with small molecule drugs is one means of inhibiting viral replication. Fusion is required for metabolism of the nucleocapsid (uncoating) (5) and release of the viral ribonucleoprotein complex into host cell cytoplasm (6). The viral RNA and associated viral proteins (including three polymerases) are transported to the nucleus (7). From here, positive‐stranded mRNA templates with poly(A) tails are synthesized (with and without host splicing) at the expense of most host protein synthesis and exported to the cytoplasm for translation (8). Newly synthesized viral ribonucleoproteins are exported to the cytoplasm for eventual virion assembly (9). Influenza A virus assembles and buds from the cell surface in a polarized fashion (eg, from apical surface of epithelial cells).
As host cell surface protein glycosylation patterns are species and location specific, the type of HA also substantially determines in which species and organs the virus can efficiently replicate. Avian strains of IAV preferentially bind α2,3‐linked sialic acids prevalent in avian gut epithelium, while mammalian strains prefer α2,6‐linked sialic acids prevalent in human upper respiratory epithelium and α2,3‐linked sialic acids prevalent in human lower respiratory tract 25, 26. After binding the cell surface (Figure 1.2), endocytosis (Figure 1.2 and 1.3) and fusion with low pH (endosomal) compartment (Figure 1.4) is required for release of the viral ribonucleoprotein complex into host cell cytoplasm (Figure 1.5 and 1.6) 67. Blocking acidification of the virion with small molecule drugs is one means of inhibiting viral replication.
After fusion, the virion's nucleocapsid is metabolized (uncoated), and viral RNA and associated viral proteins (including three polymerases) are transported to the nucleus (Figure 1.7). From here, positive‐stranded mRNA templates with 5′ caps and poly(A) tails are synthesized (with and without host splicing) at the expense of most host protein synthesis and exported to the host cytoplasm for translation (Figure 1.8). Newly formed viral ribonucleoproteins are also released to the cytoplasm (Figure 1.9) for virion assembly and budding.
IAV assembles and buds from the plasma membrane (Figure 1.10) in a polarized fashion (eg, from apical surface of epithelial cells). This polarized budding may account in part for the limited systemic spread of IAV and its concentration within the lung and gut 32, 120. While HA and NA are incorporated in the envelope in a ratio of 4:1 to 5:1, only the most abundant viral protein underlying the envelope, matrix protein 1 (M1), is required for viral particle formation 17, 43, 85.
Critical to evolution of IAV is packaging each of the eight viral RNA segments into the individual virion. If done randomly, less than 1% of virus particles would be infectious 31. While there is some degree of selection and sorting, this is not a perfect process and incorporation of genes from other subtypes of IAV superinfecting individual cells leads to genetic reassortment (and consequent “Shifts,” see below). After virion assembly, NA is crucial for viral release and virion movement through an environment filled with mucous and replete with sialic acid binding sites 91, 92.
Immune response
Of course, the host mounts a variety of innate and adaptive defenses to block viral infection and replication. Although we are unable to fully describe the immune response here, a general overview will be summarized. As the first barrier of defense, soluble proteins in mucosal secretions such as mucins, gp‐340, pentraxins, collectins, natural IgM, complement and defensins promote IAV aggregation, clearance and reduce infectivity 126. At the onset of cellular infection, IAV is detected by pattern recognition receptors TLR3 or TLR7 located in endosomal compartments. Single‐stranded viral RNA triggers TLR7 signaling and secretion of type I interferons (IFNα/β) [indeed the discovery of IFN was the result of examining heat inactivated IAV 54 ] 29. Type I IFNs induce a variety of antiviral responses including augmenting innate and adaptive immunity (eg, natural killer and B‐cell proliferation, dendritic cell maturation, T‐cell survival and activation), induction of antiviral genes such as activating protein kinase R (pkr/eif2ak2), ADAR, oas1, rnase l and mxa that lead to inhibition of various steps of virus replication, and initiation of a cascade of cytokine secretion (IL6, IL8, TNFα, among others) 39, 96. Not surprisingly, IAV has evolved a protein [nonstructural protein 1 (NS1)] that abrogates IFN synthesis in epithelial cells and conventional dendritic cells; however, plasmacytoid dendritic cells are resistant to the effects of NS1 29.
In addition to arresting primary IAV infection, the initial innate immune response induces an adaptive immune response to clear virus and more quickly and effectively block future IAV infection. The humoral arm of the adaptive immune response is principally focused on surface envelope proteins NA and especially HA 27. Resistance to subsequent infection and illness correlates with HA and NA antibody titers. Mutations and reassortment in HA or NA genes are thus key to IAV's capacity to reinfect host populations and periodically cause epidemics and pandemics. The role of cellular immunity in clearing viral infection is less clear, but it does limit severity and transmission of IAV infection. Antiviral‐specific CD4 T cells help B cells produce neutralizing antibodies and secrete Th1‐type cytokines such as IFN‐γ, IL2 and TNFα to direct T‐cell responses, but may also directly control IAV infection 15, 71. The major targets of T‐cell immunity in IAV are epitopes in M1 and nucleoprotein. IAV‐specific CD8 T cells contribute to IAV clearance and control through mechanisms such as cytotoxic effector delivery, proinflammatory cytokine secretion (eg, TNFα and IFN‐γ) and expression of FasL and TRAIL death domain receptors 71. T‐cell responses generally recognize more conserved IAV epitopes than the antibody response, but viral escape variants may exist.
Drift vs. shift
Epidemic flu
As with other infectious agents (eg, malaria), the ability of HA and NA glycoproteins in the coat of influenza virus accounts for much of IAV's capacity to elude immune detection and clearance and guarantees the pathogen's survival in the host population. The diversity of HAs and NAs in nature is evidenced by the observation that all but two known HA (H1–H16) and NA (N1–N9) subtypes can be found in sylvatic avian species, with the other subtypes (H17–18 and N10–N11) found only in New World bats 84, 116, 117. Adapted to the aquatic bird host, IAV is under limited selective pressure from an immune response. When transmitted to land‐based birds or mammals, adaptive immune pressure leads to selection of mutations within HA and NA genes and a slow antigenic drift. At any one time, multiple viral strains circulate in human and other mammalian populations. While self‐sustaining, this ecosystem is characterized by periodic epidemics. Every few years, the “drift” in amino acid mutations mediates enough antigenic change that the virus escapes neutralization by antibodies specific to previous strains and can mediate new infections. This leads to a new epidemic in the host population. Immunization against newly emergent strains is one attempt to mitigate the severity of these epidemics.
Pandemic flu
More precarious to humans is introduction of a new IAV HA subtype (with or without a novel NA subtype) that the population has not seen before and for which the population has insufficient adaptive immunity. This much more rare event is termed antigenic shift and occurs when there is direct introduction of IAV from other mammal or avian hosts or when two IAV subtypes infect the same host (mixing vessel) at the same time in the same cell. In this setting, gene segments are exchanged between different subtypes (eg, HA of one strain is exchanged with HA of another strain leading to a new HA–NA combination) and entirely new viral subtypes or reassortants are generated that with some minor adaptations of transmission efficiency can mediate pandemics in populations immunologically naive to the new agent. While swine are frequently sited as the “mixing vessel” for this reassortment to occur, pigs and barnyard avian species (such as ducks or geese) intermingle and can mix influenza viruses between the two species and create novel influenza viruses. This mechanism may lead to introduction of novel influenza subtypes into the human population.
Pandemics of IAV emerge killing significant percentages of the human population and even changing the course of history 112. Forty to fifty percent of the human population is infected in the course of a pandemic with an increase in the number of expected deaths 104. The severity of such a pandemic is not well appreciated by current citizens of the developed world. The “Spanish Flu” of 1918–1919 (H1N1) killed 25–50 million people or more worldwide 58 and decreased average life expectancy in the United States by 10 years 44. Infection mortality during the 1918 pandemic changed from the usual 0.1% of infected individuals to 2.5% 79. Perversely rather than afflicting infants and elderly, pandemic infections are particularly lethal to young adults 68. Subsequent pandemics in 1957, 1968 and 2009 have resulted from reassortment events generating novel H2N2, H3N2 and H1N1 viral strains. For a variety of reasons, including great efforts to immunize the world population, these pandemics were more benign than Spanish Flu was.
Avian flu
The restriction of IAV to infecting lung with only rare reports of systemic spread would suggest this virus has little relevance to neurological disease. However, in 1997 an outbreak of lethal avian flu (H5N1) occurred in humans in Hong Kong 19. This outbreak was the result of direct avian‐to‐human transmission without an intermediate host, and because the virus showed only limited capacity to spread from human to human, it was not able to achieve epidemic proportions and was halted relatively quickly. Since this initial outbreak, avian H5N1 IAV has become endemic in Asia, the Middle East, Europe and Africa with 826 confirmed human cases, 440 of which were fatal 128. Systemic spread, including CNS involvement, has been reported in humans infected with H5N1 IAV and detailed in mammalian models (described below). In addition, the past few years have seen an increase in the number of reports detailing direct transmission of avian influenza viruses to humans 42, 73, 107 including strains such as H9N2 and H7N9. To date, none of these avian transmissions resulted in new strains of virus capable of spreading efficiently between human contacts; however, recent cases of H7N9 virus in China suggest human‐to‐human transmission can occur but so far is not sustained. In part, human transmission requires capacity to replicate at lower temperatures of the upper airways and the ability of the HA molecule to bind to the human epithelial glycosylation pattern of sialic acid with α2,6 linkage 51. Before taking solace in this barrier to pandemic spread, it should be noted that mutation of only four additional amino acids is required to effectively support mammal‐to‐mammal spread 51, 74.
Treatment
With detailed knowledge of IAV replication strategy and pathogenesis, it has been possible to block infection and transmission either through drug or immune therapy. Drug therapies have focused on two viral functions critical to replication and spread. The first involves blocking the viral ion channel M2 protein in the virion that is essential for acidification of the endocytosed viral compartment and shedding of the viral coat. Amantadine and rimantadine inhibit acidification of the inner virion and thus prevent viral release into the host cell cytoplasm. A second class of drugs competitively inhibits viral NA activity, preventing viral particle release and spread within the mucous‐rich environment. Other drugs in use in other countries target different aspects of the viral replication cycle such as inhibition of target membrane fusion of IAV and host cells (arbidol) 72 and inhibition of the HA cleavage by inhibiting trypsin and related proteolytic enzymes (aprotinin).
Of course, one of the major advances in modern medicine has been the introduction of vaccinations. The effectiveness of vaccinations to prevent severe illness and death is no more evident than in the case of IAV. Inactivated whole or subcomponent IAV vaccines are able to generate protective antibodies to HA and NA 21. Live attenuated vaccines that replicate at lower temperatures of the upper airways have the additional advantage of inducing local immunity and potentially priming cellular immunity 57. Because of constant antigenic drift and occasional antigenic shift in IAV's HA and NA, the tricky part to flu vaccination is creating a vaccine for the strain that will circulate when flu season begins.
Neurological disease associated with avian influenza
Autopsies of H5N1 virus‐infected humans have been very limited; but commensurate with clinical syndrome, H5N1 virus infection mediates more of a gastrointestinal illness and most importantly a systemic infection. This systemic infection is particularly notable for severe CNS symptoms and infection (ie, encephalitis). Influenza outbreaks have long been associated with neurological sequelae. Influenza encephalopathies, encephalitis, encephalitis lethargica and Reye's syndrome are rare, but are serious CNS diseases that manifest with influenza infection, especially with young children 118.
What is necessary to change a traditionally self‐limited pulmonic disease into lethal encephalitis? At least three conditions need to be met in order for influenza to directly mediate neurological disease. First, the active virus needs to escape local control at the site of primary replication to reach the brain compartment. The selective polarity of viral budding to the epithelial surface weighs against viremia and systemic spread, but some zoonotic influenza strains have shown to be highly virulent in their nonnatural human hosts and may even trigger an overcharged innate immune response 70. As discussed above, highly pathogenic strains generally have HAs that can be cleaved and activated by ubiquitously expressed proteases such as furin‐like proteins and thus readily capable of systemic spread. IAV could enter the brain by direct infection of nerve endings or via hematogenous routes. Neurovirulent H5N1 IAV strains have been shown to spread to the CNS by infecting nerve endings of olfactory, vagus, trigeminal and sympathetic nerves 93. Other neurovirulent viruses such as poliovirus and rabies virus spread to the CNS by infecting motor neurons at neuromuscular junctions, while others such as HIV enter the CNS when infected leukocytes (eg, monocytes/macrophages) traverse the blood–brain barrier 60, 70, 127. Many RNA viruses (eg, West Nile virus and hepatitis C virus) can infect brain microvascular endothelium after loss of effective clearance in peripheral sites leads to viremia. In brains of ferrets infected with H5N1 IAV, we have observed patterns of both hematogenous spread and entry via olfactory epithelium 11. Once in the CNS, IAV needs to infect neurons and spread. Most CNS neurons have abundant expression of α2,3‐linked sialic acid glycosylation pattern 86, 129, so IAV is thus able to bind to neuronal cells. Given the ubiquity of glycosylated proteins on the surface of mammalian cells, this is not a particularly high threshold to clear. While a map of neuroglial glycosylation linkages is not available, certainly neurons are infectable by either binding avian IAV and/or through endocytosis. If IAV directly binds neurons, there are plenty of furin‐like proteases present in the brain to cleave IAV HA for neuronal entry. After entry, IAV particles must undergo retrograde transport to co‐opt replication machinery in the neuron 70. The complex and unusual distribution of neuronal cellular machinery in axons, dendrites and the cell body might facilitate IAV replication. The virus must then bud and spread through synapses or intimate cell‐cell processes. Finally, the neuropathogenic strain needs to be able to subvert intrinsic immune responses. Clearance of virus from infected neurons is a big hurdle as immune responses tend to be slower (eg, IFNγ secretion and antibody‐mediated clearance rather than cytolytic mechanisms). If IAV already escaped the antibody response to spread to the CNS, this final restriction also does not present a high barrier to disease.
New neuropathology of avian influenza
H5N1 virus‐infected cells can be detected in the brains of infected birds, mice, ferrets and humans 10, 11, 37, 38, 45, 77, 95, 125. But before the virus can infect the brain, it first has to escape the immune response of respiratory epithelium. Animal models of H5N1 virus infection have been employed to chronicle this systemic spread. Because of similarities between ferret and human respiratory system glycosylation patterns, ferrets are the animal model of choice for pathogenesis studies 3, 80. However, immunological reagents to study the ferret model are more limited than traditional mouse models; therefore, depending upon the experimental question, either rodent model has been used. Initial infection with either high or low pathogenic strains of IAV follows a similar course. Inhalation of the virus is followed by rapid binding to epithelial cell surface receptors, endocytosis and explosive replication. Different animal hosts express different glycosylation patterns in different regions of their respiratory and GI system. Similar glycosylation patterns in the human and ferret respiratory system lead to binding of α2‐6 linkage‐tropic viruses in the upper respiratory system (nasal epithelium and bronchi) and binding of α2‐3 linkage‐tropic viruses in lower respiratory system (terminal bronchioles and alveoli). Early innate immune response evolves to a cytokine “storm” that accounts for most of the early clinical symptoms of fever and malaise but is presumably crucial for limiting viral spread prior to the development of adaptive immunity 113. Within the first day, viral replication is robust and the host rapidly disseminates the virus through coughs and sneezes. Peak viral replication in the respiratory system occurs within 48–72 h and then is rapidly suppressed such that by 7–8 days infected cells are difficult to detect 10, 11.
Five days after infection, the clinical course of highly pathogenic and low pathogenic strains diverges. Low pathogenic strains are cleared from the lung and physiological function returns. With high pathogenic strains, lung damage is more severe and complicated by secondary bacterial infections, which mediate the majority of the morbidity and mortality 59.
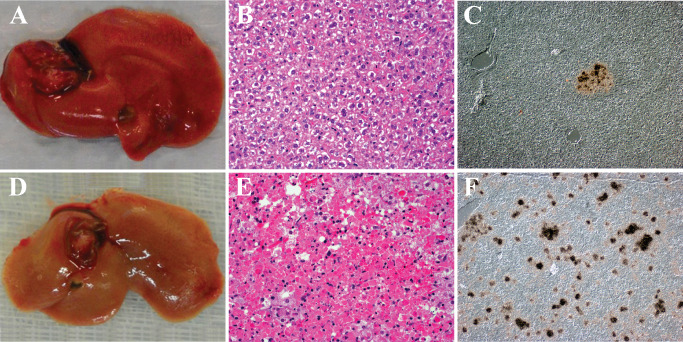
The recent IAV pandemic in 2009 was the result of introduction of a novel strain of H1N1 (H1N1pdm09) to the human population that replaced the circulating H1N1 seasonal strains 12. This novel strain disproportionately affected children and young adults 22 but was not associated with widespread systemic dissemination. Humans infected with H1N1pdm09 virus exhibit severe bronchopneumonia (Figure 2A,B). Ferrets also show bronchopneumonia (Figure 2C) with infected cells found mostly in bronchi with occasional infected alveolar cells (Figure 2D) and prominent submucosal gland involvement (Figure 2E). H1N1pdm09 virus is able to spread to the gastrointestinal tract where it infects cells in the lamina propria (Figure 2F). Thus, compared with avian H5N1 viruses, H1N1pdm09 is an IAV with restricted systemic spread. Comparing the systemic spread of different IAV strains in ferrets shows all strains (H5N1, H1N1pdm09 and H3N2 viruses) infect the lung and gut lamina propria, whereas only H5N1 virus is able to spread to the brain and liver (Figure 3). Interestingly, prior infection with H1N1pdm09 virus protects against H5N1 virus infection, while prior infection with H3N2 virus protects against lung infection but leads to systemic infection of the CNS and liver 11.
Figure 2.
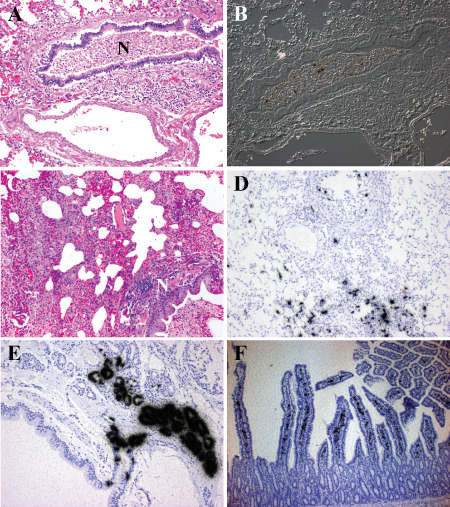
H1N1 virus infection of human and ferret. A,B. Histopathology of the lung from a patient who succumbed to H1N1pdm09 virus infection. A. Hematoxylin & eosin (H&E)‐stained paraffin section demonstrates a severe bronchopneumonia. Necrotic debris (N) fills the lumen of a moderate size bronchiole. Surrounding alveolar tissue shows edema and severe inflammation. B. Differential interference contrast and in situ hybridization for influenza matrix protein RNA (black grains) demonstrates infected cells within the necrotic debris. At this late stage of infection, the immune response has cleared the majority of virus. C–E. Histopathology of lungs from ferrets inoculated with H1N1 virus intranasally. C. H&E‐stained paraffin section illustrates the severe bronchopneumonia at 5 days post‐infection (DPI). Necrotic debris (N) fills the lumen of a moderate size bronchiole. Surrounding alveolar tissue shows edema and severe inflammation. D. In situ hybridization for influenza matrix protein RNA (black grains) (counterstained with hematoxylin) shows infected cells in epithelial cells of the bronchi and alveoli at 3 DPI. More viruses are detected in the ferret lung suggesting the animal was sacrificed at an earlier stage of infection than when the human case died. By 8 DPI, no virus is detected in the ferret lung. E. In situ hybridization for influenza matrix protein RNA (black grains) (counterstained with hematoxylin) illustrates the severity of submucosal gland involvement as early as 1 DPI. F. The histopathology of small bowel from a ferret infected with H1N1 virus 14 days previously. In situ hybridization for influenza matrix protein RNA (black grains) (counterstained with hematoxylin) demonstrates infected cells within the lamina propria at a time when virus cannot be detected anywhere else systemically.
Figure 3.
Distribution and quantitation of influenza infection in the ferret at different time points after infection. Throughout the time course of infection with H1N1pdm09 virus, viral‐infected cells are restricted to the respiratory tract except for a late chronic infection of the gut lamina propria. Infection with H5N1 virus (VN04) follows an entirely different course. While beginning in the lung, H5N1 virus infection quickly spreads to systemic organs. H5N1 virus can be detected in the liver by 2 days post‐infection (DPI) and as early as 4 DPI in the brain. At the terminal stage of infection (marked by X on the line chart), the vast majority of virus can be detected within the brain, while infection in the lung has begun to abate. Ferrets infected first with H1N1pdm09 or H3N2 virus (Vic11) followed by H5N1 virus (VN04) challenge 3 months later have different outcomes as well. Prior infection with H1N1pdm09 virus protects the ferret from H5N1 virus infection except for a late chronic infection of the gut lamina propria and liver. Prior infection with H3N2 virus leads to systemic spread of H5N1 virus to the brain and liver with lethal encephalitis by 6 DPI (marked by X on line chart).
But some highly pathogenic strains have an additional attribute that can contribute to worse clinical outcome: the capacity to spread systemically. Much of this capacity to spread systemically is associated with enzymatic stability of the HA molecule 78 and the ability to escape containment by the host immune response. As described above, maturation of newly synthesized virus into an infectious agent requires cleavage of virion HA. The HA of highly pathogenic strains is more readily cleaved by ubiquitous furin‐like proteases and thus capable of maturing in any body compartment rather than being limited to extracellular spaces like those in the respiratory and gastrointestinal system. With systemic spread, infectious loci are distributed throughout the host contributing to a secondary viremia. Prior to the capacity of the adaptive immune system to control this systemic spread, IAV finds its way into the CNS, which for multiple known and unknown reasons is susceptible to infection.
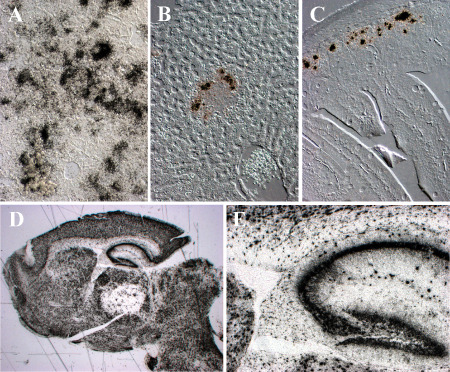
Direct indication of IAV‐infected neurons or CNS support cells in humans is limited 38, 45, but in ferret and murine models the neuron is the predominant infected cell type observed in the CNS 10, 11, 37, 56, 77. Macrophages are thought to be infected by influenza virus 88, and there are reports suggesting microglia are infected 37, 56, but others have not observed microglia infection 10, 11, 77. In mice, IAV is first detected in the brain stem and olfactory cortex at 4 days post‐infection (DPI) 10. Within a few days, it spreads throughout the cortex, largely sparing the striatum. Interestingly, the infection does not appear to be uniform, but rather occurs in small foci throughout the brain suggesting hematogenous dissemination. A similar distribution is seen in ferrets (Figure 4C) 11. Some but not all H5N1 virus‐infected ferrets show a severe bronchopneumonia (Figure 4A) characterized by abundant lower lung alveoli infection (Figure 4B). The virus spreads to the liver, spleen, gut and brain (Figure 4C–F). The brain shows infection throughout the CNS in olfactory cortex, cerebral cortex, deep gray nuclei and brainstem (Figure 4C). Neurons are the predominant infected cell (Figure 4F). Occasional ferrets show infection of ependymal cells lining the ventricles or cells within the leptomeninges, suggesting dissemination through the cerebrospinal fluid (CSF).
Figure 4.
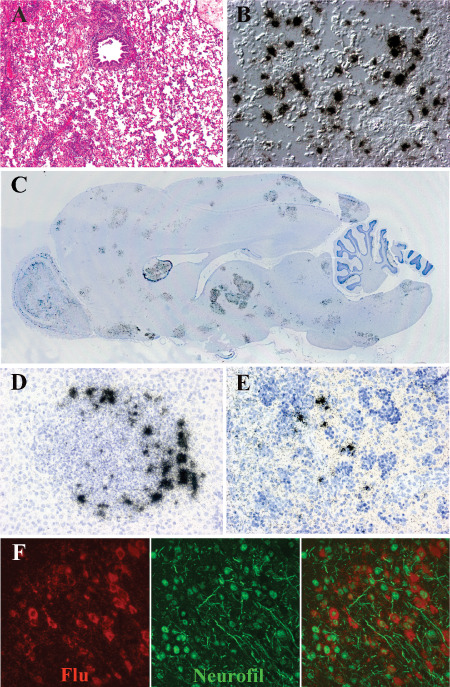
H5N1 virus infection of the ferret. A,B. The histopathology of the lung from a ferret infected with H5N1 virus 5 days previously. A. Hematoxylin & eosin (H&E)‐stained paraffin section demonstrates a severe broncho‐ and alveolar pneumonia. B. Differential interference contrast and in situ hybridization for influenza matrix protein RNA (black grains) demonstrates infected alveolar cells in lower airway at 2 days post‐infection (DPI). C. Whole mount of the ferret brain 6 DPI with H5N1 virus hybridized with radioactive probes to influenza matrix protein RNA (black grains) demonstrates multifocal infection in olfactory cortex, cerebral cortex, deep gray nuclei and brainstem. D. In situ hybridization for influenza matrix protein RNA (black grains) (counterstained with hematoxylin) shows infected cells in liver surrounding intense inflammatory nodules. E. In situ hybridization for influenza matrix protein RNA (black grains) (counterstained with hematoxylin) illustrates infected cells in splenic red pulp at 18 DPI. F. Double label in situ hybridization for influenza matrix protein RNA (red) and immunohistochemistry for neurofilament (green) shows infection of neuronal elements.
It has been reported that C57B6 mice infected with a strain of H5N1 isolated from the CSF of a Vietnamese boy showed evidence of chronic microglial activation and loss of dopaminergic neurons approximately 50 days following viral clearance 56. This suggests that H5N1 brain infection can lead to long‐lasting effects that could contribute to pathological processes observed in diseases such as Parkinson's disease. Recently, this same group has reported microglial activation in the CNS of mice infected with pandemic 2009 strains of H1N1 IAV in the absence of CNS IAV infection 101. This may be a common outcome after systemic infection with any virus without neurotropism. Whether the microglial activation is neuroprotective or contributes to neurodegenerative processes is a matter of debate.
Why do RNA viruses devastate the brain once they obtain access to the CNS? As discussed above, the brain generally does not mount the full armamentarium of viral clearance strategies upon infection. This limited immune response has been thought to curtail death of a cell population with limited regenerative capacity. However, recent studies have revealed that the brain is unique not only in its massive expression of numerous genes but also in its overall processing of RNA 4, 131. Critical to the brain's very function is the capacity to modulate RNA processing in an intricate and spatially refined way. While systemic organs like the gut and lung can utilize generic innate immune responses that entail shutting down RNA processing, such a defense might not be available to the CNS. The inability of the brain to defend itself from viral infection may reside in the importance of meticulous control of RNA processing necessary for brain functionality. In the absence of an effective means of suppressing viral RNA replication, the brain rapidly becomes a sea of virus unparalleled in other organs.
Fortunately, humoral immunity provides an effective barrier to CNS invasion by RNA viruses. But this immunity is fine grained and needs to be approached comprehensively when designing immunization strategies. The immune response following natural infection is broad based and recognizes numerous viral epitopes. Infection with one strain of IAV can generate an immune response that protects from modestly different strains of IAV. Called heterosubtypic immunity, this is the product of a robust immune response to an active infection. Immune responses to most vaccines are more limited and focused on antigens chosen in the vaccine design and generated in body compartments distal from the site of natural exposure. Vaccines may provide enough immunity to block severe acute systemic disease without providing sterilizing immunity. This could open the possibility of spread to the CNS. We have observed precisely this scenario in examining vaccines to Rift Valley fever virus (RVFV).
Rift Valley fever virus
Virology
The taxonomic family of Bunyaviridae was established in 1975 87. Four genera of the family infect animals after transmission from arthropods. An important exception to that mode is Hantavirus, which is transmitted by rodent excreta. Infection of arthropod tissue varies depending on the arthropod and virus, but infected tissues include salivary glands and gonads that permit the viruses to be transmitted horizontally, vertically or venereally 8.
Like influenza, the Bunyaviridae is enveloped and has a segmented genome. The tripartite single‐stranded RNA genome segments are named on the basis of their size (large, medium and small). Rather than being universally negative sense as in the case of influenza, Bunyaviridae is negative sense or a combination of negative sense and antisense 67 (eg, the small RNA segment of Phlebovirus and Tospovirus genera has message transcripts generated in opposite orientations). For purposes here, other than the ambisense segments that necessitate two rounds of transcription to occur, Bunyaviridae replication is similar to that of IAV with the following exceptions:
-
(i)
Viral surface envelope attachment proteins (Gn and/or Gc) attach to host cell receptors 2, 30, 62, which for some viral strains are integrins or DC‐SIGN but for most family members are not known 40, 75.
-
(ii)
Cellular entry requires endocytosis and vesicle acidification as with IAV; however, RNA transcription occurs in the cytoplasm without the addition of poly(A) tails 47.
-
(iii)
Most viral assembly occurs in the Golgi with transport and exocytosis at the plasma membrane (although some alternate assembly at the plasma membrane does occur).
-
(iv)
Viral replication is generally cytolytic in vertebrate cells but not in arthropod cells 14.
Many members of the Bunyaviridae family are important human and livestock pathogens. Some of these like California encephalitis and La Crosse are notorious for mediating neurological disease; however, the following description will be limited to RVFV which was first isolated in 1930 28.
Epidemiology and natural history
As suggested by its name, members of the Phlebovirus genera are transmitted by a diverse group of blood‐sucking arthropods. RVFV is transmitted principally by the Aedes and Culex mosquitoes 94, 103, 119, 121. In 1977, an Egyptian outbreak of 200 000 cases (approximately 600 deaths) followed completion of dams that led to increased mosquito populations and greater livestock infection 52, 81.
After transmission of the virus through a mosquito bite, local replication takes place in regional draining lymph nodes, followed by viremia and secondary amplification in the reticuloendothelial system, liver, adrenals, lung and kidney tissue 67. Infection of livestock is signaled by increased incidence of abortions (not a feature of human disease) 7, 110. In humans, the incubation period is approximately 3–6 days 99 and results in fever, headache and myalgia with complete recovery in most individuals. Less than 1% of infected individuals experience severe disease including fulminant hepatitis, retinitis, encephalitis and hemorrhagic syndromes 34, 41.
Alternate routes of transmission to humans via contact with tissues or fluids from infected animals can lead to particularly severe outcomes. Individuals involved in animal husbandry are at high risk of infection through the shearing or butchering of animals during epizootic infections. With such unconventional, usually aerosolized exposure, the resulting disease is more severe consisting of hemorrhagic fever, encephalitis and retinitis. With dissemination via aerosolization, easy access to natural isolates during epizootics in endemic areas and easy amplification in cell culture or infected animals, there is concern that RVFV could be used as a bioterror agent 98.
Immunization
Vaccination of livestock is probably the most effective way of preventing human infections. A live attenuated vaccine is highly effective at controlling epizootic livestock infections; however, it is associated too with abortions in some pregnant animals 7. Formalin‐inactivated vaccines are available but provide only short‐lived immunity. Currently, there are no approved vaccines or therapeutics to ameliorate human disease. Attempts to create human vaccines depend upon developing reliable nonhuman primate models 48.
New neuropathology
RVFV is highly infectious through the aerosol route with numerous reports of infections of laboratory personnel 16, 36, 52, 61. We have studied aerosol exposure of BALB/c mice to evaluate the pathogenesis of RVFV infection and the capacity of vaccines to protect mice from disease 98.
While most mammals are susceptible to RVFV infection, certainly the murine system is the most commonly used laboratory model. The natural route of infection via mosquito bite is readily mimicked by subcutaneous, intramuscular or intraperitoneal inoculation of mice. Comparison of live attenuated vaccine strain (MP12) vs. wild‐type strain (ZH501) shows dramatic differences in survival and pathogenesis. Intraperitoneal infection of young BALB/c mice with the live attenuated vaccine strain MP12 readily reproduces most human mosquito‐borne infections. Over the first 5 DPI, mice show acute signs of infection including ruffled fur and lethargy with mild weight loss. A low‐level viremia is established but this is cleared within a week. Beginning at 3 DPI, very low levels of virus appear in the liver (Figure 5A–C); but by 5 DPI, virus has been cleared from the liver and the animals are well on their way to recovery. No other organ shows evidence of infection.
Figure 5.
Intraperitoneal (IP) infection of the mouse with pathogenic and nonpathogenic RVFV. IP infection of mice with nonpathogenic (MP12) RVFV leads to no significant weight loss over 4 days, while infection with pathogenic RVFV (ZH501) leads to rapid weight loss and lethal infection within 4 days. A,D. Gross photographs of livers of mice infected with MP12 and ZH501 strains of RVFV. A. MP12 infection shows no significant gross pathology at 4 days post‐infection (DPI), while livers of mice infected with pathogenic RVFV (ZH501) are pale in color (D). B,E. Hematoxylin & eosin (H&E)‐stained paraffin sections from livers of mice infected with MP12 and ZH501 strains of RVFV. B. Mice infected with nonpathogenic RVFV (MP12) show normal histology, while sections from livers of mice infected with pathogenic RVFV (ZH501) demonstrate widespread necrosis (E). C,F. Differential interference contrast and in situ hybridization for RVFV RNA (black grains) in liver. C. Mice infected with nonpathogenic RVFV MP12 show rare foci of parenchymal infection, while similar studies of mice infected with pathogenic RVFV ZH501 show multifocal necrosis of abundant viral‐infected hepatocytes (F).
In contrast, infection with the clinical isolate and pathogenic RVFV strain ZH501 is much more dramatic. Viral infection of the liver occurs earlier by 1 DPI and is massive by 2 DPI with abundant viral replication and hepatocyte necrosis (Figure 5D–F). At 2 DPI, virus appears in the white pulp of the spleen and within another day rapidly inundates this organ with a pan‐splenic infection. Because of liver failure, most mice require sacrifice by 3 DPI. The brain does not show evidence of infection at this early time point. The value of immunization is clear by the observation that prior vaccination with MP12 completely protects mice from subsequent intraperitoneal challenge with ZH501.
Unconventional exposure to RVFV, such as that which might be experienced by abattoir workers or individuals potentially exposed to aerosolized biological weapons, has a very different clinical outcome. With aerosol exposure of BALB/c mice, death from ZH501 infection takes longer and is associated with widespread systemic dissemination. By 1 DPI, systemic infection has spread to many organs including the liver, spleen, lung, heart and gut. Compared with the liver (Figure 6A), the other organs show relatively low levels of virus infection. The brain can show infection as early as 1 DPI, but by 6–7 DPI it is heavily involved. Mice develop neurological symptoms including circling the cage and hind limb paralysis. The pathogenesis of such broad dissemination is not known but is most likely hematogenous.
Figure 6.
Aerosol infection of non‐immunized and previously immunized mice with pathogenic RVFV. Non‐immunized mice show a severe hepatitis after aerosol exposure to ZH501 RVFV, while previously immunized mice show only mild hepatitis after aerosol exposure. A–E. Differential interference contrast and in situ hybridization for RVFV RNA (black grains). A. Non‐immunized mouse shows severe hepatic infection at 6 days post‐infection (DPI). Infection of the liver is delayed with aerosol infection compared with intraperitoneal infection. B. Mice immunized with alphavirus replicons expressing the Gn glycoprotein of RVFV show occasional infected foci in the liver 3 DPI. Virus is cleared from the liver by 6 DPI. C. Three days post‐infection after aerosol exposure to RVFV, enteric infection is observed in epithelial cells at the depths of small bowel crypts in mice immunized with alphavirus replicons expressing the Gn glycoprotein of RVFV. D,E. Despite modest systemic infection in mice previously immunized with DNA plasmids expressing Gn glycoprotein of RVFV fused to three copies of complement protein (C3d), aerosol exposure to RVFV ZH501 leads to lethal encephalitis 7–10 days later. D. The brain of terminally ill mice illustrates that the vast majority of neurons are infected. E. High power image of the hippocampus of (D).
Unfortunately, protection conferred by live attenuated vaccine strains or DNA/replicon RVFV GN protein vaccines is limited to conventional exposure (ie, mosquito bite) as demonstrated in the mouse intraperitoneal challenge model 6. When challenged through the aerosol route, prior immunization provides protection of the heart and spleen and limited liver infection with clearance of virus by 8 DPI (Figure 6B). Infection of the gut epithelial crypt cells is more persistent (Figure 6C). RVFV aerosol challenge results in delayed infection kinetics of the brain. Around 10 DPI, RVFV mediates a lethal panencephalitis (Figure 6C,D). Protection from liver disease but death from late‐developing encephalitis has been noted in RVFV‐infected mice treated with ribavirin or infected with the M847‐A strain 83.
How does RVFV get to the brain and why is the brain not protected from fulminant infection? The presence of neutralizing antibody in the serum would argue against hematogenous spread to the CNS 6. Infection of the olfactory bulb raises the possibility that virus could ascend directly from the nasal neuroepithelium; however, early olfactory bulb infection does not appear to be a feature of this disease. The frequent observation of gut epithelial cell infection raises the possibility that virus may pass through gut innervation to the brainstem followed by subsequent transsynaptic spread throughout the brain.
Regardless of how RVFV reaches the CNS, why does neuroinfection proceed unabated? The brain has been variably described as “immunologically privileged” (ie, in some ways sequestered from systemic immune surveillance). Certainly immunoglobulin, a key component to neutralizing RVFV, does not normally pass into the CNS, but once infection appears in the CNS the blood–brain barrier integrity is disrupted and immunoglobulin passes more freely. Additionally, astrocytes, microglia and endothelial cells in the CNS quickly release cytokines leading to rapid ingress of CD8 cytotoxic lymphocytes. Nevertheless, after aerosol exposure to RVFV, the brain is consumed by viral infection. It remains to be determined why RVFV replication cannot be suppressed in the CNS, but a potential hypothesis is that the CNS cannot respond to RNA viral infections like other organ systems. Because key brain functionality requires intricate RNA processing and transcription, perhaps immune response options in the CNS are more limited.
Human Parechovirus
Virology
Human parechoviruses (HPeVs) are a newly classified genus of the family. Picornaviruses are small (∼30 nm), non‐enveloped, single‐stranded RNA viruses of positive polarity that have been recognized as human and animal pathogens for over 100 years. The most notorious of these, poliovirus, was isolated at the turn of the previous century. There is a continually growing number of Picornaviridae genera (currently 26) 50, 69. Originally classified in the Enterovirus genus as echoviruses 22 and 23, HPeVs were later classified into their own genus based upon their distinct biological and genomic sequence characteristics (sharing less than 30% amino acid identity with other picornaviruses) and a unique capsid structure consisting of three rather than four capsid proteins 106. The enteric forms including parechoviruses are notable for significant acid stability 67. As a single‐stranded RNA molecule, the picornaviruses have a complex 5′‐noncoding region that facilitates ribosomal entry and translation of a single polypeptide protein. Cleavage of this polyprotein releases both enzymatic and structural proteins.
There are a wide variety of known (not to mention unknown) cellular receptors for picornavirus capsids. But not all cells possessing receptors are infectable because other host factors are required for viral replication or host factors in certain cells/tissues can inhibit viral replication. For example, tropism of poliovirus is further regulated by the capacity of the host cell to respond to type I IFN, and as the CNS lacks sufficient IFN responsiveness, it is targeted by poliovirus 53. After binding to the cell surface receptor, replication proceeds as described above for IAV with the following exceptions:
-
(i)
Only a subset of picornaviruses requires acidification to release the viral genome into the cytoplasm.
-
(ii)
After uncoating, viral transcription depends upon binding of ribosomal subunits by a sequence in the 5′‐noncoding region 76.
-
(iii)
Following polypeptide synthesis and cleavage, virions are assembled in the cytoplasm around newly synthesized positive‐stranded genomic RNA for release from the cell by cytolysis 67, 115.
Like other RNA viruses, there is a substantially high virion‐to‐infectious virion ratio (two to three orders of magnitude). HPeVs are different from most picornaviruses because they do not shut off host cell protein synthesis during replication and have limited proteolysis of their capsid proteins 105, 106.
Because the pathogenesis of enteroviral infection has been best established with polio, it will be used here to describe the paradigmatic infection. Fecal shedding of virus occurs over weeks supporting oral‐oral route of transmission. With exposure, polio replicates initially in the tonsils or Peyer's patches of the intestine 13. This leads to minor viremia followed by a more diffuse infection of the reticuloendothelial system and robust viremia. It is at this stage that the CNS can be invaded [although in the immune intact host, CNS invasion is distinctly uncommon occurring in less than 1:200 infections 13, 67, 100 ]. There is some controversy regarding the relative role of hematogenous spread to the CNS vs. transaxonal retrograde spread to select sites such as anterior horn cells of spinal cord 89. Regardless of its systemic route of entry, polio binds to a ubiquitous host receptor, CD155 82. The principal neurological disease of anterior horn motor neurons and brainstem neurons that follows is thought to be a product of neural cells having a diminished type I IFN response 53 or inability to suppress viral‐directed RNA processing. It is unclear why poliovirus infection rarely spreads to cerebral and cerebellar cortex (exception of motor cortex) and the basal ganglia.
Epidemiology
While most enteroviral infections are asymptomatic, there are estimates of 10–15 million symptomatic infections per year in the United States 108, suggesting that most humans are infected at least once a year. In the immune intact host, symptoms are mostly mild; however, as a group, enteroviruses are the most common cause of meningitis 49. The even more rare infection of CNS parenchyma was first appreciated by Charcot in his studies of spinal cord poliomyelitis 20, that today only afflicts non‐immunized hosts.
Echoviruses were first isolated in 1951 and received their name because they were Enteric isolates that mediated a Cytopathic effect mostly in Human cells but were unassociated with a human disease (“Orphans”) 24. With improvements in molecular sequencing, it became evident that echoviruses 22 and 23 were genetically distinct from other enteroviruses and thus reclassified into a new genus Parechovirus, of which there are now 16 strains 50, 97. Serologic studies have shown that HPeV infections are ubiquitous, with seroconversion occurring the first year of life 1, 111. Even more recently, HPeV3 was first identified in a 1999 stool sample 55. In the United Kingdom, virological screening of 3415 CSFs found that HPeV3 was the most prevalent picornavirus present 49. Phylogenetic analysis of HPeV members indicates the family may be a recently evolved (emergent) agent 18.
Immune response
Little has been reported about HPeV‐specific immunity, so a brief review of general immune responses to enterovirus infection will be described here. As with all viral infections, innate immunity is key to controlling viral replication and dissemination. After infection, enteroviruses are recognized by Toll‐like receptors and retinoic acid‐inducible gene I (RIG‐I)‐like receptors, which trigger cytokines, chemokines and proteins that can directly attack viral replication (eg, PKR and type I IFNs). IFNs and virus‐specific IgA are increased in saliva and respiratory and gastrointestinal tracts during acute infection 130. Antibody responses are essential for protection against enteroviruses; without them, hosts are unable to clear the virus, even with an intact CD8 T‐cell response 64, 67. In fact, many CD8 T‐cell responses are weak or undetectable and can be inhibited by viral mediated suppression of major histocompatibility complex (MHC) class I expression 63. This may explain why shedding of virus from the gut can persist for weeks 23 and why some level of chronic infection has been documented following enteroviral infection [eg, CVB3 in mice 66 ].
Treatment
Treatment of most enterovirus infections is generally limited to physiological support. However, there are a variety of potential therapies for picornavirus infections including IFN, pooled immunoglobulin and antiviral drugs (eg, pleconaril) that bind to hydrophobic sites in the viral capsid, thus blocking cell attachment 35, 114. Vaccines for picornaviruses are limited to poliovirus and hepatitis A virus 35. Poliovirus vaccines have been available for over 50 years. Intramuscular inactivated polio vaccine is administered at 2, 4, 6–18 and 48–72 months and is associated with lifelong humoral immunity. Thus, the role of oral live attenuated vaccine has diminished. For years we have been on the cusp of eliminating polio throughout the globe, and while a potential historical accomplishment, would eliminate only one of many picornavirus CNS pathogens. There are currently no vaccines in the pipeline for HPeVs.
Neurological disease associated with HPeV infection
HPeV infections are common but are usually asymptomatic. Nevertheless, HPeV isolates have been recovered from patients (most frequently neonates) presenting with a variety of nonspecific disorders including sepsis, gastrointestinal and respiratory infections, and meningitis 1, 5, 124. CSF analyses of patients with CNS HPeV3 infection show an atypical noninflammatory profile. This makes the range of clinical CNS HPeV disease difficult to define. A survey of 10 infants presenting with seizures and diagnosed with HPeV CNS infection identified viral RNA in the CSF of seven, in the blood of three, and in one each from the nasopharynx and stool 123. Abnormal periventricular white matter was observed after evaluation with imaging methods in all 10 infants. None of the infants succumbed to infection, but clinical follow‐up revealed no to varying neurological sequelae including: cerebral palsy, epilepsy, and learning disabilities that were worse in preterm vs. term infants 123. Similar patients with white matter abnormalities have been reported more recently 46.
New neuropathology
Autopsy descriptions of CNS HPeV infection are limited as death associated with this viral infection is rarely documented 9, 33, 102, 122. As predicted by the clinical and imaging findings (Figure 7A), HPeV3 infection of the neonatal brain is associated with histopathological findings of classical periventricular leukomalacia (PVL) (Figure 7B,C) 9, 122. Tissue cavitation and severe reactive gliosis are observed in the absence of a cellular immune response (Figure 7A–E). Additionally, the leptomeninges demonstrate a moderate cellular reaction. Localizing a role of HPeV3 for this pleomorphic and multifocal pathology was challenging. Probes to detect infected cells demonstrated an intriguing distribution of virus and pathogenesis of tissue damage 9. Rather than infecting CNS parenchymal cells, HPeV was localized to blood vessel smooth muscle cells in the leptomeninges and pulmonary vasculature (Figure 8). This suggests that much of the severe CNS tissue damage (PVL) is an indirect effect of vascular compromise to metabolically active regions.
Figure 7.
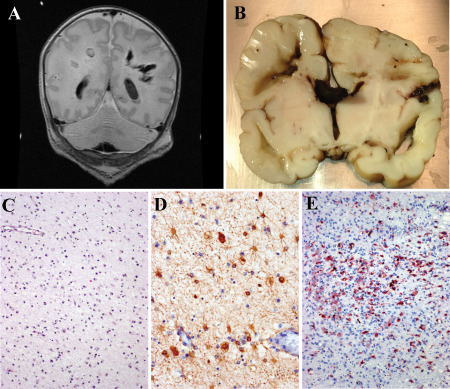
HPeV3 infection of neonates. A. T1‐weighted non‐contrast magnetic resonance imaging (MRI) from an HPeV3‐infected infant. The child was healthy at birth but developed “neonatal sepsis” after exposure to an ill adult 30 days after delivery. Initial radiologic studies were normal; but after developing seizures, subsequent scans demonstrated cavitary deep white matter lesions. The infant died the following day. B. Gross coronal section of the infant's brain confirms the presence of deep‐seated periventricular cavitary lesions with associated hemorrhage. C. Hematoxylin & eosin (H&E)‐stained sections adjacent to the cavitary lesions demonstrate a bland gliosis with mineralization and no adaptive immune cell infiltration. D. Immunohistochemistry for glial fibrillary acidic protein confirms perilesional astrocytosis, whereas immunohistochemistry for CD68 confirms perilesional microglial activation (E).
Figure 8.
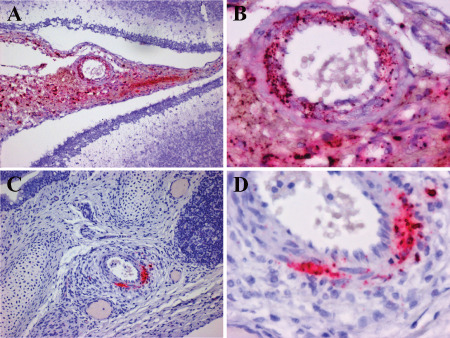
HPeV3 infects blood vessel smooth muscle cells in leptomeninges and pulmonary vasculature. A,B. Paraffin‐embedded cerebellum and overlying leptomeninges probed for HPeV3 RNA using in situ hybridization (red) (counterstained with hematoxylin). Abundant HPeV3 viral RNA is confined to the modestly hypercellular leptomeninges with no evidence of infection of the brain parenchyma. Higher power (B) image of (A) confirms the presence of HPeV3 RNA in leptomeningeal cells and particularly in smooth muscle cells of blood vessel walls. C,D. Paraffin‐embedded lung probed for HPeV3 RNA using in situ hybridization (red) (counterstained with hematoxylin). HPeV3 RNAs are confined to the modestly hypercellular pulmonary arteries without evidence of lung parenchymal infection. Higher power (D) of (C) confirms the presence of HpeV3 RNA in smooth muscle cells of blood vessel walls. These observations suggest that damage noted in severe periventricular leukoencephalopathy is an indirect effect of vascular compromise to metabolically active regions.
Conclusions
Review of these three examples of emergent neurological infections defines four important themes to consider for the future:
-
(i)
As demonstrated by influenza and HPeV, there are abundant opportunities for infectious agents to genetically evolve in real time. It is not necessary to review history books to detect emergence of pathogenic viral strains. Some degree of vigilance is required to identify these new agents when they arise so that we can quickly respond with effective preventative and therapeutic strategies.
-
(ii)
Vaccines have proven to be highly effective means of protecting the human population from infectious agents of high morbidity or mortality. But it is clear that we have been fortunate in creating vaccines to what proved to be easy targets. Many of these vaccines were developed at a time when not only did we have little understanding of the immunology, but in the case of some diseases, when we did not even know they were infectious agents (eg, Jenner's original cowpox vaccine). The original polio vaccine worked because humans were the only natural host and there were only three strains of pathogenic polio and all three were incorporated into an effective vaccine. Within the genera of enteroviruses alone, there are many more candidates that may emerge as important human pathogens for which a comprehensive analysis of strains will be necessary before creating a successful vaccine. Furthermore, as clearly demonstrated by RVFV, vaccines that confer protection from systemic disease do not necessarily confer protection for the brain when exposed through unconventional routes.
-
(iii)
Something about aerosol transmission is associated with uncontrolled neurological infection for agents that are usually spread by other routes. Agents notorious for causing systemic disease (eg, enteroviruses) when delivered through a “natural” route for unknown reasons become highly neurotropic when delivered by aerosol. Perhaps this relates to some selective hole in our natural immune response to inhaled agents that for whatever reason such exposure is not associated with effective immunity for the brain. Such a hole can be readily targeted, if not by nature, then by malign members of our own species.
-
(iv)
The brain is a marvelously complex organ with phenomenal functionality, but in part this complex functionality depends upon intricate RNA processing. Innate immune responses like those precipitated by IFN with shutting down of viral RNA processing may be incompatible with brain function. This implies that our best immune defense could come at the cost of mutual destruction of brain functionality.
Future
How do we protect ourselves in general, and our brains in particular, from viral infections? First, we need to know what agents in our environment are potential threats to our brain. This requires vigilance in monitoring and detecting agents as they emerge in zoonotic populations and taking these threats seriously (eg, henipaviruses in pigs and arboviruses in avian populations). Second, we need to know what natural immunological mechanisms protect the CNS and how agents circumvent these barriers. With this knowledge in hand, the creation of vaccines and pharmacological therapies can be efficiently designed to target these weaknesses.
While we cannot prevent microbes from mutating and evolving into more lethal pathogens, we can stop trying to give them a helping hand by developing biological weapons and unique ways to expose our fellow man through evolutionarily novel routes. We should also give serious thought to our blithe disregard for evolutionarily ancient ecosystems we destroy on whim. There appears to be no part of this globe that is not subjected to dense human monoculture. We could not intentionally design a more precarious setting ripe for exploitation by highly infectious pathogens. Perhaps a segment of our outbred population with its tremendous genetically diverse immunity will be protected from the next plague, but it is clear our species is toeing a thin line of survival with some highly lethal agents.
Acknowledgments
We would like to thank Guoji Wang for help assembling the figures. This work was supported in part with federal funds from the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (U01AI111598).
References
- 1. Abed Y, Boivin G (2006) Human parechovirus types 1, 2 and 3 infections in Canada. Emerg Infect Dis 12:969–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arikawa J, Schmaljohn AL, Dalrymple JM, Schmaljohn CS (1989) Characterization of Hantaan virus envelope glycoprotein antigenic determinants defined by monoclonal antibodies. J Gen Virol 70:615–624. [DOI] [PubMed] [Google Scholar]
- 3. Belser JA, Katz JM, Tumpey TM (2011) The ferret as a model organism to study influenza A virus infection. Dis Model Mech 4:575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Belzil VV, Gendron TF, Petrucelli L (2013) RNA‐mediated toxicity in neurodegenerative disease. Mol Cell Neurosci 56:406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benschop KS, Schinkel J, Minnaar RP, Pajkrt D, Spanjerberg L, Kraakman HC et al (2006) Human parechovirus infections in Dutch children and the association between serotype and disease severity. Clin Infect Dis 42:204–210. [DOI] [PubMed] [Google Scholar]
- 6. Bhardwaj N, Heise MT, Ross TM (2010) Vaccination with DNA plasmids expressing Gn coupled to C3d or alphavirus replicons expressing Gn protects mice against Rift Valley fever virus. PLoS Negl Trop Dis 4:e725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bird BH, Nichol ST (2012) Breaking the chain: Rift Valley fever virus control via livestock vaccination. Curr Opin Virol 2:315–323. [DOI] [PubMed] [Google Scholar]
- 8. Bishop DHL (1996) Biology and molecular biology of bunyaviruses. In: The Bunyaviridae. Elliott RM (ed.), pp. 19–62. Springer Science+Business Media: New York. [Google Scholar]
- 9. Bissel SJ, Auer R, Chiang CH, Kofler J, Murdoch GH, Nix WA et al (2015) Human parechovirus 3 meningitis and fatal leukoencephalopathy. J Neuropathol Exp Neurol 74(8):767–777. [DOI] [PubMed] [Google Scholar]
- 10. Bissel SJ, Giles BM, Wang G, Olevian DC, Ross TM, Wiley CA (2012) Acute murine H5N1 influenza A encephalitis. Brain Pathol 22:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bissel SJ, Wang G, Carter DM, Crevar CJ, Ross TM, Wiley CA (2014) H1N1, but not H3N2, influenza A virus infection protects ferrets from H5N1 encephalitis. J Virol 88:3077–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blyth CC, Kelso A, McPhie KA, Ratnamohan VM, Catton M, Druce JD et al (2010) The impact of the pandemic influenza A(H1N1) 2009 virus on seasonal influenza A viruses in the southern hemisphere, 2009. Euro Surveill 15:19631. [PubMed] [Google Scholar]
- 13. Bodian D (1955) Emerging concept of poliomyelitis infection. Science 122:105–108. [DOI] [PubMed] [Google Scholar]
- 14. Briese T, Calisher CH, Higgs S (2013) Viruses of the family Bunyaviridae: are all available isolates reassortants? Virology 446:207–216. [DOI] [PubMed] [Google Scholar]
- 15. Brown DM, Roman E, Swain SL (2004) CD4 T cell responses to influenza infection. Semin Immunol 16:171–177. [DOI] [PubMed] [Google Scholar]
- 16. Brown JL, Dominik JW, Morrissey RL (1981) Respiratory infectivity of a recently isolated Egyptian strain of Rift Valley fever virus. Infect Immun 33:848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burleigh LM, Calder LJ, Skehel JJ, Steinhauer DA (2005) Influenza a viruses with mutations in the m1 helix six domain display a wide variety of morphological phenotypes. J Virol 79:1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Calvert J, Chieochansin T, Benschop KS, McWilliam Leitch EC, Drexler JF, Grywna K et al (2010) Recombination dynamics of human parechoviruses: investigation of type‐specific differences in frequency and epidemiological correlates. J Gen Virol 91:1229–1238. [DOI] [PubMed] [Google Scholar]
- 19. Chan PK (2002) Outbreak of avian influenza A(H5N1) virus infection in Hong Kong in 1997. Clin Infect Dis 34:S58–S64. [DOI] [PubMed] [Google Scholar]
- 20. Charcot JM, Joffroy A (1870) Cas de paralysie infantile spinale avec lesins des cornes anterieures de la substance grise de la moelle epiniere. Arch Physiol Norm Pathol 3:134–152. [Google Scholar]
- 21. Chiu C, Ellebedy AH, Wrammert J, Ahmed R (2015) B cell responses to influenza infection and vaccination. Curr Top Microbiol Immunol 386:381–398. [DOI] [PubMed] [Google Scholar]
- 22. Chowell G, Bertozzi SM, Colchero MA, Lopez‐Gatell H, Alpuche‐Aranda C, Hernandez M, Miller MA (2009) Severe respiratory disease concurrent with the circulation of H1N1 influenza. N Engl J Med 361:674–679. [DOI] [PubMed] [Google Scholar]
- 23. Chung PW, Huang YC, Chang LY, Lin TY, Ning HC (2001) Duration of enterovirus shedding in stool. J Microbiol Immunol Infect 34:167–170. [PubMed] [Google Scholar]
- 24. Committee on the ECHO Viruses (1955) ENTERIC cytopathogenic human orphan (ECHO) viruses. Science 122:1187–1188. [PubMed] [Google Scholar]
- 25. Connor RJ, Kawaoka Y, Webster RG, Paulson JC (1994) Receptor specificity in human, avian, and equine H2 and H3 influenza virus isolates. Virology 205:17–23. [DOI] [PubMed] [Google Scholar]
- 26. Couceiro JN, Paulson JC, Baum LG (1993) Influenza virus strains selectively recognize sialyloligosaccharides on human respiratory epithelium; the role of the host cell in selection of hemagglutinin receptor specificity. Virus Res 29:155–165. [DOI] [PubMed] [Google Scholar]
- 27. Couch RB (2003) An overview of serum antibody responses to influenza virus antigens. Dev Biol (Basel) 115:25–30. [PubMed] [Google Scholar]
- 28. Daubney R, Hudson JR, Garnham PC (1931) Enzootic hepatitis or Rift Valley fever. An undescribed virus disease of sheep, cattle and man from East Africa. J Pathol Bacteriol 34:545–579. [Google Scholar]
- 29. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C (2004) Innate antiviral responses by means of TLR7‐mediated recognition of single‐stranded RNA. Science 303:1529–1531. [DOI] [PubMed] [Google Scholar]
- 30. Elliott RM, Schmaljohn CS, Collett MS (1991) Bunyaviridae genome structure and gene expression. Curr Top Microbiol Immunol 169:91–141. [DOI] [PubMed] [Google Scholar]
- 31. Enami M, Sharma G, Benham C, Palese P (1991) An influenza virus containing nine different RNA segments. Virology 185:291–298. [DOI] [PubMed] [Google Scholar]
- 32. Feldmann A, Schafer MK, Garten W, Klenk HD (2000) Targeted infection of endothelial cells by avian influenza virus A/FPV/Rostock/34 (H7N1) in chicken embryos. J Virol 74:8018–8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fischer TK, Midgley S, Dalgaard C, Nielsen AY (2014) Human parechovirus infection, Denmark. Emerg Infect Dis 20:83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Flick R, Bouloy M (2005) Rift Valley fever virus. Curr Mol Med 5:827–834. [DOI] [PubMed] [Google Scholar]
- 35. Ford Siltz LA, Viktorova EG, Zhang B, Kouiavskaia D, Dragunsky E, Chumakov K et al (2014) New small‐molecule inhibitors effectively blocking picornavirus replication. J Virol 88:11091–11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Francis T, Magill TP (1935) Rift Valley fever: a report of three cases of laboratory infection and the experimental transmission of the disease to ferrets. J Exp Med 62:433–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gao P, Watanabe S, Ito T, Goto H, Wells K, McGregor M et al (1999) Biological heterogeneity, including systemic replication in mice, of H5N1 influenza A virus isolates from humans in Hong Kong. J Virol 73:3184–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao R, Dong L, Dong J, Wen L, Zhang Y, Yu H et al (2010) A systematic molecular pathology study of a laboratory confirmed H5N1 human case. PLoS ONE 5:e13315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garcia‐Sastre A (2006) Antiviral response in pandemic influenza viruses. Emerg Infect Dis 12:44–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gavrilovskaya IN, Brown EJ, Ginsberg MH, Mackow ER (1999) Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J Virol 73:3951–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gerdes GH (2004) Rift Valley fever. Rev Sci Tech 23:613–623. [DOI] [PubMed] [Google Scholar]
- 42. Gillim‐Ross L, Subbarao K (2006) Emerging respiratory viruses: challenges and vaccine strategies. Clin Microbiol Rev 19:614–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gomez‐Puertas P, Albo C, Perez‐Pastrana E, Vivo A, Portela A (2000) Influenza virus matrix protein is the major driving force in virus budding. J Virol 74:11538–11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grove RD, Hetzel AM (1968) Vital Statistics Rates in the United States: 1940–1960. National Center for Health Statistics (ed.), Government Printing Office: Washington, DC. [Google Scholar]
- 45. Gu J, Xie Z, Gao Z, Liu J, Korteweg C, Ye J et al (2007) H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet 370:1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gupta S, Fernandez D, Siddiqui A, Tong WC, Pohl K, Jungbluth H (2010) Extensive white matter abnormalities associated with neonatal Parechovirus (HPeV) infection. Eur J Paediatr Neurol 14:531–534. [DOI] [PubMed] [Google Scholar]
- 47. Guu TS, Zheng W, Tao YJ (2012) Bunyavirus: structure and replication. Adv Exp Med Biol 726:245–266. [DOI] [PubMed] [Google Scholar]
- 48. Hartman AL, Powell DS, Bethel LM, Caroline AL, Schmid RJ, Oury T, Reed DS (2014) Aerosolized rift valley fever virus causes fatal encephalitis in African green monkeys and common marmosets. J Virol 88:2235–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harvala H, Calvert J, Van Nguyen D, Clasper L, Gadsby N, Molyneaux P et al (2014) Comparison of diagnostic clinical samples and environmental sampling for enterovirus and parechovirus surveillance in Scotland, 2010 to 2012. Euro Surveill 19:20772. [DOI] [PubMed] [Google Scholar]
- 50. Harvala H, Simmonds P (2009) Human parechoviruses: biology, epidemiology and clinical significance. J Clin Virol 45:1–9. [DOI] [PubMed] [Google Scholar]
- 51. Herfst S, Imai M, Kawaoka Y, Fouchier RA (2014) Avian influenza virus transmission to mammals. Curr Top Microbiol Immunol 385:137–155. [DOI] [PubMed] [Google Scholar]
- 52. Hoogstraal H, Meegan JM, Khalil GM, Adham FK (1979) The Rift Valley fever epizootic in Egypt 1977–78. 2. Ecological and entomological studies. Trans R Soc Trop Med Hyg 73:624–629. [DOI] [PubMed] [Google Scholar]
- 53. Ida‐Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T et al (2005) The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol 79:4460–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Isaacs A, Lindenmann J (1957) Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147:258–267. [PubMed] [Google Scholar]
- 55. Ito M, Yamashita T, Tsuzuki H, Takeda N, Sakae K (2004) Isolation and identification of a novel human parechovirus. J Gen Virol 85:391–398. [DOI] [PubMed] [Google Scholar]
- 56. Jang H, Boltz D, Sturm‐Ramirez K, Shepherd KR, Jiao Y, Webster R, Smeyne RJ (2009) Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A 106:14063–14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jin H, Subbarao K (2015) Live attenuated influenza vaccine. Curr Top Microbiol Immunol 386:181–204. [DOI] [PubMed] [Google Scholar]
- 58. Johnson NP, Mueller J (2002) Updating the accounts: global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull Hist Med 76:105–115. [DOI] [PubMed] [Google Scholar]
- 59. Joseph C, Togawa Y, Shindo N (2013) Bacterial and viral infections associated with influenza. Influenza Other Respir Viruses 7:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury and apoptosis in HIV‐associated dementia. Nature 410:988–994. [DOI] [PubMed] [Google Scholar]
- 61. Keefer GV, Zebarth GL, Allen WP (1972) Susceptibility of dogs and cats to Rift Valley fever by inhalation or ingestion of virus. J Infect Dis 125:307–309. [DOI] [PubMed] [Google Scholar]
- 62. Keegan K, Collett MS (1986) Use of bacterial expression cloning to define the amino acid sequences of antigenic determinants on the G2 glycoprotein of Rift Valley fever virus. J Virol 58:263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kemball CC, Harkins S, Whitmire JK, Flynn CT, Feuer R, Whitton JL (2009) Coxsackievirus B3 inhibits antigen presentation in vivo, exerting a profound and selective effect on the MHC class I pathway. PLoS Pathog 5:e1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kew OM, Nottay BK, Rico‐Hesse R, Pallansch MA (1990) Molecular epidemiology of wild poliovirus transmission. In: Virus Variability, Epidemiology, and Control. Kurstak E, Marusyk RG, Murphy FA, van Regenmortel MHV (eds), pp. 199–222. Plenum Medical Book Company: New York. [Google Scholar]
- 65. Klenk HD, Garten W (1994) Host cell proteases controlling virus pathogenicity. Trends Microbiol 2:39–43. [DOI] [PubMed] [Google Scholar]
- 66. Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R (1992) Ongoing enterovirus‐induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci U S A 89:314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Knipe DM, Howley PM (2013) Fields Virology, 6th edn. Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA. [Google Scholar]
- 68. Knobler S, Institute of Medicine (U.S.) , Forum on Microbial Threats , Institute of Medicine (U.S.) , Board on Global Health (2005) The Threat of Pandemic Influenza: Are We Ready? Workshop Summary. National Academies Press: Washington, DC. [PubMed] [Google Scholar]
- 69. Knowles NJ, Delwart E, Gorbalenya AE, Hovi T, Hyypia T, King AMQ et al (2014) Picorniviridae: 26 genera, 46 species and growing… In: Abstract Europic 18th International Picornavirus Meeting. p. 98. Blankenberge, Belgium.
- 70. Koyuncu OO, Hogue IB, Enquist LW (2013) Virus infections in the nervous system. Cell Host Microbe 13:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. La Gruta NL, Turner SJ (2014) T cell mediated immunity to influenza: mechanisms of viral control. Trends Immunol 35:396–402. [DOI] [PubMed] [Google Scholar]
- 72. Leneva IA, Russell RJ, Boriskin YS, Hay AJ (2009) Characteristics of arbidol‐resistant mutants of influenza virus: implications for the mechanism of anti‐influenza action of arbidol. Antiviral Res 81:132–140. [DOI] [PubMed] [Google Scholar]
- 73. Lewis DB (2006) Avian flu to human influenza. Annu Rev Med 57:139–154. [DOI] [PubMed] [Google Scholar]
- 74. Lipsitch M, Plotkin JB, Simonsen L, Bloom B (2012) Evolution, safety, and highly pathogenic influenza viruses. Science 336:1529–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lozach PY, Kuhbacher A, Meier R, Mancini R, Bitto D, Bouloy M, Helenius A (2011) DC‐SIGN as a receptor for phleboviruses. Cell Host Microbe 10:75–88. [DOI] [PubMed] [Google Scholar]
- 76. Macejak DG, Sarnow P (1991) Internal initiation of translation mediated by the 5′ leader of a cellular mRNA. Nature 353:90–94. [DOI] [PubMed] [Google Scholar]
- 77. Maines TR, Lu XH, Erb SM, Edwards L, Guarner J, Greer PW et al (2005) Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J Virol 79:11788–11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mair CM, Ludwig K, Herrmann A, Sieben C (2014) Receptor binding and pH stability—how influenza A virus hemagglutinin affects host‐specific virus infection. Biochim Biophys Acta 1838:1153–1168. [DOI] [PubMed] [Google Scholar]
- 79. Marks G, Beatty WK (1976) Epidemics. Scribner: New York. [Google Scholar]
- 80. Matsuoka Y, Lamirande EW, Subbarao K (2009) The ferret model for influenza. Curr Protoc Microbiol 13:15G.2.1–15G.2.29. [DOI] [PubMed] [Google Scholar]
- 81. Meegan JM, Hoogstraal H, Moussa MI (1979) An epizootic of Rift Valley fever in Egypt in 1977. Vet Rec 105:124–125. [DOI] [PubMed] [Google Scholar]
- 82. Mendelsohn CL, Wimmer E, Racaniello VR (1989) Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56:855–865. [DOI] [PubMed] [Google Scholar]
- 83. Morrill JC, Ikegami T, Yoshikawa‐Iwata N, Lokugamage N, Won S, Terasaki K et al (2010) Rapid accumulation of virulent Rift Valley fever virus in mice from an attenuated virus carrying a single nucleotide substitution in the m RNA. PLoS ONE 5:e9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Munster VJ, Baas C, Lexmond P, Waldenstrom J, Wallensten A, Fransson T et al (2007) Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog 3:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nayak DP, Hui EK, Barman S (2004) Assembly and budding of influenza virus. Virus Res 106:147–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nelli RK, Kuchipudi SV, White GA, Perez BB, Dunham SP, Chang KC (2010) Comparative distribution of human and avian type sialic acid influenza receptors in the pig. BMC Vet Res 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nichol ST, Beaty BJ, Elliott RM, Goldbach R, Plyusnin A, Schmaljohn CS, Tesh RB (2005) Bunyaviridae. In: Virus Taxonomy: VIIIth Report of the International Committee on Taxonomy of Viruses. Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (eds), pp. 695–716. Elsevier Academic Press: London. [Google Scholar]
- 88. Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL et al (2007) Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med 13:147–149. [DOI] [PubMed] [Google Scholar]
- 89. Ohka S, Nihei C, Yamazaki M, Nomoto A (2012) Poliovirus trafficking toward central nervous system via human poliovirus receptor‐dependent and ‐independent pathway. Front Microbiol 3:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Oshinsky DM (2005) Polio: an American Story. Oxford University Press: Oxford, New York. [Google Scholar]
- 91. Palese P, Compans RW (1976) Inhibition of influenza virus replication in tissue culture by 2‐deoxy‐2,3‐dehydro‐N‐trifluoroacetylneuraminic acid (FANA): mechanism of action. J Gen Virol 33:159–163. [DOI] [PubMed] [Google Scholar]
- 92. Palese P, Tobita K, Ueda M, Compans RW (1974) Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61:397–410. [DOI] [PubMed] [Google Scholar]
- 93. Park CH, Ishinaka M, Takada A, Kida H, Kimura T, Ochiai K, Umemura T (2002) The invasion routes of neurovirulent A/Hong Kong/483/97 (H5N1) influenza virus into the central nervous system after respiratory infection in mice. Arch Virol 147:1425–1436. [DOI] [PubMed] [Google Scholar]
- 94. Patrican LA, Bailey CL (1989) Ingestion of immune bloodmeals and infection of Aedes fowleri, Aedes mcintoshi, and Culex pipiens with Rift Valley fever virus. Am J Trop Med Hyg 40:534–540. [DOI] [PubMed] [Google Scholar]
- 95. Perkins LE, Swayne DE (2002) Pathogenicity of a Hong Kong‐origin H5N1 highly pathogenic avian influenza virus for emus, geese, ducks, and pigeons. Avian Dis 46:53–63. [DOI] [PubMed] [Google Scholar]
- 96. Perry AK, Chen G, Zheng D, Tang H, Cheng G (2005) The host type I interferon response to viral and bacterial infections. Cell Res 15:407–422. [DOI] [PubMed] [Google Scholar]
- 97. Picornaviridae.com (2015) Parechovirus A. Available at: http://www.picornaviridae.com/parechovirus/parechovirus_a/parechovirus_a.htm (accessed 8 May 2015).
- 98. Ross TM, Bhardwaj N, Bissel SJ, Hartman AL, Smith DR (2012) Animal models of Rift Valley fever virus infection. Virus Res 163:417–423. [DOI] [PubMed] [Google Scholar]
- 99. Rudolph KE, Lessler J, Moloney RM, Kmush B, Cummings DA (2014) Incubation periods of mosquito‐borne viral infections: a systematic review. Am J Trop Med Hyg 90:882–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sabin AB (1956) Pathogenesis of poliomyelitis; reappraisal in the light of new data. Science 123:1151–1157. [DOI] [PubMed] [Google Scholar]
- 101. Sadasivan S, Zanin M, O'Brien K, Schultz‐Cherry S, Smeyne RJ (2015) Induction of microglia activation after infection with the non‐neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS ONE 10:e0124047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sedmak G, Nix WA, Jentzen J, Haupt TE, Davis JP, Bhattacharyya S et al (2010) Infant deaths associated with human parechovirus infection in Wisconsin. Clin Infect Dis 50:357–361. [DOI] [PubMed] [Google Scholar]
- 103. Seufi AM, Galal FH (2010) Role of Culex and Anopheles mosquito species as potential vectors of Rift Valley fever virus in Sudan outbreak, 2007. BMC Infect Dis 10:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Simonsen L (1999) The global impact of influenza on morbidity and mortality. Vaccine 17:S3–S10. [DOI] [PubMed] [Google Scholar]
- 105. Stanway G, Hyypia T (1999) Parechoviruses. J Virol 73:5249–5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Stanway G, Kalkkinen N, Roivainen M, Ghazi F, Khan M, Smyth M et al (1994) Molecular and biological characteristics of echovirus 22, a representative of a new picornavirus group. J Virol 68:8232–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Stephenson I, Nicholson KG, Wood JM, Zambon MC, Katz JM (2004) Confronting the avian influenza threat: vaccine development for a potential pandemic. Lancet Infect Dis 4:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Strikas RA, Anderson LJ, Parker RA (1986) Temporal and geographic patterns of isolates of nonpolio enterovirus in the United States, 1970–1983. J Infect Dis 153:346–351. [DOI] [PubMed] [Google Scholar]
- 109. Suzuki Y, Nei M (2002) Origin and evolution of influenza virus hemagglutinin genes. Mol Biol Evol 19:501–509. [DOI] [PubMed] [Google Scholar]
- 110. Swanepoel R, Struthers JK, Erasmus MJ, Shepherd SP, McGillivray GM, Erasmus BJ, Barnard BJ (1986) Comparison of techniques for demonstrating antibodies to Rift Valley fever virus. J Hyg (Lond) 97:317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Takao S, Shimazu Y, Fukuda S, Noda M, Miyazaki K (2001) Seroepidemiological study of human Parechovirus 1. Jpn J Infect Dis 54:85–87. [PubMed] [Google Scholar]
- 112. Taubenberger JK, Morens DM (2009) Pandemic influenza—including a risk assessment of H5N1. Rev Sci Tech 28:187–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F et al (2011) Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146:980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Thibaut HJ, De Palma AM, Neyts J (2012) Combating enterovirus replication: state‐of‐the‐art on antiviral research. Biochem Pharmacol 83:185–192. [DOI] [PubMed] [Google Scholar]
- 115. Thompson SR, Sarnow P (2000) Regulation of host cell translation by viruses and effects on cell function. Curr Opin Microbiol 3:366–370. [DOI] [PubMed] [Google Scholar]
- 116. Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM et al (2012) A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci U S A 109:4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M et al (2013) New world bats harbor diverse influenza A viruses. PLoS Pathog 9:e1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Toovey S (2008) Influenza‐associated central nervous system dysfunction: a literature review. Travel Med Infect Dis 6:114–124. [DOI] [PubMed] [Google Scholar]
- 119. Tourre YM, Lacaux JP, Vignolles C, Ndione JA, Lafaye M (2008) Mapping of zones potentially occupied by Aedes vexans and Culex poicilipes mosquitoes, the main vectors of Rift Valley fever in Senegal. Geospat Health 3:69–79. [DOI] [PubMed] [Google Scholar]
- 120. Tucker SP, Compans RW (1993) Virus infection of polarized epithelial cells. Adv Virus Res 42:187–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Turell MJ, Gargan TP 2nd, Bailey CL (1985) Culex pipiens (Diptera: Culicidae) morbidity and mortality associated with Rift Valley fever virus infection. J Med Entomol 22:332–337. [DOI] [PubMed] [Google Scholar]
- 122. van Zwol AL, Lequin M, Aarts‐Tesselaar C, van der Eijk AA, Driessen GA, de Hoog M, Govaert P (2009) Fatal neonatal parechovirus encephalitis. BMJ Case Rep 2009:1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Verboon‐Maciolek MA, Groenendaal F, Hahn CD, Hellmann J, van Loon AM, Boivin G, de Vries LS (2008) Human parechovirus causes encephalitis with white matter injury in neonates. Ann Neurol 64:266–273. [DOI] [PubMed] [Google Scholar]
- 124. Verboon‐Maciolek MA, Krediet TG, Gerards LJ, de Vries LS, Groenendaal F, van Loon AM (2008) Severe neonatal parechovirus infection and similarity with enterovirus infection. Pediatr Infect Dis J 27:241–245. [DOI] [PubMed] [Google Scholar]
- 125. Wang X, Zhao J, Tang S, Ye Z, Hewlett I (2010) Viremia associated with fatal outcomes in ferrets infected with avian H5N1 influenza virus. PLoS ONE 5:e12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. White MR, Doss M, Boland P, Tecle T, Hartshorn KL (2008) Innate immunity to influenza virus: implications for future therapy. Expert Rev Clin Immunol 4:497–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB (1986) Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci U S A 83:7089–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. World Health Organization (2015) Cumulative number of confirmed human cases for avian influenza A (H5N1) reported to WHO, 2003–2015. Available at: http://www.who.int/influenza/human_animal_interface/EN_GIP_201503031cumulativeNumberH5N1cases.pdf?ua=1 (accessed 11 May 2015).
- 129. Yao L, Korteweg C, Hsueh W, Gu J (2008) Avian influenza receptor expression in H5N1‐infected and noninfected human tissues. FASEB J 22:733–740. [DOI] [PubMed] [Google Scholar]
- 130. Yin‐Murphy M, Almond JW (1996) Picornaviruses. Chapter 53. In: Medical Microbiology. Baron S (ed.), University of Texas Medical Branch at Galveston: Galveston, TX. [PubMed] [Google Scholar]
- 131. Zaghlool A, Ameur A, Cavelier L, Feuk L (2014) Splicing in the human brain. Int Rev Neurobiol 116:95–125. [DOI] [PubMed] [Google Scholar]