


Abstract
The N-terminal acidic transactivation domain (TAD) of ERM/ETV5 (ERM38–68), a PEA3 group member of Ets-related transcription factors, directly interacts with the ACID/PTOV domain of the Mediator complex subunit MED25. Molecular details of this interaction were investigated using nuclear magnetic resonance (NMR) spectroscopy. The TAD is disordered in solution but has a propensity to adopt local transient secondary structure. We show that it folds upon binding to MED25 and that the resulting ERM–MED25 complex displays characteristics of a fuzzy complex. Mutational analysis further reveals that two aromatic residues in the ERM TAD (F47 and W57) are involved in the binding to MED25 and participate in the ability of ERM TAD to activate transcription. Mutation of a key residue Q451 in the VP16 H1 binding pocket of MED25 affects the binding of ERM. Furthermore, competition experiments show that ERM and VP16 H1 share a common binding interface on MED25. NMR data confirms the occupancy of this binding pocket by ERM TAD. Based on these experimental data, a structural model of a functional interaction is proposed. This study provides mechanistic insights into the Mediator–transactivator interactions.
INTRODUCTION
Transcription factors regulate gene expression by both binding specific DNA sequences and serving as an interaction platform for proteins that control transcription. This latter property is mainly supported by the transactivation domains (TADs) that exhibit binding sites for a multitude of interacting co-regulators in order to assemble transcriptional complexes at promoters and enhancers. Classical TAD-interacting proteins are components of the general transcription factors such as TFIIB, TFIIH and TFIID, histone modifying enzymes and subunits of the Mediator complex (1). Given its large size (∼1.5 MDa) and multi-subunit composition, the Mediator complex provides multiple interfaces for protein–protein interactions and studies over the past two decades reveal that transcription factors are important Mediator-interacting partners (2–6).
The human Mediator complex subunit MED25 interacts through its ACID (Activator Interacting Domain)/PTOV (Prostate Tumor OVerexpressed) domain with the viral transactivators VP16 (7,8), IE62 (9,10) and Lana-1 (11), the endoplasmic reticulum stress-responsive transcription factor ATF6α (12) and the Arabidopsis transcriptional regulator Dreb2a (13,14). We have recently shown that the N-terminal TAD of human ERM/ETV5 (ERM38–68), a PEA3 subfamily member of Ets transcription factors, also specifically binds to the ACID/PTOV domain of MED25 (15). This region of ERM/ETV5 contains a number of acidic amino acids as well as several hydrophobic/aromatic residues. Notably, mutating phenylalanine 47 to leucine (F47L mutant) within the TAD of Ets-related molecule (ERM) affects its binding to MED25 resulting in reduced transactivation activity (15). Besides ERM, it is noteworthy that all these transcriptional regulators described above, possess acidic/hydrophobic-rich domains in their TADs.
Structural studies of the human Mediator MED25 ACID/PTOV domain reveal, unlike other known activator protein partners, an original arrangement as a seven-stranded closed β-barrel surrounded by three α-helices (16–18). Previous studies by us and others showed that the ACID/PTOV domain of MED25 provides two distinct surfaces for interaction with the Herpes simplex VP16 TAD (16–18). The VP16 subdomain H1 binds in a hydrophobic groove formed at the interface between strands β2–β1–β3 and helix α3 (17,18), whereas the VP16 subdomain H2 binds in a groove located on the opposite face of the MED25 β-barrel and delimited by helix α1 and strands β4–β7–β6 (16–18).
To date, due to conformational heterogeneity, the structure of human MED25 ACID/PTOV domain in complex with an acidic TAD is still not available. Here we report the molecular basis of the ERM TAD interaction with the ACID/PTOV domain of MED25, which has been shown to be key for ERM transactivation (15). Despite the inherent difficulties in characterizing this type of interaction, we use a combination of biophysical methods and mutagenesis analysis to define key properties of the interaction. Based on our data, we propose a model in which the N-terminal TAD of ERM binds the MED25 VP16 H1 binding pocket in a helical conformation.
MATERIALS AND METHODS
Plasmid construction
The pET24d expression vectors encoding the human ACID/PTOV domain (residues 391 to 548) has been described previously (16). The ERM activation domain derivatives (38–68, 38–68 F47L, 38–68 W57L and 38–68 F47/W57L) were subcloned into pBIND (Gal4 DNA-binding domain) vector (Promega). The bacterial expression plasmid encoding Halo-Tag ERM38–68 (pET24d-HaloTag ERM38–68) was generated by co-ligating the polymerase chain reaction (PCR) amplified Halo-Tag (template pFN22K (Promega) and ERM38–68 (template pSG5 hERM (19)). To insert point mutation into our respective vectors (MED25 ACID/PTOV Q451E and R466E, ERM38–68 F47L, W57L, F47L/W57L, F47A/W57A, A41C and P64C), PCR site-directed mutagenesis was used. The VP16 H1 (amino acids 413–452) and VP16 H2 (amino acids 453–490) peptides were subcloned into pGEX-6P1 (Promega). All clones were verified by sequencing. Primers sequences and detailed procedures are available upon request.
Protein expression, purification and modification
All proteins were expressed in Escherichia coli strain BL21 (DE3). Bacteria were grown in LB medium at 37°C to an optical density of 0.6 at 600 nm and expression was induced with IPTG for 18 h at 20°C. For labelling experiments, LB was replaced by minimal M9 medium supplemented with 15NH4Cl and/or 13C-glucose. The MED25 ACID/PTOV domain was purified as previously described (16) and the same purification procedure was applied for the Q451E and R446E mutants. Recombinant Halo-Tag, Halo-Tag ERM38–68 and derivatives (F47L, W57L, F47L/W57L, F47A/W57A, A41C and P64C) were expressed in E. coli. Briefly, Halo-tag ERM38–68 fusion peptides were purified using ion exchange chromatography HiTrap Q HP (GE) using a gradient to 1 M NaCl. To remove the fusion tag, the peptides were cleaved by Tobacco Etch Virus (TEV) protease and finally purified using size exclusion chromatography Superdex 30 Hiload 16/60 (GE). Recombinant VP16 H1 and VP 16 H2 peptides were purified as described in (20). For spin labelling, 250 μM of A41C ERM38–68 or P64C ERM38–68 in 50 mM Ammonium Bicarbonate was incubated with 1 mM Tris (hydroxymethyl) phosphine (THP) for 1 h at room temperature. A total of 5 mM of 3-(2-iodoacetamido)-PROXYL (Sigma) in dimethyl sulfoxide were next added and incubation continued for 3 h. Excess of spin-label was removed by desalting on a G25 resin (PD10 column, GE Healthcare) into the nuclear magnetic resonance (NMR) buffer. ERM38–68 wild-type and mutants, VP16 H1 and VP16 H2 peptides were labelled with TAMRA-NHS ester (Pierce) at the N-terminal free NH2 group following indications from the manufacturers.
Fluorescence polarization and isothermal titration calorimetry
TAMRA-labelled VP16 H1, VP16 H2 and ERM38–68-derived peptides at 4 nM were titrated with a range of MED25 ACID/PTOV concentrations in phosphate buffered saline buffer containing 0.1 mg/ml bovine serum albumin. Reaction mixtures were incubated for 15 min at room temperature before measurements were made on a PherastarFS (BMG Labtech) plate reader with excitation at 540 nm and emission at 590 nm, with correction for buffer background, using 384-well optiplates (Greiner). Binding data were fitted to a 1:1 non-linear binding model (Y = Bmax * X/(Kd + X) where Bmax is the maximum polarization and X is the concentration of MED25) using Prism 6.05 (Graphpad software Inc.). In the competition experiments, 4 nM of TAMRA-labelled ERM38–68, 0.2 μM of ACID/PTOV and varying concentrations of unlabelled ERM38–68, VP16 H1 and VP16 H2 peptides were used for IC50 determination. Isothermal titration calorimetry (ITC) titrations were performed as described (15). All titrations fit a single-binding site mechanism with a 1:1 stoichiometry.
Transactivation assay and western-blot
U2OS cells were cultured in Dubbelco's Modified Eagle Medium supplemented with 10% Fetal Calf Serum (FCS) (Gibco BRL). In 24-well plates, 5 × 104 cells/well were plated and the next day, transfections were performed using PEI Exgen 500 procedure (Euromedex, France) with 200 ng total DNA per well including 50 ng reporter plasmid (Gal4)5-E1B-Luc (21). The activities of Gal4-ERM derivatives were then assayed using the dual-luciferase reporter assay system (Promega) as described previously (15). For protein expression experiments, cells were transfected as above in 12-well plates with 200 ng of expression plasmid and lysed in sodium dodecyl sulphate electrophoresis loading buffer. Immunoblot analyses were performed with anti-Gal4 DBD antibody (Santa Cruz). Halo-Tag pull-down were performed as described previously (15).
NMR data acquisition
The NMR samples were prepared in 100 mM NaP Phosphate buffer at pH 6.5, 25 mM NaCl, 2.5 mM ethylenediaminetetraacetic acid, 1 mM Dithiothreitol, 1 mM D4-TMSP (TriMethyl Silyl Propionate) as proton chemical shifts internal reference and 5% D2O. NMR spectra were recorded at 298K on a Bruker DMX 600MHz and Avance III 900MHz spectrometers equipped with a triple resonance cryogenic probehead (Bruker, Karlsruhe, Germany). Two-dimensional (2D) HSQC spectra were acquired on 75–125 μM MED25. Classical pairs of Three-dimensional (3D) HNCACB, HN(CO)CACB and HN(CA)NNH were processed using Bruker TOPSPIN 2.1 (Bruker, Karlsruhe, Germany) in order to assign backbone resonances from ERM38–68 (Supplementary Table S1). Additional 2D-Noesy and 2D-Dipsy pair was recorded on a 1.5 mM solution of ERM38–68 to assign 1H resonances of ERM38–68 and to observe nuclear Overhauser effect (NOE) contacts between 1H resonances.
NMR data analysis
Peak picking and intensity measurements were performed using Sparky (22). Secondary chemical shifts correspond to the deviation of the experimental difference 13Cα–13Cβ chemical shift values for each residue in the protein and the reference value for the corresponding amino acid residue found in databases of chemical shifts compiled from strictly disordered proteins. Secondary chemical shifts 13Cα–13Cβ were here calculated as indicators for residual local secondary structure based on the random coil values database (ncIDP) from Tamiola et al. (23) that are neighbour-corrected. The secondary structure propensity (SSP) analysis compares the Cα and Cβ values not only to the random coil values expected in a strictly disordered protein but also to the Cα and Cβ values expected in a fully formed α-helix or β-strand, taking additionally into account the propensity of a given amino acid to adopt such a secondary structure. The results correspond for each amino acid residue to the fraction of the conformer distribution to adopt a secondary structure. SSP scores (24) were calculated using the refDB random coil database (25). Consecutive positive values of both secondary 13Cα–13Cβ chemical shifts and structure propensity are related to a local helical tendency while negative values indicate an extended conformation. Calculations of the co-linear chemical shift deviations (CCSD) are based on the detailed protocol in (26). A CCSD line was first defined between the resonance in free MED25 and in the ERM38–68-bound MED25 as chemical shift deviations (CSD) in ppm = ((Hshift)2 + (0.2*Nshift)2)1/2. A scalar projection on this line of the CSD for resonances corresponding to the set of complexes was next calculated. The CCSD corresponds to the ratio of the scalar projection on the CCSD line, expressed as percentage.
HADDOCK docking
HADDOCK is a docking program to model protein interactions using experimental data as restraints (27). The ERM peptide in the modelization consisted in residues L50 to A61 in helical conformation, based on the data of the SSP (Figure 1B). PyMOL (28)was used to build the helical conformation peptide ERM 50–61. Active residues were chosen as L53, W57 and E60, consisting of the residues on the face of the peptide containing the W57. The protein active residues were based on peak intensity decreases and chosen to represent three secondary structures that showed the strongest intensity perturbations upon ERM binding to MED25 VP16 H1 binding site: I449 and Q451 in strand β3, V534, I537 and R538 in helix α3 and K413 and Pro414 in the β1–β2 loop of MED25. All calculations were run through the WeNMR Web portal (29) with the prediction interface. The software defined the passive residues based on the active ones and took into account interface prediction by CPORT (30). Each calculation generated 1000/200/200 models for the different it0/it1/water refinement stages of HADDOCK. A total of 195 final models were clustered based on their interface-ligand RMSD, using a cutoff of 5 Å. The final overall score of each cluster (Supplementary Table S2) is based on the four highest HADDOCK scores of the models in that cluster.
Figure 1.
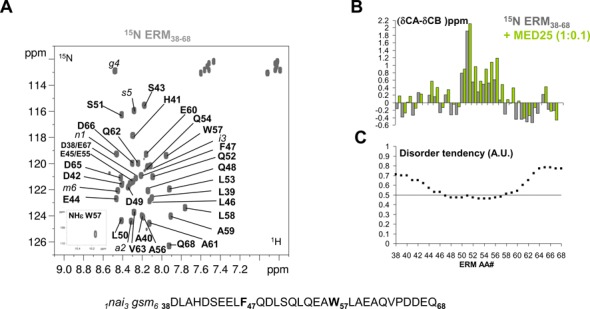
Conformation of ERM38–68. (A) Annotated 2D [1H, 15N] HSQC spectrum of ERM38–68. Sequence of ERM38–68 is shown below the spectrum, with aromatic residues F47 and W57 highlighted in bold. Residual amino acids after cleavage from the N-terminal fusion Halo-Tag are annotated in italic and numbered from 1 to 6. (B) Histogram of measured Cα−Cβ secondary chemical shifts along the ERM38–68 sequence (scale in ppm), grey bars are for the free ERM38–68, green bars for the MED25-bound ERM38–68 (stoichiometry 1 ERM/0.1 MED25). (C) Disorder prediction for each amino acid in the sequence (arbitrary units A.U.). The 0.5 lower threshold for disorder prediction is drawn as a line. metaPrDOS integrates the results of different prediction methods.
RESULTS
Transient secondary structure in the disordered ERM transactivation domain
We recently identified an acidic segment within the N-terminal TAD of ERM (ERM38–68) that interacts with the MED25 ACID/PTOV domain (15). The structural properties of ERM TAD were investigated using NMR spectroscopy. The 2D 1H-15N HSQC spectrum of ERM38–68 is characteristic of an unstructured peptide with all amide proton chemical shifts confined to 7.7–8.5 ppm (Figure 1A). Complete peptide backbone assignments were obtained using conventional 3D experiments on a 15N-13C double-labelled sample (Figure 1A and Supplementary Table S1). These resonance assignments were used to probe with a per residue resolution the propensity of the peptide to adopt a transient secondary structure in solution. By comparing the measured Cα and Cβ chemical shift values of the amino acid residues along the ERM38–68 sequence with the predicted corresponding random coil values (23), secondary chemical shifts can be calculated that are empirically correlated with the local backbone structures (grey bars in Figure 1B). Though ERM TAD lacks a well-defined globular structure, positive secondary chemical shifts are observed between amino acid L50 and W57, indicative of the formation of a transient α-helix in this peptide region. Consistent with our previous reports (31,32), disordered prediction using the metaPrDOS algorithm (33) returns a disorder tendency for most of the ERM38–68 amino acid sequence, except for residues F47 to W57 (Figure 1C). Moreover, SSP analysis (24) confirms this region as having a 20–30% propensity to adopt defined conformation in solution (grey bars in Supplementary Figure S1A). Finally, NOEs resonances in a 15N-filtered 1H-1H NOESY spectrum consisted mainly of sequential proximities although a i-2 NOE cross peak between HN-W57 and Hβ-E55 is clearly detected (Supplementary Figure S1B). These data suggest that the ERM TAD is not entirely disordered but populated with transiently ordered regions.
Interaction of MED25 ACID/PTOV domain with ERM TAD
To investigate the structural basis for ERM38–68 interaction with MED25, NMR spectra of the 15N-labeled ERM38–68 were monitored with increasing concentrations of unlabelled MED25 (Figure 2 and Supplementary Figure S2). At a molar ratio of 1:2, a drastic change in the chemical shift dispersion of the [1H,15N]-ERM38–68 resonances is observed in the 2D spectrum (Figure 2B and Supplementary Figure S2B), suggesting that a conformational rearrangement accompanies binding. Assignment of the bound ERM peptide was hindered by the line broadening that precluded acquisition of additional 3D spectra. In presence of 0.1 and 0.2 molar ratios of MED25 ACID/PTOV, the resonances of many residues of ERM38–68 in the region between L46 and V63 showed CSD corresponding to an upfield shift in both the 1H and 15N dimensions (Figure 2A and Supplementary Figure S2A). These resonances in ERM38–68 displayed gradual chemical shift changes at sub-stoichiometric ratios of MED25 ACID/PTOV (1:0.1 and 1:0.2 molar ratios), with minor perturbation of the resonance intensities (Figure 2A and Supplementary Figure S2B). Thus, the trends in secondary chemical shift changes observed at the 1:0.1 ERM38–68/MED25 molar ratio were used to investigate how the ERM38–68 structure changes upon binding. An increase helical tendency is observed for residue L50 to A59 upon ERM38–68 binding to MED25 as compared to ERM38–68 free in solution (green bars in Figure 1B, Supplementary Figure S1A and S1C).
Figure 2.
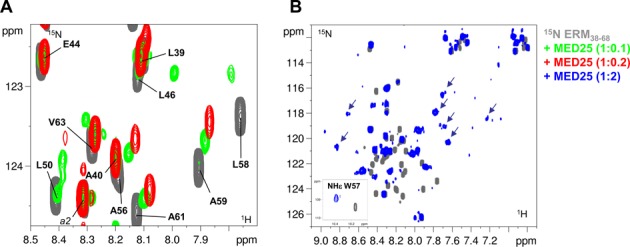
Interaction of ERM38–68 with MED25. (A) Superimposed enlarged view of 2D [1H, 15N] HSQC spectra of 15N-ERM38–68 (in grey), 15N-ERM38–68/unlabelled MED25 1/0.1 molar ratio (overlayed in green) and 15N-ERM38–68/unlabelled MED25 1/0.2 molar ratio (overlayed in red). (B) Superimposed 2D [1H, 15N] HSQC spectra of 15N-ERM38–68 (in grey) and 15N-ERM38–68/unlabelled MED25 1/2 molar ratio (overlayed in blue). Inlayed are HNϵ from the side chain of W57. The arrows indicate broadened resonances with chemical shift typical of a folded peptide.
The gradual chemical shift change observed at low stoichiometry of ERM38–68/MED25 in the titration is indicative of a fast exchange on the NMR time scale between the free and the bound forms (for a detailed explanation see Supplementary Figure S3A). However, our recent ITC assays rather revealed tight binding of ERM38–68 to MED25 ACID/PTOV with a Kd of 0.6 μM (15). Such a discrepancy is indicative of an additional mode of interaction of weak affinity (34).
Mutational analysis of ERM binding to MED25
Hydrophobic and aromatic residues in acid rich TADs were found in many studies to be critical for TADs-target contacts (35–40). We have indeed previously shown that the ERM F47L mutation reduced binding to MED25 (15). In order to develop this study, W57 of ERM38–68 was replaced by a leucine and a double mutant, F47L/W57L, was also constructed (Figure 3A). ERM mutants were first tested for their capacity to bind endogenous MED25 by incubating nuclear extracts of DAMI cells with purified Halo-Tag ERM38–68 wild-type and derivatives (F47L, W57L and F47L/W57L) coupled to magnetic beads (Figure 3B). Extending our previous observations with ERM1–72 (15), endogenous MED25 could be almost completely depleted from the nuclear extracts after incubation with Halo-Tag ERM38–68 but not with Halo-Tag alone (Figure 3B). Importantly, the F47L and W57L single ERM mutants showed a reduced capacity to capture MED25, whereas the double mutant failed to associate with MED25 under the experimental conditions.
Figure 3.
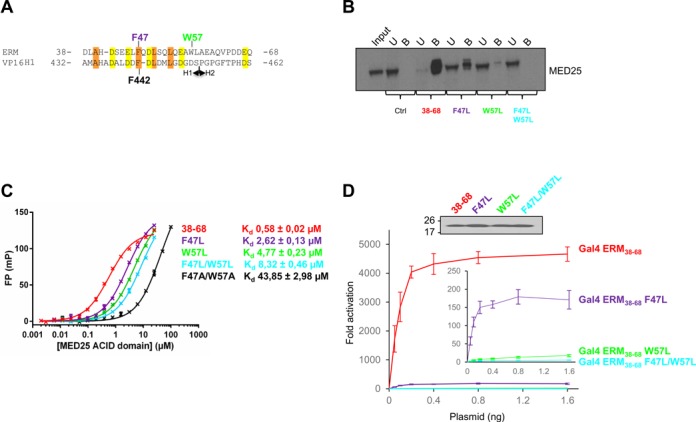
Mutational analysis. (A) Sequence alignment of ERM38–68 with the Herpes simplex VP16 H1 subdomain. Conserved hydrophobic residues are boxed in orange, acidic residues in yellow. F47 (purple) and W57 (green) of ERM and F442 of VP16 H1 are indicated. (B) Endogenous MED25 binds specifically to ERM38–68. Nuclear extracts from DAMI cells were incubated with immobilized Halo-Tag, Halo-Tag ERM38–68, Halo-Tag ERM36–68 F47L, Halo-Tag ERM36–68 W57L and Halo-Tag ERM38–68 F47L/W57L. MED25 not associated with ERM wild-type and derivatives present in the supernatant was saved as the unbound fraction (U). Samples were separated by SDS-PAGE and MED25 was detected by western-blot. Input lane and unbound fraction represents 10% of total extract used in the pull-down, Bound fractions (B, 100%). (C) Fluorescence polarization (FP) peptide binding assay for ERM38–68, ERM38–68 F47L, ERM38–68 W57L, ERM38–68 F47L/W57L and ERM38–68 F47A/W57A. Measured Kd are indicated. TAMRA-labeled peptides were at 4 nM. (D) Effects of ERM TAD mutations on transcriptional activation. U2OS cells were transfected with increasing amounts of Gal4-ERM38–68 wild-type and derivatives together with a (Gal4)5-E1B-Luc reporter construct. In the inset, the data for ERM38–68 has been excluded and the scale expanded (Upper panel). The level of expression of the Gal4 derivatives was monitored by western-blot using an anti-Gal antibody.
Fluorescence polarization (FP) binding assays were subsequently performed in order to determine the dissociation constant (Kd) between MED25 ACID/PTOV and the different mutants of ERM38–68 (Figure 3C). Native ERM38–68 binds to MED25 ACID/PTOV with a Kd of 0.6 μM, in agreement with our previous studies by ITC (15). Importantly, the F47L, W57L and F47L/W57L mutants bind to MED25 with Kd of respectively 2.6, 4.8 and 8.3 μM, showing a 4.5-, 8.2- and 14.3-fold drop in affinity relative to wild-type ERM38–68 (Figure 3C). Alanine substitutions at these positions (F47A/W57A) had the most dramatic effect, showing a 75-fold drop in affinity (Figure 3C). These results pinpoint a critical role for F47 and W57 and suggest that aromatic/hydrophobic character is an important criterion for effective binding of ERM to MED25.
Finally, we assessed the effect of these mutations on the ability of ERM to activate transcription. For this assay (Figure 3D), ERM38–68 and the three mutants (F47L, W57L and F47L/W57L) were fused to the DNA-binding domain of Gal4 and their activity for a luciferase reporter gene was measured relative to Gal4 alone. As shown in Figure 3D, the F47L or W57L mutations drastically reduced the ability of ERM to activate transcription (3.9 and 0.3% residual transactivation, respectively). The double mutant F47L/W57L was even more severely affected, with a residual transcriptional activity less than 0.1%. Collectively, all these data indicate that the affinity of ERM TAD-MED25 ACID/PTOV interaction strongly correlates with ERM transcriptional activity and demonstrate a crucial role of the hydrophobic residues F47 and W57.
The ERM38–68 TAD-MED25 ACID/PTOV interface
To map the ERM38–68 TAD binding site on MED25 ACID/PTOV, 1H-15N HSQC titration experiments of unlabelled ERM against 15N-labelled MED25 were performed. Addition of an equimolar amount of ERM38–68 to MED25 resulted in large CSD and severe line broadening of most of the resonances (Figure 4A). As for the titration of ERM38–68 by MED25, both slow and fast exchange behaviours are observed for the resonances of MED25 as the concentration of ERM38–68 is increased (Supplementary Figure S3). A similar result has been previously observed for the binding of the KIX domain of CBP to the phosphorylated kinase-inducible activation domain of CREB which suggests the existence of an ensemble of transient encounter complexes in protein–protein association (34). In accordance, the global modification of the spectrum associated with the line broadening suggests a large interaction interface associated with conformational heterogeneity. However, this makes the localization of the binding site by the titration experiments inconclusive. Thus, in order to circumvent the inherent difficulties of studying fuzzy complexes and be able to better define the intermolecular interface, the mutants of ERM38–68 with altered affinity for MED25 were used.
Figure 4.
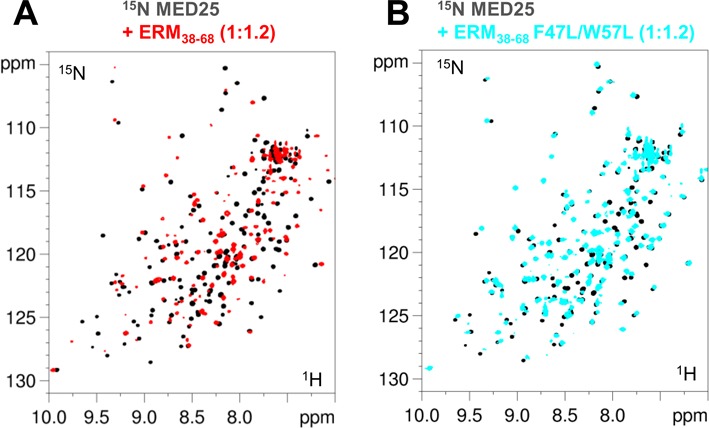
Spectra of MED25. 2D [1H, 15N] HSQC spectra of 100μM 15N,13C-MED25 free in solution (black) or with 1.2 molar amount of (A) ERM38–68 (overlaid in red) and (B) F47L/W57L-ERM38–68 (overlaid in blue).
F47L, W57L and F47L/W57L ERM peptides were separately titrated into 15N-labeled MED25 ACID/PTOV. We anticipated that ERM mutants with lower affinity for MED25 would improve the quality of the ERM38–68 bound MED25 NMR spectra and indeed, found this to be the case (compare Figure 4A with 4B). Given that NMR experiments were carried out at MED25 concentrations 100-fold above the Kd, direct interaction between MED25 and mutants ERM can still be observed (Figure 4B). Comparison of 2D spectra of MED25 bound to the various mutated ERM38–68 showed that the resonances corresponding to a MED25 amino acid residue in each of these complexes has a linear pattern of CSD (Figure 5 and Supplementary Figure S4). Given the Kd that were measured using FP experiments and the relatively high concentration used for the NMR experiments, we expect full saturation of MED25 at a 1/1 ratio for the whole set of ERM peptides. The CCSD thus suggests a range of affinities and their analysis (26) indeed shows that the CSD of each mutant at saturation of MED25 is linearly correlated to its Kd (Supplementary Figure S4). From these data, we can conclude that the ERM mutants sample the same conformational ensemble corresponding to the bound state, although with different weighted populations and importantly that all mutant ERM peptides have a common binding interface for MED25. This linear behaviour was additionally used to link most of the resonances in ERM38–68-bound MED25 [1H, 15N] HSQC to the assignment of free MED25 given that the observed line broadening excluded further acquisition of 3D spectra. The global intensity decrease of the MED25 resonances due to the interaction were inversely correlated with the Kd of ERM mutants for MED25 (Figure 5).
Figure 5.
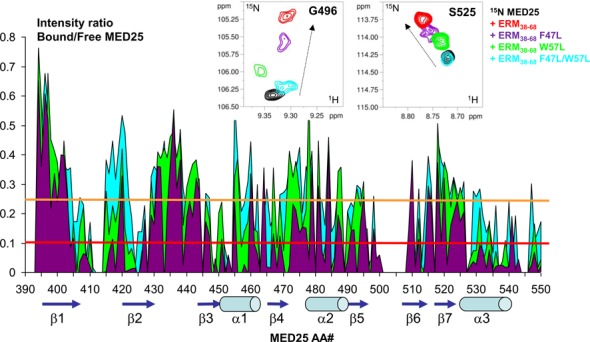
Mapping of the MED25 interaction site with ERM38–68 based on resonance line broadening in 2D spectra. (Upper panel) Details of overlaid 2D [1H, 15N] HSQC spectra of 125μM 15N-MED25 free in solution (black) or with 1.2 molar amount of ERM38–68 (red), F47L-ERM38–68 (purple), F47L/W57L-ERM38–68 (cyan) or 1.6 molar amount of W57L-ERM38–68 (green). (Lower panel) Graphic representation of relative intensity ratio I/I0 of corresponding resonances in [1H,15N] HSQC spectra of MED25 with 1.2 molar amount of F47L/W57L-ERM38–68 (cyan) and F47L-ERM38–68 (purple) or 1.6 molar amount of W57L-ERM38–68 (green), versus free in solution. Resonances between amino acid residues 501 and 508 are weak and were not included in the analysis, as well as the last 551–556 residues. The intensity profile for W57L-ERM38–68 interaction (green) was normalized on the terminal residue to correct for concentration of the sample. The orange threshold corresponds to the average I/I0 intensity ratio for the F47L/W57L-ERM38–68, the red one to the corresponding I/I0 average minus one I/I0 standard deviation. Peak picking was performed on [1H, 15N] HSQC spectra recorded as in Figure 4. Secondary structure elements are represented below the graphic as defined in the NMR structure of MED25 (PBD ID: 2L23, (16)). Numbering provided below the graphic corresponds to the ACID/PTOV domain in full length MED25.
The location on MED25 of the ERM38–68 binding site was thus mapped via the signal intensity loss between the free and bound MED25 (Figure 5, Supplementary Figures S5 and S6). When visualized onto the structure of MED25 ACID/PTOV, the most significant drop in HN peak intensity occurs for two distinct surfaces on opposite faces of the β-barrel (Figure 6, Supplementary Figures S5 and S6). One binding site involves strands β2, β1, β3 and helix α3 and the strongest effects were observed for amino acids in β3 and α3 (Figure 6A and C). This site overlaps with the previously identified VP16 H1 binding site (17). The other site, which is located on the opposite face of the MED25 ACID/PTOV β-barrel, is formed by helix α1 and strands β6, β7, β4 (Figure 6B and D). This site constitutes part of the MED25/VP16 H2 binding site (16,18).
Figure 6.
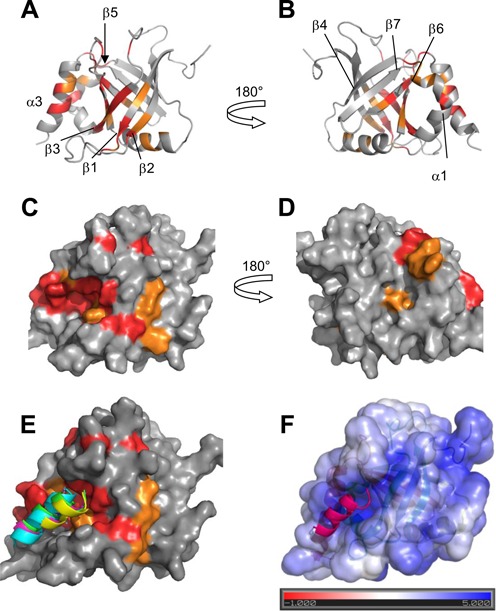
MED25 interaction site with ERM38–68. (A) and (B) Ribbon representation of MED25 (PBD ID: 2L23, (16)) with labelling of secondary structures. (C) and (D) Molecular surface of MED25. (A) and (C) correspond to VP16 H1 binding site and (B) and (D) to VP16 H2 binding site, as defined in results. (B) and (D) representations were obtained by a 180° rotation around the vertical axis from (A) and (C), respectively. Colour coding is orange for residues with I/I0 below the average I/I0 and red, below the I/I0 average minus I/I0 standard deviation as defined from measurements shown in Figure 5 for interaction with F47L/W57L. Representations were created using Pymol. (E) Structural models of the ERM50–61 peptide bound to the ACID/PTOV domain. Surface view of the MED25 protein with CSD as in (C). Superimposed peptides are represented as ribbons and correspond to the four highest score models of cluster 2 that best fit the experimental data. (F) Structural model of one ERM50–61 peptide bound to the ACID/PTOV domain. The solvent-accessible surface of MED25 is coloured according to the electrostatic potential calculated using the APBS (Adaptative Poisson-Boltzman Solver) tools within Pymol (41) The colour scale ranges from red at −1 kT/e to blue at 5 kT/e. The dielectric constants were set at 2 for the protein and 80 for the solvent.
To obtain additional structural information on the ERM38–68 peptide location at the MED25 surface, paramagnetic relaxation enhancement experiments were used. Mutation of Ala41 or Pro64 into a cysteine residue allowed the attachment of a spin label (nitroxide) at the N- or C-terminus of the ERM38–68 peptide. The spin label enhances the relaxation rate of the nuclei in a sphere of influence of about 30 Å radius, causing resonance broadening. Most of the detected resonances in the ERM38–68/MED25 complex are affected by the spin label, showing again that the interaction surface is extensive (Supplementary Figure S7). Comparison of the 2D spectra of MED25 in presence of ERM38–68 spin-labelled at its N or C-terminus however showed a preferential occupation of the MED25 surface by the C-terminus of the peptide, as the global intensity decrease is larger with the nitroxide label at the C-terminus of ERM (Supplementary Figure S7, compare A and B).
MED25 ACID/PTOV mutants selectively abrogate the ERM–MED25 interaction
Since ERM38–68 interacts with the VP16 H1 binding site but shows additional contacts with the VP16 H2 site on MED25 ACID/PTOV, we finally confirmed the preferred mode of interaction using MED25 point mutants and peptide competition assay. Guided by both our experimentally observed chemical shift changes on the MED25 ACID/PTOV domain (Figures 5 and 6) and previous studies done by us and others (15–18), Q451E (located at the centre of the VP16 H1 binding site) and R466E (located at the centre of the VP16 H2 binding site) mutants were chosen for further analysis.
In FP assays, the Q451E MED25 mutant displayed a 5.6-fold decrease in affinity for ERM38–68 while the R466E mutant showed binding affinity similar to that of the wild-type ACID/PTOV domain (Figure 7A). Subsequent NMR analyses confirmed that the Q451E MED25 mutant barely interacts with ERM F47L/W57L (Supplementary Figure S8). We next carried out a series of competition experiments to investigate whether VP16 H1 and VP16 H2 peptides can compete with ERM38–68 for binding to MED25. Binding of these two peptides was first determined by both FP and ITC assays (Figure 7B). In FP, VP16 H1 bound to MED25 with an apparent Kd of 0.15 μM and VP16 H2 bound to MED25 with 13 times lower affinity (Kd of 2 μM) compared with VP16 H1 (Figure 7B). The binding affinities of VP16 H1 and VP16 H2 peptides obtained in our ITC assays (0.2 and 2.6 μM, respectively) are in very good agreement with those obtained by FP (Figure 7B). In the competition assays, we found that unlabelled ERM38–68 and VP16 H1 can efficiently displace TAMRA-ERM38–68 from MED25 ACID/PTOV with IC50 value of 1.5 and 0.3 μM, respectively (Figure 7C). VP16 H2, which binds with much weaker affinity to the opposite face of MED25 (Figure 7B), was able to disrupt the ERM38–68/MED25 complex but only at much higher concentrations, with an IC50 value of ∼21 μM (Figure 7C). This is consistent with our previous findings that VP16 H2 can also bind the H1 pocket with low affinity (16). We thus conclude that the specific interaction between ERM38–68 and MED25 ACID/PTOV overlapped with the previously identified VP16 H1 binding site.
Figure 7.
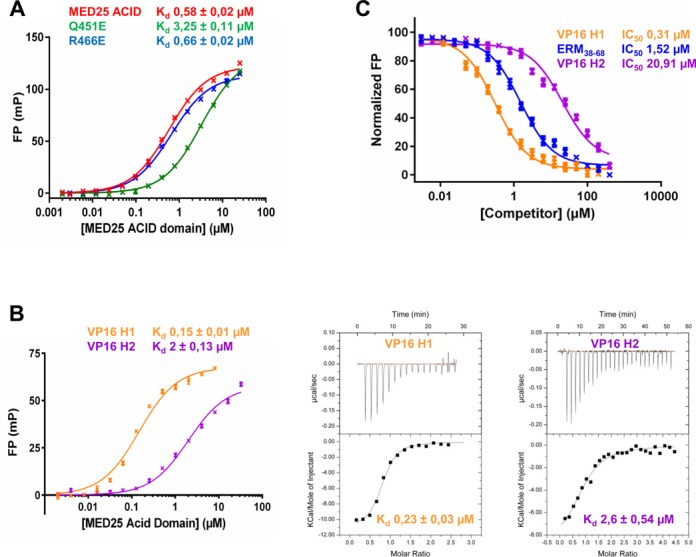
Competition between ERM38–68, VP16 H1 and VP16 H2 for binding to MED25 ACID/PTOV. (A) Comparison of the dissociation constant (Kd) values for the binding of MED25 ACID/PTOV domain and its mutants (Q541E and R466E) to ERM38–68. (B) (left) FP experiments and (right) ITC data of a titration of MED25 ACID/PTOV with VP16 H1 or VP16 H2 peptides to measure the binding affinity. (C) IC50 values for the displacement of TAMRA-ERM38–68 (4 nM) from MED25 ACID/PTOV (0.2 μM) by unlabelled ERM38–68 (LogEC50 = 0.18 ± 0.03), VP16 H1 (LogEC50 = −0.51 ± 0.04) and VP16 H2 (LogEC50 = 1.32 ± 0.04).
Structural model of ERM TAD in complex with MED25 ACID/PTOV domain
Because our data suggest that there is not a single mode of interaction between ERM TAD and MED25 ACID/PTOV, the structure of the complex cannot be determined strictly based on experimental data. Furthermore, given the important broadening of the resonances in the complex, no intermolecular NOEs were detected. Structural models which are meant to be representative of ERM specific binding but are not exclusive, were thus generated using ambiguous interaction restraints. Experimental data from NMR spectroscopy were used to construct a helical ERM model peptide for the interaction (Figure 1B and Supplementary Figure S1A) and to define a set of distance restraints for the docking simulation (Figure 5, Supplementary Figures S5 and S6). The model of ERM L50-A61 peptide was used as a starting structure for docking calculations into the VP16 H1 MED25 ACID/PTOV binding site by the HADDOCK program (27,30). The four highest score structures of the two top clusters (on three clusters) that showed good statistics (Supplementary Table S2) were analyzed. Peptide orientation of cluster 2 best fitted the experimental constraints (Figure 6E). The peptide is located along a positively charge groove ((41), Figure 6F). The consistence between the binding pocket defined using the CSD and the charged pocket (compare Figure 6E and 6F) indeed argues in favour of the involvement of electrostatic interactions in the ERM/MED25 specific complex formation. In addition, the models (Figure 6E, 6F and Supplementary Figure S9) predict that the side chain of W57 is buried in the previously described hydrophobic furrow ((17), Supplementary Figure S9A). Side chain of W57 is also in close proximity to H499 (Supplementary Figure S9B), a residue that was shown to be important for the VP16/MED25 interaction (17). Finally, the indole ring of W57 is in close proximity to the amide group of Q451 residue whose mutation into E451 decreases the interaction with ERM. Such amino–aromatic interactions are reported, corresponding to van der Waals contacts with the π electron clouds of the aromatic residues (42).
DISCUSSION
The results presented here reveal how the TAD of ERM, a PEA3 group member of the Ets family, binds to the PTOV domain of MED25 at the molecular level. This is the first study reporting detailed molecular insights into MED25 interaction with a human transcription factor, which is of specific interest as overexpresssion of PEA3 transcription factors has oncogenic properties (43).
The first feature that is observed for ERM binding to MED25 is a folding upon binding mechanism (Figures 1B and 2B, Supplementary Figures S1A and S2B). Several studies, particularly those with NMR spectroscopy, revealed that TADs of many transcription factors are disordered in solution but become structured upon binding to their interaction partners. This has been shown for example in VP16 binding to TFIIB (44) and Tfb1 subunit of TFIIH (38), or RelA (45) and Hif-1α (46) binding to the TAZ1 domain of CBP/p300 and the TAD of Gcn4 in complex with Gal11/MED15 (35). We additionally observed an important broadening of numerous resonances in the 2D 1H-15N spectrum of ERM-bound MED25, indicative of multiple bound conformations. Many eukaryotic TADs were also reported to form ‘fuzzy complexes’ with their binding partners (35,47–51) i.e. the interface cannot be described by a single conformation. Our results strongly suggest that the ERM/MED25 complex is another example of a fuzzy complex. In addition, our NMR titration results show that cross-peaks of several ERM amino acid residues shift gradually at sub-stoichiometric amounts of MED25, indicating that different ERM conformations are in fast exchange on the NMR time scale between free and bound forms (Figure 2 and Supplementary Figure S3A). At late titration points of MED25, a drastic spectral change of ERM is observed that shows many characteristics of a folded peptide (Supplementary Figure S2). The reverse phenomenon is observed for the titration of MED25 by ERM, with slow exchange regime for sub-stoichiometry of ERM and fast exchange regime above the 1 to 1 saturation ratio for all the resonances affected by the interaction. The same NMR spectral characteristics were reported during titration of the KIX domain to phospho-KID TAD (34). Our data are in agreement with the model of an encounter complex (34–35,52–53) in which the ERM38–68 peptide scans the MED25 surface, in an unspecific manner driven by weak electrostatic interactions. Another interpretation of the data could be the presence of a second additional binding site for ERM of weak affinity. These two types of potential additional binding would correspond to the weak affinity mode of interaction (gradual chemical shift indicative of fast exchange) that we detect in our NMR titrations of MED25, and reversely ERM (Figure 2, Supplementary Figures S2 and S3).
In this work, we have used NMR methods to map the binding site of MED25 for ERM38–68. Technically, however, the study of the fuzzy complexes such as ERM/MED25 remains challenging (49). Here, to obtain structural information despite the limitations imposed by the complex dynamics, we took advantage of the gradual decrease in affinity of ERM for MED25 obtained by mutating its aromatic residues into Leucine (Figure 3). The residues in the ERM-bound MED25 with all of the ERM mutants being tested exhibited CCSD, showing that each complex is part of the same dynamic population ensemble. This provides additional evidence that the bound conformation of ERM is a rapidly inter-converting ensemble of conformations. The interface is conserved within all of the complexes but the decreased affinity allows to highlight the core of the binding pocket(s). The CSD observed between free and bound MED25 ACID/PTOV however affected resonances assigned to the two previously described binding sites on MED25 for VP16 H1 and H2. Similar observations were made by probing the interaction of the isolated VP16 H1 or H2 TAD domains with MED25 (16–18). Given the redundancy of all the TAD sequences and their promiscuity, it might not be surprising that both binding sites can be occupied by the ERM TAD. For example, the N-terminal TAD of the tumour suppressor p53 directly contacts TBP (54), TAF9 (TAFII31) (55), the p62/tfb1 subunit of TFIIH (56), the Mediator complex subunit MED17 (57) and p300 (58).
Mutation of residue Q451E in the H1 VP16 binding site was however shown to decrease the affinity of ERM to MED25, indicating its contribution to the binding (Figure 7). Moreover, we have previously shown that this ERM-binding defective MED25 Q451E mutant was unable to efficiently rescue ERM transactivation in MED25-depleted cells (15). Based on these data, a functional interaction of ERM with VP16 H1 binding site can be deduced. On the contrary the direct interaction of ERM with VP16 H2 binding site cannot be proven as none of the mutations affecting this binding site appeared sufficient to affect the Kd of the ERM/MED25 interaction (R466E and data no shown). Moreover, given that ERM38–68 and VP16 H1 peptides efficiently displace ERM38–68 from MED25, it can be deduced that they target a common binding site with comparable affinities (Figure 7). Further clarification might await the identification of additional proteins that could occupy the VP16 H2 binding site of MED25 or play in allosteric regulation as seen for example by accommodation by the KIX domain of the CREB-binding protein of two TAD domains with two distinct binding pockets (59).
Our investigation suggested that the binding sites on MED25 for ERM and VP16 may overlap but the interactions with the TAD of ERM appear slightly different than those seen with VP16. Notably, the ERM TAD is 30-amino-acid-long as compared to that of VP16 (80-residue fragment) and does not contain a VP16-like bipartite binding motif for MED25. The overall affinity for the full-length VP16 TAD is significantly higher (50 nM) than for binding of either of the isolated H1 (∼0.2 μM) or H2 (∼2 μM) subdomains suggesting that the two motifs bind cooperatively to MED25 (17,18). We note, however, that our VP16 H1 binds to MED25 with a higher affinity than what has been previously reported (17). In comparison, ERM38–68 binding to MED25 is weaker (0.6 μM), suggesting that the ERM TAD only reproduces the binding affinity of the isolated VP16 subdomains H1 or H2. Another key feature observed for ERM is that two hydrophobic amino acids (F47 and W57) are essential for high-affinity binding and transcriptional activity of ERM. Interestingly we and others previously noted that the TADs of ERM (15), ATF6α (12,60–61) and Dreb2a (13) exhibit some sequence similarity to the VP16 H1 subdomain. Of particular note, mutation of just the phenylalanine residue within these regions (F47 in ERM, F442 in VP16 H1 and F62 in ATF6α) strongly impaired MED25 recruitment. Our experimental NMR data of ERM38–68 peptide in complex with MED25 also indicate that W57 is crucial for this interaction and this is supported by our mutational analysis. The insertion of this extra hydrophobic residue could contribute to the high affinity of ERM for MED25 and its efficient transactivation activity. Insertion of bulky hydrophobic residues was indeed demonstrated to create efficient activation domains for Gcn4 TAD (62).
In conclusion, close examination of the molecular mechanisms by which individual Mediator subunits are recruited by TADs has been reported for only a few cases (MED15/SREBP, VP16/MED25, GCN4/MED15) (13,16–18,35,63–64). We have revealed here the first molecular insights into MED25 interaction with the human oncogenic ERM transcription factor. It will be of interest in the future to obtain additional data to examine if the dynamic character of the TAD interaction with Mediator subunits is a parameter that sustains the very function of activation.
Supplementary Material
Acknowledgments
US Open Science Grid (OSG) are acknowledged for the use of web portals, computing and storage facilities. We thank Prof. A. Bonvin (Utrecht University, Netherlands) for his support in using the Haddock Web interface, Dr F. Bontems (CNRS, France) for preliminary NMR measurements of ERM38–68 and Dr G. Lippens (CNRS, France) for helpful discussions. We thank the anonymous reviewers for their challenging and lively comments on the initial manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Région Nord; CNRS; Pasteur Institute of Lille; European Community (FEDER); French Research Ministry; University of Sciences and Technologies of Lille I (to NMR facilities); TGE RMN THC FR-3050, France (to NMR facilities); ‘Projets Emergents’ Nord-Pas-de-Calais Regional Council; Ligue Nationale contre le Cancer (comité Pas-de-Calais); WeNMR project (European FP7 e-Infrastructure grant, contract no. 261572, www.wenmr.eu), supported by the European Grid Initiative (EGI) through the national GRID Initiatives of Belgium, France, Italy, Germany, the Netherlands, Poland, Portugal, Spain, UK, South Africa, Malaysia,Taiwan, the Latin America GRID infrastructure via the Gisela project. Funding for open access charge: CNRS.
Conflict of interest statement. None declared.
REFERENCES
- 1.Green M.R. Eukaryotic transcription activation: right on target. Mol. Cell. 2005;18:399–402. doi: 10.1016/j.molcel.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 2.Borggrefe T., Yue X. Interactions between subunits of the Mediator complex with gene-specific transcription factors. Semin. Cell Dev. Biol. 2011;22:759–768. doi: 10.1016/j.semcdb.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 3.Poss Z.C., Ebmeier C.C., Taatjes D.J. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 2013;48:575–608. doi: 10.3109/10409238.2013.840259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balamotis M.A., Pennella M.A., Stevens J.L., Wasylyk B., Belmont A.S., Berk A.J. Complexity in transcription control at the activation domain-mediator interface. Sci. Signal. 2009;2:ra20. doi: 10.1126/scisignal.1164302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malik S., Roeder R.G. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat. Rev. Genet. 2010;11:761–772. doi: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taatjes D.J., Marr M.T., Tjian R. Regulatory diversity among metazoan co-activator complexes. Nat. Rev. Mol. Cell. Biol. 2004;5:403–410. doi: 10.1038/nrm1369. [DOI] [PubMed] [Google Scholar]
- 7.Mittler G., Stuhler T., Santolin L., Uhlmann T., Kremmer E., Lottspeich F., Berti L., Meisterernst M. A novel docking site on Mediator is critical for activation by VP16 in mammalian cells. EMBO J. 2003;22:6494–6504. doi: 10.1093/emboj/cdg619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang F., DeBeaumont R., Zhou S., Naar A.M. The activator-recruited cofactor/Mediator coactivator subunit ARC92 is a functionally important target of the VP16 transcriptional activator. Proc. Natl. Acad. Sci. U.S.A. 2004;101:2339–2344. doi: 10.1073/pnas.0308676100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto S., Eletsky A., Szyperski T., Hay J., Ruyechan W.T. Analysis of the varicella-zoster virus IE62 N-terminal acidic transactivating domain and its interaction with the human mediator complex. J. Virol. 2009;83:6300–6305. doi: 10.1128/JVI.00054-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang M., Hay J., Ruyechan W.T. Varicella-zoster virus IE62 protein utilizes the human mediator complex in promoter activation. J. Virol. 2008;82:12154–12163. doi: 10.1128/JVI.01693-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roupelieva M., Griffiths S.J., Kremmer E., Meisterernst M., Viejo-Borbolla A., Schulz T., Haas J. Kaposi's sarcoma-associated herpesvirus Lana-1 is a major activator of the serum response element and mitogen-activated protein kinase pathways via interactions with the Mediator complex. J. Gen. Virol. 2010;91:1138–1149. doi: 10.1099/vir.0.017715-0. [DOI] [PubMed] [Google Scholar]
- 12.Sela D., Conkright J.J., Chen L., Gilmore J., Washburn M.P., Florens L., Conaway R.C., Conaway J.W. Role for human mediator subunit MED25 in recruitment of mediator to promoters by endoplasmic reticulum stress-responsive transcription factor ATF6alpha. J. Biol. Chem. 2013;288:26179–26187. doi: 10.1074/jbc.M113.496968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aguilar X., Blomberg J., Brannstrom K., Olofsson A., Schleucher J., Bjorklund S. Interaction studies of the human and Arabidopsis thaliana Med25-ACID proteins with the herpes simplex virus VP16- and plant-specific Dreb2a transcription factors. PLoS One. 2014;9:e98575. doi: 10.1371/journal.pone.0098575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blomberg J., Aguilar X., Brannstrom K., Rautio L., Olofsson A., Wittung-Stafshede P., Bjorklund S. Interactions between DNA, transcriptional regulator Dreb2a and the Med25 mediator subunit from Arabidopsis thaliana involve conformational changes. Nucleic Acids Res. 2012;40:5938–5950. doi: 10.1093/nar/gks265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verger A., Baert J.L., Verreman K., Dewitte F., Ferreira E., Lens Z., de Launoit Y., Villeret V., Monte D. The Mediator complex subunit MED25 is targeted by the N-terminal transactivation domain of the PEA3 group members. Nucleic Acids Res. 2013;41:4847–4859. doi: 10.1093/nar/gkt199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bontems F., Verger A., Dewitte F., Lens Z., Baert J.L., Ferreira E., Launoit Y.D., Sizun C., Guittet E., Villeret V., et al. NMR structure of the human Mediator MED25 ACID domain. J. Struct. Biol. 2011;174:245–251. doi: 10.1016/j.jsb.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 17.Milbradt A.G., Kulkarni M., Yi T., Takeuchi K., Sun Z.J., Luna R.E., Selenko P., Naar A., Wagner G. Structure of the VP16 transactivator target in ARC/Mediator. Nat. Struct. Biol. 2011;18:410–415. doi: 10.1038/nsmb.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vojnic E., Mourao A., Seizl M., Simon B., Wenzeck L., Lariviere L., Baumli S., Baumgart K., Meisterernest M., Sattler M., et al. The mediator MED25 activator interaction domain: structure and cooperative binding of VP16 subdomains. Nat. Struct. Biol. 2011;18:404–409. doi: 10.1038/nsmb.1997. [DOI] [PubMed] [Google Scholar]
- 19.Baert J.L., Monte D., Musgrove E.A., Albagli O., Sutherland R.L., de Launoit Y. Expression of the PEA3 group of ETS-related transcription factors in human breast-cancer cells. Int. J. Cancer. 1997;70:590–597. doi: 10.1002/(sici)1097-0215(19970304)70:5<590::aid-ijc17>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 20.Nedialkov Y.A., Shooltz D.D., Triezenberg S.J. Purification and protein interaction assays of the VP16C transcription activation domain. Methods Enzymol. 2003;370:522–535. doi: 10.1016/S0076-6879(03)70044-5. [DOI] [PubMed] [Google Scholar]
- 21.Blais A., Monté D., Pouliot F., Labrie C. Regulation of the human cyclin-dependent kinase inhibitor p18INK4c by the transcription factors E2F1 and Sp1. J. Biol. Chem. 2002;277:31679–31693. doi: 10.1074/jbc.M204554200. [DOI] [PubMed] [Google Scholar]
- 22.Goddard T.D., Kneller D. G. SPARKY 3. San Francisco, CA: University of California; [Google Scholar]
- 23.Tamiola K., Acar B., Mulder F.A. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 2010;132:18000–18003. doi: 10.1021/ja105656t. [DOI] [PubMed] [Google Scholar]
- 24.Marsh J.A., Singh V.K., Jia Z., Forman-Kay J.D. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H., Neal S., Wishart D.S. RefDB: a database of uniformly referenced protein chemical shifts. J. Biomol. NMR. 2003;25:173–195. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 26.Stollar E.J., Lin H., Davidson A.R., Forman-Kay J.D. Differential dynamic engagement within 24 SH3 domain: peptide complexes revealed by co-linear chemical shift perturbation analysis. PLoS One. 2012;7:e51282. doi: 10.1371/journal.pone.0051282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Vries S.J., van Dijk M., Bonvin A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 28.DeLano W.L. The PyMOL Molecular Graphics System (2002) on World Wide Web. 2002 http://www.pymol.org. [Google Scholar]
- 29.Wassenaar T.A., van Dijk M., Loureiro-Ferreira N., van der Schot G., De Vries S.J., Schmitz C., van der Zwan J., Boelens R., Giachetti A., Ferella L., et al. WeNMR: structural biology on the grid. J. Grid. Comp. 2012;10:743–767. [Google Scholar]
- 30.de Vries S.J., Bonvin A.M. CPORT: a consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS One. 2011;6:e17695. doi: 10.1371/journal.pone.0017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Defossez P.A., Baert J.L., Monnot M., de Launoit Y. The ETS family member ERM contains an alpha-helical acidic activation domain that contacts TAFII60. Nucleic Acids Res. 1997;25:4455–4463. doi: 10.1093/nar/25.22.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lens Z., Dewitte F., Monté D., Baert J.L., Bompard C., Sénéchal M., Van Lint C., de Launoit Y., Villeret V., Verger A. Solution structure of the N-terminal transactivation domain of ERM modified by SUMO-1. Biochem. Biophys. Res. Commun. 2010;399:104–110. doi: 10.1016/j.bbrc.2010.07.049. [DOI] [PubMed] [Google Scholar]
- 33.Ishida T., Kinoshita K. Prediction of disordered regions in proteins based on the meta approach. Bioinformatics. 2008;24:1344–1348. doi: 10.1093/bioinformatics/btn195. [DOI] [PubMed] [Google Scholar]
- 34.Sugase K., Dyson H.J., Wright P.E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 35.Brzovic P.S., Heikaus C.C., Kisselev L., Vernon R., Herbig E., Pacheco D., Warfield L., Littlefield P., Baker D., Klevit R.E., et al. The acidic transcription activator gcn4 binds the mediator subunit gal11/med15 using a simple protein interface forming a fuzzy complex. Mol. Cell. 2011;44:942–953. doi: 10.1016/j.molcel.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chabot P.R., Raiola L., Lussier-Price M., Morse T., Arseneault G., Archambault J., Omichinski J.G. Structural and functional characterization of a complex between the acidic transactivation domain of EBNA2 and the Tfb1/p62 subunit of TFIIH. PLoS Pathog. 2014;10:e1004042. doi: 10.1371/journal.ppat.1004042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jennings B.H., Pickles L.M., Wainwright S.M., Roe S.M., Pearl L.H., Ish-Horowicz D. Molecular recognition of transcriptional repressor motifs by the WD domain of the Groucho/TLE corepressor. Mol. Cell. 2006;22:645–655. doi: 10.1016/j.molcel.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 38.Langlois C., Mas C., Di Lello P., Jenkins L.M., Legault P., Omichinski J.G. NMR structure of the complex between the Tfb1 subunit of TFIIH and the activation domain of VP16: structural similarities between VP16 and p53. J. Am. Chem. Soc. 2008;130:10596–10604. doi: 10.1021/ja800975h. [DOI] [PubMed] [Google Scholar]
- 39.Piskacek S., Gregor M., Nemethova M., Grabner M., Kovarik P., Piskacek M. Nine-amino-acid transactivation domain: establishment and prediction utilities. Genomics. 2007;89:756–768. doi: 10.1016/j.ygeno.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Sullivan S.M., Horn P.J., Olson V.A., Koop A.H., Niu W., Ebright R.H., Triezenberg S.J. Mutational analysis of a transcriptional activation region of the VP16 protein of herpes simplex virus. Nucleic Acids Res. 1998;26:4487–4496. doi: 10.1093/nar/26.19.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burley S.K., Petsko G.A. Amino-aromatic interactions in proteins. FEBS Lett. 1986;203:139–143. doi: 10.1016/0014-5793(86)80730-x. [DOI] [PubMed] [Google Scholar]
- 43.Oh S., Shin S., Janknecht R. ETV1, 4 and 5: An oncogenic subfamily of ETS transcription factors. Biochim. Biophys. Acta. 2012;1826:1–12. doi: 10.1016/j.bbcan.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jonker H.R., Wechselberger R.W., Boelens R., Folkers G.E., Kaptein R. Structural properties of the promiscuous VP16 activation domain. Biochemistry. 2005;44:827–839. doi: 10.1021/bi0482912. [DOI] [PubMed] [Google Scholar]
- 45.Mukherjee S.P., Behar M., Birnbaum H.A., Hoffmann A., Wright P.E., Ghosh G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-kappaB-driven transcription. PLoS Biol. 2013;11:e1001647. doi: 10.1371/journal.pbio.1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dames S.A., Martinez-Yamout M., De Guzman R.N., Dyson H.J., Wright P.E. Structural basis for Hif-1 alpha /CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuxreiter M., Tompa P., Simon I., Uversky V.N., Hansen J.C., Asturias F.J. Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol. 2008;4:728–737. doi: 10.1038/nchembio.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu J., Perumal N.B., Oldfield C.J., Su E.W., Uversky V.N., Dunker A.K. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tompa P., Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 50.Warfield L., Tuttle L.M., Pacheco D., Klevit R.E., Hahn S. A sequence-specific transcription activator motif and powerful synthetic variants that bind Mediator using a fuzzy protein interface. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E3506–E3513. doi: 10.1073/pnas.1412088111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andresen C., Helander S., Lemak A., Fares C., Csizmok V., Carlsson J., Penn L.Z., Forman-Kay J.D., Arrowsmith C.H., Lundstrom P., et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 2012;40:6353–6366. doi: 10.1093/nar/gks263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scanu S., Foerster J.M., Ullmann G.M., Ubbink M. Role of hydrophobic interactions in the encounter complex formation of the plastocyanin and cytochrome f complex revealed by paramagnetic NMR spectroscopy. J. Am. Chem. Soc. 2013;135:7681–7692. doi: 10.1021/ja4015452. [DOI] [PubMed] [Google Scholar]
- 53.Schneider R., Maurin D., Communie G., Kragelj J., Hansen D.F., Ruigrok R.W., Jensen M.R., Blackledge M. Visualizing the molecular recognition trajectory of an intrinsically disordered protein using multinuclear relaxation dispersion NMR. J. Am. Chem. Soc. 2015;137:1220–1229. doi: 10.1021/ja511066q. [DOI] [PubMed] [Google Scholar]
- 54.Chang J., Kim D.H., Lee S.W., Choi K.Y., Sung Y.C. Transactivation ability of p53 transcriptional activation domain is directly related to the binding affinity to TATA-binding protein. J. Biol. Chem. 1995;270:25014–25019. doi: 10.1074/jbc.270.42.25014. [DOI] [PubMed] [Google Scholar]
- 55.Buschmann T., Lin Y., Aithmitti N., Fuchs S.Y., Lu H., Resnick-Silverman L., Manfredi J.J., Ronai Z., Wu X. Stabilization and activation of p53 by the coactivator protein TAFII31. J. Biol. Chem. 2001;276:13852–13857. doi: 10.1074/jbc.M007955200. [DOI] [PubMed] [Google Scholar]
- 56.Di Lello P., Jenkins L.M., Jones T.N., Nguyen B.D., Hara T., Yamaguchi H., Dikeakos J.D., Appella E., Legault P., Omichinski J.G. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell. 2006;22:731–740. doi: 10.1016/j.molcel.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 57.Meyer K.D., Lin S.C., Bernecky C., Gao Y., Taatjes D.J. p53 activates transcription by directing structural shifts in Mediator. Nat. Struct. Mol. Biol. 2010;17:753–760. doi: 10.1038/nsmb.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Avantaggiati M.L., Ogryzko V., Gardner K., Giordano A., Levine A.S., Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- 59.Bruschweiler S., Konrat R., Tollinger M. Allosteric communication in the KIX domain proceeds through dynamic repacking of the hydrophobic core. ACS Chem. Biol. 2013;8:1600–1610. doi: 10.1021/cb4002188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sela D., Chen L., Martin-Brown S., Washburn M.P., Florens L., Conaway J.W., Conaway R.C. Endoplasmic reticulum stress-responsive transcription factor ATF6alpha directs recruitment of the Mediator of RNA polymerase II transcription and multiple histone acetyltransferase complexes. J. Biol. Chem. 2012;287:23035–23045. doi: 10.1074/jbc.M112.369504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thuerauf D.J., Morrison L.E., Hoover H., Glembotski C.C. Coordination of ATF6-mediated transcription and ATF6 degradation by a domain that is shared with the viral transcription factor, VP16. J. Biol. Chem. 2002;277:20734–20739. doi: 10.1074/jbc.M201749200. [DOI] [PubMed] [Google Scholar]
- 62.Warfield L., Tuttle L.M., Pacheco D., Klevit R.E., Hahn S. A sequence-specific transcription activator motif and powerful synthetic variants that bind Mediator using a fuzzy protein interface. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E3506–E3513. doi: 10.1073/pnas.1412088111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang F., Vought B.W., Satterlee J.S., Walker A.K., Jim Sun Z.Y., Watts J.L., DeBeaumont R., Saito R.M., Hyberts S.G., Yang S., et al. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature. 2006;442:700–704. doi: 10.1038/nature04942. [DOI] [PubMed] [Google Scholar]
- 64.Jedidi I., Zhang F., Qiu H., Stahl S.J., Palmer I., Kaufman J.D., Nadaud P.S., Mukherjee S., Wingfield P.T., Jaroniec C.P., et al. Activator Gcn4 employs multiple segments of Med15/Gal11, including the KIX domain, to recruit mediator to target genes in vivo. J. Biol. Chem. 2010;285:2438–2455. doi: 10.1074/jbc.M109.071589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.