

ABSTRACT
Both HIV-1 virions and infected cells use their surface regulators of complement activation (RCA) to resist antibody-dependent complement-mediated lysis (ADCML). Blockage of the biological function of RCA members, particularly CD59 (a key RCA member that controls formation of the membrane attack complex at the terminal stage of the complement activation cascades via all three activation pathways), has rendered both HIV-1 virions and infected cells sensitive to ADCML mediated by anti-Env antibodies (Abs) or sera/plasma from patients at different stages of viral infection. In the current study, we used the well-characterized anti-HIV-1 neutralizing Abs (nAbs), including 2G12, 2F5, and 4E10, and non-nAbs, including 2.2C, A32, N5-i5, and N12-i15, to investigate whether the enhancement of ADCML by blockage of CD59 function is mediated by nAbs, non-nAbs, or both. We found that all nAbs and two non-nAbs (N5-i5 and A32) strongly reacted to three HIV-1 laboratory strains (R5, X4, and R5/X4), six primary isolates, and provirus-activated ACH-2 cells examined. In contrast, two non-nAbs, 2.2C and N12-i15, reacted weakly and did not react to these targets, respectively. After blockage of CD59 function, the reactive Abs, regardless of their neutralizing activities, significantly enhanced specific ADCML of HIV-1 virions (both laboratory strains and primary isolates) and provirus-activated latently infected cells. The ADMCL efficacy positively correlated with the enzyme-linked immunosorbent assay-reactive intensity of those Abs with their targets. Thus, blockage of RCA function represents a novel approach to restore activities of both nAbs and non-nAbs in triggering ADCML of HIV-1 virions and provirus-activated latently infected cells.
IMPORTANCE There is a renewed interest in the potential role of non-nAbs in the control of HIV-1 infection. Our data, for the first time, demonstrated that blockage of the biological function of RCA members rendered both HIV-1 virions and infected cells sensitive to ADCML mediated by not only nAbs but also non-nAbs. Our results are significant in developing novel immune-based approaches to restore the functions of nAbs and non-nAbs in the circulation of HIV-1-infected individuals to specifically target and clear HIV-1 virions and infected cells. Our data also provide new insights into the mechanisms by which HIV-1 virions and infected cells escape Ab-mediated immunity and could aid in the design and/or development of therapeutic HIV-1 vaccines. In addition, a combination of antiretroviral therapy with RCA blockage, provirus activators, and therapeutic vaccines may represent a novel approach to eliminate HIV-1 reservoirs, i.e., the infected cells harboring replication-competent proviruses and residual viremia.
INTRODUCTION
In HIV-1-infected patients, the virus-specific antibody (Ab) response is vigorous at all stages of infection. Within a few weeks of infection, Abs against the viral envelope (Env, gp160, or gp120 plus gp41), core (Gag), and matrix (p17) become detectable in the plasma of HIV-1-positive individuals (1–6). Ab levels mount in response to the gradual increase in viral load and appear to be maintained at high levels throughout the disease (7, 8). However, this vigorous and sustained Ab response has a limited effect on controlling virus proliferation or protecting the patients from developing AIDS (7, 9–11). This puzzling and frustrating phenomenon has been explained by the lack of sufficient neutralizing Abs (nAbs), i.e., the vast majority of Abs generated in natural HIV-1 infection are non-neutralizing Abs (non-nAbs) which are unable to prevent and contain HIV-1 infection (12). While numerous studies suggest that nAbs may impact HIV-1 replication at the acute stage of the viral infection (13–15), the effect of these nAbs in mediating effector functions and limiting viral spread remain uncertain. In particular, it is still unclear whether nAbs have a substantial role in the control of chronic, established HIV-1 infection. In addition, the lack of sufficient nAbs cannot fully clarify the Ab dysfunction because (i) almost all HIV-1-infected individuals develop Abs capable of neutralizing their own viruses (autologous neutralization) in vitro (16), (ii) recent studies have demonstrated that ca. 25% of chronically infected individuals (infected for at least 1 year) have moderate to broadly reactive nAb responses (9, 10), (iii) ca. 1% of these chronically infected individuals have nAbs with unusually potent activities against a majority of HIV-1 clades (17–22), and (iv) Abs with potent neutralizing activity against a broad range of HIV-1 strains across HIV-1 clades have been found in HIV-1-infected individuals, but passive immunization with a cocktail of these nAbs conferred no or only modest clinical benefits to HIV-1-infected subjects (23). In addition, non-nAbs in other viral infections can have substantial impact on anti-virus immunity through clearing virus particles and infected cells via complement activation, opsonization and phagocytosis, and antibody-dependent cell-mediated cytotoxicity (ADCC) (24–27). Thus, it appears that the immune system in HIV-1-infected individuals has the ability to generate nAbs, and that broadly nAb (bNAb) activities are developed over time by chronic antigen exposure during infection (9, 16, 28, 29). However, HIV-1 virions and infected cells can escape Ab immunity, regardless of nAb-mediated neutralization and nAb/non-nAb-mediated complement attack, opsonization, and phagocytosis, and ADCC.
Recent studies have suggested that resistance of HIV-1 virions and infected cells to antibody-dependent complement-mediated lysis (ADCML) is dependent on regulators of complement activation (RCA), particularly CD59, a glycosylphosphatidylinositol-anchored protein. CD59 is a key RCA member since it controls the formation of the membrane attack complex (MAC) at the terminal stage of the complement activation cascades via all three activation pathways. Recent studies have shown that HIV-1 virions incorporate and hijack human CD59 (hCD59) on their surfaces at levels that protect them from ADCML mediated by anti-HIV-1 Env (gp160 or gp120/gp41) monoclonal or polyclonal Abs (30–33). Similarly, hCD59 also provides resistance for HIV-1-infected cells from ADCML by sera from HIV-1-infected patients (34). Both experimental and clinical evidences indicate that HIV-1 viral proteins and anti-HIV-1 Abs activate the early complement cascades but may not fully complete the complement activation, since MAC pores are not formed, leading to viral escape. As a consequence, incomplete activation of complement causes C3 deposition on HIV-1 virions, enhancing viral transmission to cells bearing C3 receptors. Therefore, blockage of hCD59 function with specific Abs or removal of these molecules by treatment with phosphatidylinositol-specific phospholipase C could render virions and infected cells sensitive to complement attack (30–34).
Current antiretroviral therapy (ART) can successfully control plasma levels of HIV-1 RNA below the limits of detection, but cannot eliminate infected cells and the trace levels of free virions. The efficacy of ART is also limited by cost, toxicity, lifelong treatment adherence, and development of drug resistance. Therefore, an innovative therapeutic strategy is urgently needed to restore immune functions of HIV-1-infected patients and to help limit the use of ART. Blockage of hCD59 function can render complement-resistant HIV-1 virions and infected cells sensitive to ADCML by Abs against HIV-1 Env or sera/plasma from HIV-1-infected patients (30–34). We have recently reported that blockage of hCD59 renders provirus-activated ACH-2 cells and primary human CD4+ T cells that were latently infected with HIV-1 sensitive to ADCML by anti-HIV-1 polyclonal Abs or plasma from HIV-1-infected patients (35). However, it is unclear whether the enhancement of ADCML is mediated by nAbs, non-nAbs, or both. In the present study, we use the well-characterized anti-HIV-1 nAbs and non-nAbs to address these issues. We found that blockage of hCD59 function restored activities of both nAbs and non-nAbs in triggering ADCML of HIV-1 virions and provirus-activated latently infected cells. Thus, blockage of RCA function may represent a novel approach to restore activities of both nAbs and non-nAbs in triggering ADCML of HIV-1 virions and provirus-activated latently infected cells toward an HIV-1 cure. Our results also provide a better understanding of anti-HIV-1 Ab immunity, especially the roles of nAb and non-nAbs in combating HIV-1 infection.
MATERIALS AND METHODS
Anti-HIV-1 nAbs and non-nAbs.
Numerous monoclonal Abs (MAbs) with potent neutralizing activity against a broad range of HIV-1 strains across HIV-1 clades have been obtained from HIV-1-infected individuals. Among these nAbs, 2G12, 2F5, and 4E10 are three of the best-characterized MAbs isolated from individuals chronically infected with HIV-1 subtype (or clade) B strains. These neutralizing MAbs were either obtained from the National Institutes of Health (NIH) AIDS Reagent Program (Germantown, MD) or purchased from the Polymun Scientific (Vienna, Austria). 2G12 (IgG1) recognizes a unique mannose-dependent epitope in a carbohydrate-rich region on the outer domain of gp120 (20, 36–39). 2F5 and 4E10 recognize adjacent but distinct epitopes in the well-defined cluster of the membrane-proximal external region (MPER) of gp41 ectodomain (19, 36, 40, 41). 2F5 (IgG1) binds to the ELDKWA motif on the ectodomain of gp41 (36, 40, 41), whereas 4E10 (IgG1) recognizes the epitope NWFDIT slightly upstream from the 2F5-binding site (19) (Table 1).
TABLE 1.
Characterization of the well-documented anti-HIV-1 Env nAbs and non-nAbsa
| nAbs and non-Abs | Antigen (epitope) | Isotype | Neutralizing activity | Isolated from HIV-1 clade | Reference(s) |
|---|---|---|---|---|---|
| nAbs | |||||
| 2G12 | gp120 (C3/V4 domain) | IgG1, κ | Yes | B | 20, 36–39 |
| 2F5 | gp41 (ELDKWAS) | IgG1, κ | Yes | B | 36, 40, 41 |
| 4E10 | gp41 (NVVFDIT) | IgG1, κ | Yes | B | 19 |
| Non-nAbs | |||||
| 2.2C | gp120 | IgG1, κ | No | B | 43, 44 |
| A32 | gp120 (E/K)VGKAMYAPP | IgG1, λ | No | B | 20, 44–46 |
| N5-i5 | gp120 | IgG1, λ | No | B | 44 |
| N12-i15 | gp120 | IgG1, κ | No | B | 44 |
All nAbs were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: 2G12, 2F5, and 4E10 were all from H. Katinger. All non-nAbs were obtained from Y. Guan at the University of Maryland and J. Robinson at Tulane University.
Four other human MAbs (clones 2.2C, A32, N5-i5, and N12-i15) that specifically react with HIV-1 gp120 but have no neutralizing activity (42–46) were also used in the study. A32 (IgG1) binds to a highly conserved epitope of HIV-1 gp120, and A32-gp120 binding enhances binding of CD4-induced (CD4i) Abs (45, 46). 2.2C and N5-i5 bind to overlapping but distinct epitopes of gp120 (47) and exhibit potent ADCC in vitro (47). N12-i15 recognizes a conformational epitope of gp120 following Env binding to CD4 (44). These non-nAbs were gifts from Yongjun Guan at University of Maryland (Baltimore, MD) or James Robinson at Tulane University (New Orleans, LA) or were obtained from the NIH AIDS Reagent Program (Germantown, MD). The characterization of these nAbs and non-nAbs is shown in Table 1.
HIV-1 primary isolates.
Six primary HIV-1 isolates were generated by coculture of peripheral blood mononuclear cells (PBMCs) from healthy blood donors and PBMCs from each of six HIV-1-infected individuals as we reported previously (33). The viral stocks were divided into aliquots and stored in a liquid nitrogen tank until use. These six HIV-1 primary isolates were resistant to ADCML by polyclonal Abs against HIV-1 gp120/gp160 or sera from HIV-1-infected individuals (33). Blockage of hCD59 function on the virions of these HIV-1 primary isolates rendered them sensitive to ADCML (33). These HIV-1 primary isolates were used as target viruses to ADCML by the three nAbs (2G12, 2F5, and 4E10) and the four non-nAbs (2.2C, A32, N5-i5, and N12-i15).
ELISA.
The recombinant HIV-1 gp160 (rgp160; Sanofi-Pasteur, Toronto, Ontario, Canada) was used as an enzyme-linked immunosorbent assay (ELISA) antigen to detect reactive intensity of the nAbs (2G12, 2F5, and 4E10) and non-nAbs (2.2C, A32, N5-i5, and N12-i15). The rgp160 is a soluble recombinant hybrid Env glycoprotein, containing both the gp120 sequence from the HIV-1 BX08 strain and the gp41 sequence from the HIV-1LAI strain. This rgp160 was purified from chicken embryo fibroblasts infected with the rgp160-expressing recombinant vaccinia virus. The purification procedure included ion-exchange chromatography, followed by immunoaffinity chromatography. The final products have been used as an HIV-1 protein vaccine in clinical trials (48), indicating that the protein was highly purified. The rgp160 was coated at 1 μg/ml to 96-well microplates. After a washing step with PBS-T buffer (phosphate-buffered saline supplemented with 0.05% Tween 20) and blocking with Blocker Casein Buffer (Thermo Scientific, Rockford, IL), microplates were incubated with individual nAbs or non-nAbs at various concentrations ranging from 0 to 5 μg/ml, followed by addition of an anti-human IgG MAb conjugated with horseradish peroxidase (HRP). Human IgG1 at 0 to 5 μg/ml was used as an Ab-negative control. Absorption was read at a wavelength of 450 nm by a microplate spectrophotometer (BioTek, Winooski, VT).
Viral lysates of HIV-1 laboratory strains and primary isolates were also used as ELISA antigens to determine the reactive intensity of the nAbs (2G12, 2F5, and 4E10) and non-nAbs (2.2C, A32, N5-i5, and N12-i15). HIV-1 laboratory strains, including two R5 (JR-CSF and BAL), three X4 (LAI, IIIB, and MN) and one dual R5X4 (RF), were obtained from the NIH AIDS Reagent Program (Germantown, MD). The viruses were expanded by two or three cycles of growth on phytohemagglutinin plus interleukin-2-stimulated PBMCs from healthy blood donors. Supernatants harvested from cells infected with each of these laboratory strains were subjected to virus purification by ultracentrifugation at 32,000 rpm at 4°C for 1 h. The same approach was also used for purifying virus particles of the six primary isolates. Purified HIV-1 particles of both laboratory strains and primary isolates were lysed with 1% Triton X-100 in PBS. The viral lysates were diluted in PBS and then subjected to measurement of HIV-1 p24 using a p24 antigen ELISA kit (Perkin-Elmer, Santa Clara, CA). Viral lysates were coated at 2 μg of HIV-1 Gag/ml to 96-well microplates. After washing with PBS-T buffer and blocking with the Blocker Casein Buffer (Thermo Scientific), microplates were incubated with individual nAbs or non-nAbs at 5 μg/ml, followed by the addition of an anti-human IgG MAb conjugated to HRP. Human IgG1 at 5 μg/ml was used as an Ab-negative control. Absorption was read at a wavelength of 450 nm by a microplate spectrophotometer (Bio-Tek, Winooski, VT).
Generation and purification of F(ab′)2 fragments of hCD59-blocking antibody.
We and others have reported that BRIC229 (IBGRL, Bristal, United Kingdom), an anti-hCD59 MAb (mouse IgG2b), could completely block hCD59 function when used at a concentration of 20 μg/ml or higher (32, 33). Blockage of hCD59 function with intact BRIC229 molecules allows anti-HIV-1 gp120/gp160 Abs or Abs in the sera/plasma from HIV-1-infected individuals to restore their activity of ADCML to destroy complement-resistant HIV-1 virions and infected cells (30–34). We have recently reported that blockage of hCD59 renders provirus-activated ACH-2 cells and primary human CD4+ T cells that were latently infected with HIV-1 sensitive to ADCML by anti-HIV-1 polyclonal Abs or plasma from HIV-1-infected patients (35). However, it is unclear whether F(ab′)2 fragments of BRIC229 would retain hCD59-blocking activity of the intact Ab of BRIC229. The generation and purification of BRIC229 F(ab′)2 fragments was performed using Pierce F(ab′)2 preparation kits (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's protocol. Because the pepsin protease in the Pierce F(ab′)2 preparation kit is supplied in an immobilized form as beaded agarose resin, the digestion reaction was easily stopped by removing the resin from the IgG solution, resulting in enzyme-free digest products. The protein purity of BRIC229 F(ab′)2 fragments was checked by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Coomassie blue staining. The concentration of BRIC229 F(ab′)2 fragments was determined using the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific).
Hemolytic assay.
Fresh human red blood cells (hRBCs) isolated from healthy blood donors were washed four times with PBS and then used for a hemolytic assay to determine the activity of intact BRIC229 molecules and F(ab′)2 fragments in blockage of hCD59 as previously described (33). Briefly, hRBCs (5,000 cells) were preincubated with intact BRIC229 molecules, F(ab′)2 fragments or irrelevant control Ab (IgG) at various concentrations ranging from 1 to 20 μg/ml at 37°C for 30 min to block hCD59 function. Cells were then incubated with rabbit anti-hRBC polyclonal Abs (MyBioSource, San Diego, CA) plus the pooled sera (1:5 diluted in PBS) from three healthy blood donors. Every experiment included heat-inactivated sera as a control of complement inactivation to obtain background of hemolysis, whereas total hemolysis was obtained by adding pure water to the hRBC pellet. All experiments were carried out in triplicates. The amount of hemoglobin released from lysed hRBCs was determined by the optical density of the supernatant at 414 nm (OD414), and the percent lysis was calculated as follows: [(experimental OD414 − background OD414)/(total lysis OD414 − background OD414)] × 100.
ADCML of HIV-1 virions by nAbs and non-Abs.
Blockage of CD59 function to trigger ADCML of HIV-1 virions of both laboratory strains and primary isolates by anti-Env Abs was carried out as previously described (32, 33). Briefly, HIV-1 virions (20 μl containing 50 ng of HIV-1 p24) were preincubated with or without intact BRIC229 or BRIC229 F(ab′)2 (20 μg/ml) for 30 min at 37°C, followed by the addition of individual nAbs or non-nAbs (5 μg of each Ab/ml) plus the pooled sera from three healthy blood donors as a source of complement (1:10 dilution in GVB++ buffer; Sigma-Aldrich, St. Louis, MO). Heat-inactivated sera were used as a negative control of complement activity. Virolysis of HIV-1 virions was quantified using the p24 antigen ELISA (Perkin-Elmer). The ELISA procedure was the same as the manufacturer's description, except that the lysis buffer was not used. As a consequence, we measured only the Gag released from the lysed viral particles triggered by the Ab-dependent complement-mediated virolysis. The Gag of the intact HIV-1 virions was not detected; therefore, Gag release served a parameter of virolysis. HIV-1 virions were treated with Triton X-100 or medium alone for determination of total virolysis and background of spontaneous virolysis, respectively. The percentage of virolysis was calculated as follows: [(Gag released in the presence of complement-competent sera − Gag released in the presence of heat-inactivated sera)/(Gag released from Triton X-100-treated virions − Gag released by medium only)] × 100. Experiments were conducted in duplicates, and means ± standard deviations (SD) from three experiments were compared by using a paired two-tailed Student t test.
Infectivity assay.
To test whether virus infectivity was reduced or eliminated by ADCML, we performed an HIV-1 infectivity assay as previously reported (33). Briefly, 5 μl of reaction mixture from each condition in the ADCML of HIV-1 virions described above was added to fresh H9 cells (a human T cell line permissive for HIV-1 infection and replication) at 0.2 × 106 cells per well in 200 μl of complete RPMI 1640 medium (RPMI 1640 medium supplemented with 10% fetal bovine serum [FBS], 5% penicillin-streptomycin, and 2 mM glutamine) and cultured for 7 to 10 days. The culture supernatant was subjected to HIV-1 p24 ELISA antigen assay (Perkin-Elmer) to determine viral infectivity. The lysis buffer included in the ELISA kit was used to lyse the viral particles for measuring HIV-1 core protein p24. The higher the level of p24 in the culture supernatant, the higher the number of infectious viral particles that remained in the conditioned medium of the ADCML experiments. HIV-1MN and H9 cells were used in the infectivity assay to determine the functional consequence of ADCML because H9 cells have been demonstrated to strongly support HIV-1MN infection and replication (33), thereby ensuring the sensitivity of the infectivity assay.
Western blot.
ACH-2 cells treated or untreated with phorbol 12-myristate 13-acetate (PMA; 10 ng/ml)/ionomycin (1 μg/ml) or dimethyl sulfoxide (DMSO) for 24 h were lysed in 1× cell lysis buffer (Cell Signaling Technology, Danvers, MA) plus 1× protease inhibitor cocktail (Sigma-Aldrich). Cellular lysates were cleared by centrifugation at 13,000 × g for 10 min at 4°C. The supernatants were harvested and mixed with the SDS-PAGE gel-loading buffer (Invitrogen, Carlsbad, CA), followed by heating in a boiling water bath for 5 min. The protein samples were subjected to NuPAGE Novex high-performance electrophoresis (Invitrogen). Proteins were blotted onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA). After blocking with 5% nonfat milk, the blot was incubated at room temperature for 2 h with individual nAbs (2G12, 2F5, and 4E10) or non-nAbs (2.2C, A32, N5-i5, and N12-i15) at 1 μg/ml. After washing, the blot was visualized with the appropriate HRP-conjugated secondary Ab and an enhanced chemiluminescence (ECL) detection system (Pierce, Rockford, IL). To ensure that equal amounts of cellular proteins were analyzed, the same blot was used for the detection of human β-actin protein expression after stripping, blocking, and incubation with mouse anti-human β-actin protein MAb (Abcam, Cambridge, MA), followed by incubation with HRP-conjugated anti-mouse IgG (Jackson Immunoresearch Laboratories, West Grove, PA) and analysis using the ECL detection system.
Flow cytometry.
ACH-2 or A3.01 cells were stimulated with or without PMA (10 ng/ml)/ionomycin (1 μg/ml) or DMSO for 24 h at 37°C in a 5% CO2 incubator. Cells were subjected to surface staining with an individual MAb (4 μg/ml) of nAbs (2G12, 2F5, and 4E10) or non-nAbs (2.2C, A32, N5-i5, and N12-i15) at room temperature for 30 min. An irrelevant MAb of human IgG1 was used in parallel to obtain baseline of the surface staining signals. After washing, cells were incubated with an anti-human IgG1 MAb conjugated with a fluorescent dye for 20 min at room temperature. The stained cells were washed with PBS three times before analyzing the fluorescence intensity using the BD FACSCalibur system (BD Biosciences, San Jose, CA). Flow cytometry data were analyzed using FlowJo software (Tree Star, San Carlos, CA).
Confocal microscopy.
ACH-2 cells were treated with PMA (10 ng/ml)/ionomycin (1 μg/ml) or DMSO for 24 h. Cells were fixed with 2% paraformaldehyde and then subjected to immunostaining for confocal microscopy analysis of viral protein expression and colocalization of HIV-1 Env with hCD59 and ganglioside M1 (GM1) lipid rafts on the cell surface. Cells were incubated with cholera toxin subunit B (CTB) conjugated with Alexa Fluor 488 (Life Technologies, Carlsbad, CA) at 4°C for 20 min to stain GM1 lipid rafts as previously described (49). Cells were also subjected to staining with an individual anti-HIV-1 Env MAb (1 μg/ml) of nAbs (2G12, 2F5, and 4E10), non-nAbs (2.2C, A32, N5-i5, and N12-i15), or anti-hCD59 BRIC229. After washing, cells were stained with the secondary Abs, including goat anti-human IgG conjugated with Alexa Fluor 633 and donkey anti-mouse IgG conjugated with Alexa Fluor 546. Both primary- and secondary-Ab incubations were carried out for 30 min at room temperature with 2% FBS/PBS. After staining, cells were adhered to poly-l-lysine-coated coverslips and mounted onto glass slides using ProLong Gold antifade reagent (Life Technologies), which contains DAPI (4′,6′-diamidino-2-phenylindole) dye for fluorescent staining of DNA content and nuclei. Cells were analyzed using an Olympus FV1000-MPE confocal/multiphoton microscope fitted with a ×60 objective lens. Images were processed and analyzed using the FV10-ASW 3.0 Viewer software (Olympus America, Inc., Center Valley, PA).
ADCML of provirus-activated ACH-2 cells by anti-HIV-1 nAbs and non-nAbs.
ADCML of provirus-activated ACH-2 cells by nAbs or non-nAbs was quantified using the calcein release assay, as described in our recent report (35). Briefly, ACH-2 or A3.01 cells treated or untreated with PMA/ionomycin or DMSO were labeled at 37°C for 40 min with 15 μM calcein-AM dye (Invitrogen) in 200 μl of PBS. The cells were washed three times with PBS to remove the free calcein-AM dye and then resuspended at a concentration of 106 cells per ml in the complete RPMI 1640 medium. Cells were dispensed into round-bottom 96-well microplates (100 μl containing 10,000 cells per well). Cells were preincubated for 30 min at 37°C with or without BRIC229 (20 μg/ml) plus an individual nAb or non-nAb at 5 μg of each Ab/ml. After the preincubation, 50 μl of the pooled sera from three healthy blood donors was added as a source of complement. The microplates were incubated at 37°C for 1 h, followed by centrifugation at 130 × g for 5 min. After the centrifugation, 100 μl of supernatant from each well was transferred into fresh 96-well microplates to measure the fluorescence intensity using an automated fluorescence plate reader (Molecular Devices, Sunnyvale, CA). To measure the maximum fluorescence intensity, a sample of calcein-labeled cells was lysed with 200 μl of Triton X-100 (5% in PBS). To obtain baseline of spontaneous cytolysis, parallel ADCML assays were run with heat-inactivated sera from three healthy blood donors, culture medium alone, or an irrelevant MAb (IgG1). All experiments were carried out in triplicates. The percent cytolysis of ADCML was determined as follows: % cytolysis = [(experimental cytolysis − spontaneous cytolysis)/(maximum cytolysis − spontaneous cytolysis)] × 100.
Statistical analysis.
Data were analyzed using Tukey's post hoc analysis of variance test and the Student t test. P values of <0.05 were considered statistically significant.
RESULTS
Reactivity of nAbs and non-nAbs to the Env of HIV-1 laboratory strains and primary isolates.
HIV-1 Env (gp160 or gp120/gp41) is not only the target of nAbs in preventing the initiation and spread of HIV-1 infection by blocking viral attachment to its CD4 receptor but also the target of both nAb and non-nAb in triggering ADCML of virions and infected cells. Because HIV-1 Env has a highly variable amino acid sequence and glycosylation pattern, the antigenicity of this protein may vary in reaction with previously described nAbs and non-nAbs. For this reason, we determined the reactivity of the three nAbs (2F5, 2G12, and 4E10) and four non-nAbs (2.2C, A32, N5-i5, and N12-i15) with HIV-1 Env from a variety of HIV-1 laboratory strains used worldwide and six primary isolates generated in our laboratory (33).
First, we used the recombinant HIV-1 gp160 (rgp160; Sanofi-Pasteur, Toronto, Canada) as an ELISA antigen to titrate the reactive titer of these nAbs (2G12, 2F5, and 4E10) and non-nAbs (2.2C, A32, N5-i5, and N12-i15). The ELISA results revealed that all three nAbs reacted with the rgp160 coated to the 96-well plates at 1 μg/ml in a dose-dependent manner (Fig. 1A). Non-nAbs, including A32 and N5-i5, also showed reactions at moderate levels compared to nAbs, whereas 2.2C showed a weak reaction and N12-i15 did not show any difference in reactivity compared to human IgG control (Fig. 1A). Under the experimental conditions described in Materials and Methods, reactivity of all nAbs tested to the rgp160 showed OD450 values of from <0.1 to 1.08, <0.1 to 0.43, and <0.1 to 0.26, corresponding to Ab concentrations of 5, 1, and 0.2 μg/ml, respectively (Fig. 1A). Meanwhile, the OD450 of the positive control (1:1,000 diluted plasma from the HIV-1-infected patient 4 who had a titer of anti-Env at 1:10,000) (33) was 1.7 (data not shown), and the negative control (1:1,000 diluted plasma from a healthy blood donor or 5 μg of human IgG protein/ml) was <0.1. N12-i15 MAb did not react with HIV-1 Env because this MAb recognizes a conformational gp120 epitope that is exposed only after Env binding to CD4 (44). It is pertinent to note that the reactions of all MAbs used at the highest concentration (5 μg/ml) have not reached the plateau (Fig. 1A), and 2.2C and N5-i5 can strongly react with Env if its concentration is increased (44, 47). Since our study was undertaken to clarify whether nAbs and non-nAbs triggered ADCML of HIV-1 virions and infected cells and whether the efficacy of ADCML was associated with reaction intensity of these MAbs rather than their neutralizing activities, we used the 5 μg of individual nAbs or non-nAbs/ml that generated reactions at different degrees for the rest of experiments through our study.
FIG 1.
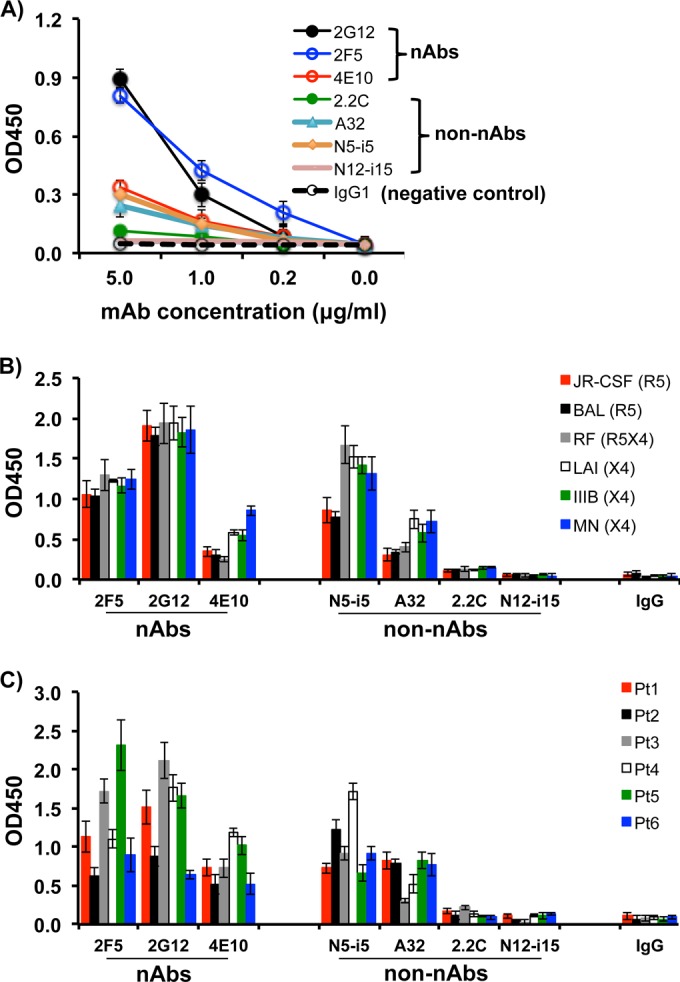
Reactivity of nAbs and non-nAbs to Env of HIV-1 laboratory strains and primary isolates. (A) The recombinant HIV-1 gp160 (rgp160) was used at 1 μg/ml as an ELISA antigen to coat 96-well microplates for titrating the reactive titer of nAbs (2G12, 2F5, and 4E10) and non-nAbs (2.2C, A32, N5-i5, and N12-i15). The rgp160 is a soluble recombinant hybrid Env glycoprotein containing the gp120 sequence from the HIV-1 BX08 strain and the gp41 sequence from the HIV-1LAI strain. (B and C) Microplates were coated with virus antigens derived from lysed virions (an input of 2 μg of HIV-1 p24/ml) from each of these strains as indicated. Individual nAbs and non-nAbs were used at 5 μg/ml. Purified human IgG1 at 5 μg/ml was used as a negative control of anti-HIV-1 Abs. The optical density of each well was read at 450 nm (OD450). All samples were tested in triplicate. All experiments were repeated three times.
Next, we tested the reactivity of these three nAbs (2G12, 2F5, and 4E10) and four non-nAbs (2.2C, A32, N5-i5, and N12-i15) with a variety of HIV-1 laboratory strains, including two R5 (JR-CSF and BAL), three X4 (LAI, IIIB and MN), and one dual R5X4 (RF) strains. ELISA data showed that all nAbs used at 5 μg/ml specifically reacted with each of the virus strains, albeit to various degrees. 2G12 showed the highest reactivity to all virus strains tested, whereas 2F5 and 4E10 showed moderate activities to all virus strains (Fig. 1B). Compared to these nAbs, N5-i5 reacted with these virus strains at degrees comparable to that of 2F5, whereas A32 showed moderate reactivity to all viruses (Fig. 1B). 2.2C showed a weak reaction and N12-i15 did not readily react with any of these virus strains (Fig. 1B). The difference in reactivity among the nAbs/non-nAbs tested was due to their relative binding affinity or degree of exposure of the antigenic epitope to those individual Abs rather than to any variation in the quality and quantity of virus antigens coated to the ELISA plates because each well was coated with the same amount of virus antigens prepared from the same batch of virus stocks.
Finally, we tested the reactivity of these three nAbs (2G12, 2F5, and 4E10) and four non-nAbs (2.2C, A32, N5-i5, and N12-i15) with all six primary isolates of HIV-1 that have been isolated in our laboratory (33). The results were similar to that of HIV-1 laboratory strains (Fig. 1C). All three nAbs tested reacted with all six primary isolates of HIV-1, albeit at various degrees (Fig. 1C). Two non-nAbs (N5-i5 and A32) reacted with all six primary HIV-1 isolates at comparable levels to nAb 2F5 (Fig. 1C). 2.2C showed a weak reaction (OD450 > 0.1, but OD450 < 0.3) and N12-i15 barely reacted to any of these primary isolates (Fig. 1C).
Blockage of CD59 function allowed both nAbs and non-nAbs to trigger ADCML of HIV-1 laboratory strains.
We and others have previously reported that blockage of hCD59 function renders complement-resistant HIV-1 virions and infected cells sensitive to ADCML by HIV-1 gp120/gp160 Abs or serum/plasma from HIV-1-infected patients (33, 34). However, it is unclear whether the enhancement of ADCML is mediated by nAbs, non-nAbs, or both. After we clarified that the anti-HIV-1 Env nAbs (2G12, 2F5, and 4E10) and two of non-nAbs (A32 and N5-i5) reacted with all six laboratory strains examined, we tested activity of these nAbs and non-nAbs in triggering complement-mediated virolysis of these laboratory strains upon blockage of CD59 function. We pretreated each of the laboratory strains with BRIC229 and exposed them to each of the nAbs or non-nAbs, followed by incubation with the pooled sera from three healthy blood donors as a source of complement. Heated-inactivated sera were also included in each experiment as a control of complement activity. We also generated BRCI229 F(ab′)2 fragments and compared the effects of BRIC229 F(ab′)2 fragments to those intact BRIC229 molecules on the blockage of hCD59 function. As shown in Fig. 2A, no contamination of intact BRIC229 IgG was observed in the purified BRIC229 F(ab′)2 fragments by non-reducing SDS-PAGE analysis. BRIC229 F(ab′)2 fragments completely blocked the binding of the intact BRIC229 IgG molecules to target cells (Fig. 2B), indicating that BRIC229 F(ab′)2 fragments compete with the intact BRIC229 molecules for the binding epitopes of hCD59 on the cell surface. We then used a hemolysis assay to determine whether BRIC229 F(ab′)2 fragments retained the functional activity of intact BRIC229 molecules in blockage of hCD59 function. Similar to intact BRIC229, BRIC229 F(ab′)2 fragments blocked hCD59 function in a dose-dependent manner. BRIC229 or BRIC229 F(ab′)2 fragments at 20 μg/ml completely blocked hCD59 function as ADCML of hRBCs reached almost 100%, whereas hRBCs were resistant to ADCML in the absence of BRIC229 or BRIC229 F(ab′)2 fragments (Fig. 2C).
FIG 2.

ADCML of HIV-1 virions of laboratory strains by nAbs and non-nAbs. (A) Generation of BRIC229 F(ab′)2 fragments and characterization of their functional activity as a blocker of hCD59. SDS-PAGE separation and Coomassie blue staining of intact BRIC229 molecules and purified BRIC229 F(ab′)2 fragments. (B) Confirmation of BRIC229 F(ab′)2 fragments binding to hCD59 on the surface of H9 cells by flow cytometry analysis. BRIC229 F(ab′)2 binding to hCD59 was not detectable by a fluorescent-label anti-Fc Ab (left), as F(ab′)2 does not contain Fc. Binding of intact BRIC229 Ab to hCD59 was detected (middle). Preincubation of BRIC229 F(ab′)2 fragments inhibits binding of intact BRIC229 molecules to H9 cells that express a high level of hCD59 (right). (C) BRIC229 F(ab′)2 fragments exhibit a similar function as intact BRIC229 Ab in blockage of hCD59 function to trigger complement-mediated hemolysis. The results are means ± SD from four independent experiments. (D) ADCML of HIV-1 virions by nAbs and non-nAbs. HIV-1 virions from six laboratory strains as indicated were preincubated with intact BRIC229 Ab or F(ab′)2 fragments (20 μg/ml) and treated with anti-HIV-1 nAbs or non-nAbs plus the pooled sera from three healthy blood donors as a source of complement. In the parallel experiments, heat-inactivated sera from the same donors were used as a negative control of complement activity. Virolysis was analyzed by ELISA titration of released viral Gag protein. In each experiment, treatments with culture medium alone or with Triton X-100 were used as blank and 100% virolysis, respectively. Means ± SD from three experiments performed in duplicate are shown. (E) Pooled virolysis data from HIV-1 virions of all six laboratory strains. (F) HIV-1 infectivity assay. Titration of produced p24 in culture supernatants from H9 cells exposed for 10 days to 5 μl of reaction mixture from each condition in the ADCML of HIV-1MN virions. The experiments were repeated twice. The results are represented by means ± SD. Statistical significance (**, P < 0.01 versus medium or IgG treatment group) is indicated by asterisks. ns, not significant.
We used intact BRIC229 molecules or BRIC229 F(ab′)2 fragments at 20 μg/ml to block hCD59 function for analyzing ADCML of HIV-1 virions as previously described (32, 33). The results showed that all reactive nAbs (2G12, 2F5, and 4E10) and two non-nAbs (A32 and N5-i5) triggered complement-mediated virolysis of each of the six laboratory strains, regardless of the Abs bearing neutralizing activity or not (Fig. 2D). The efficacy of ADCML triggered by these MAbs is positively correlated with the extent of their reactivity with HIV-1 Env (Fig. 2D and E). BRIC229 F(ab′)2 fragments retained activity of intact BRIC229 molecules in blocking hCD59 function. BRIC229 F(ab′)2 fragments restored activities of both nAbs and non-nAbs in triggering ADCML of laboratory strains to a similar degree as intact BRIC229 molecules (Fig. 2D and E). These results indicate that blockage of hCD59 function with BRIC229 or F(ab′)2 fragments restores the functions of both nAbs and non-nAbs in triggering complement-mediated virolysis of HIV-1 virions.
To clarify whether virus infectivity was reduced or eliminated by ADCML, we performed an HIV-1 infectivity assay as previously reported (33). HIV-1-permissive H9 cells were exposed for 10 days to each conditioned solution from ADCML of HIV-1MN experiments depicted in Fig. 2D (HIV-1MN panel). The culture supernatants were collected to determine HIV-1 infection intensity by measuring HIV-1 p24. Figure 2F showed that p24 was undetectable in the supernatant from H9 cells exposed to conditioned medium of Triton X-100 treatment (total lysis), indicating that potentially infective particles were totally lysed and no infectious viral particles remained. The supernatants from cells exposed for 10 days to the control conditioned medium [i.e., conditioned by the virions not treated with intact BRIC229 molecules or F(ab′)2 fragments before exposure to endogenous anti-HIV-1 Ab and complement] had high titers of p24, indicating that the viral particles were not lysed and their infectivity was preserved. In contrast, supernatants from cells exposed for 10 days to conditioned medium of intact BRIC229 molecules or F(ab′)2 fragments pretreated virions (treated with BRIC229 before exposure to endogenous anti-HIV-1 Ab and complement) showed low levels of p24 (Fig. 2F), an indication that hCD59 blockage allowed the anti-HIV-1 Abs to trigger complement-mediated virolysis and thereby reduced the infective potential of HIV-1 virions.
Blockage of CD59 function allowed both nAbs and non-nAbs to trigger ADCML of primary HIV-1 isolates.
Next, we tested the activity of these nAbs and non-nAbs in triggering complement-mediated virolysis of the primary isolates of HIV-1 upon blockage of hCD59 function. We pretreated each of the primary HIV-1 isolates with BRIC229 and exposed them to each of the nAbs or non-nAbs, followed by incubation with the pooled sera from three healthy blood donors as a source of complement. Heat-inactivated sera were also included in each experiment as a control of complement activity. The results were similar to those obtained with HIV-1 laboratory strains. All nAbs and non-nAbs that were reactive to the primary isolates triggered ADCML of each of the six primary HIV-1 isolates, whereas non-nAbs that were not reactive to primary isolates did not have any effects on the ADCML of these viruses (Fig. 3). These results indicate that blockage of hCD59 function with BRIC229 restores the functions of nAbs and non-nAbs in triggering complement-mediated virolysis of primary HIV-1 isolates, regardless of the Abs neutralizing activity.
FIG 3.

ADCML of HIV-1 virions of primary isolates by nAbs and non-nAbs. (A) HIV-1 virions of the six primary isolates were incubated with BRIC229 (20 μg/ml) to block hCD59. Nonspecific IgG was used at 20 μg/ml as a negative control of BRIC229. Virions were treated with individual nAbs or non-nAbs as indicated (5 μg/ml), followed by exposure to 10% of the pooled sera from three healthy blood donors as a source of complement. In parallel experiments, heat-inactivated sera from the same healthy blood donors were used as a negative control of complement activity. Virolysis was analyzed by ELISA titration of released HIV-1 Gag proteins. In each experiment, treatments with culture medium alone or with Triton X-100 were used as blank and 100% virolysis, respectively. Means ± SD from three experiments performed in duplicate are shown. (B) Pooled virolysis data from HIV-1 virions of all six primary isolates. Statistical significance (**, P < 0.01 versus medium or IgG treatment group) is indicated by asterisks. ns, not significant.
Reactivity of nAbs and non-nAbs to HIV-1 Env on the surface of latently infected T cells upon provirus reactivation.
ACH-2 is a human T cell line latently infected with the HIV-1LAV strain, while A3.01 is the parental uninfected cell line. Cells were treated or untreated with PMA (10 ng/ml) plus ionomycin (1 μg/ml) or DMSO for 24 h. Supernatants were subjected to HIV-1 p24 ELISA to determine virus production, and stimulated ACH-2 cells were subjected to immunostaining with each of nAbs and non-nAbs for flow cytometry analysis to determine reactivity of these MAbs to HIV-1 Env on the surface of latently infected T cells after provirus activation. As shown in Fig. 4A, PMA/ionomycin stimulated ACH-2 to produce high levels of virus production compared to medium or DMSO alone. Flow cytometry results showed that, except for one non-nAb (N12-i15), all Abs examined reacted with HIV-1 Env on the surface of ACH-2 cells after provirus activation, albeit to various degrees (Fig. 4B). Among these reactive Abs, 2G12 and 2F5 reacted to provirus-activated ACH-2 cells at the highest degrees.
FIG 4.

Reactivity of nAbs and non-nAbs to HIV-1 Env on the surface of latently infected T cells after provirus reactivation. ACH-2 cells were incubated with PMA (10 ng/ml) plus ionomycin (1 μg/ml) or DMSO for 24 h. Supernatants were subjected to HIV-1 p24 ELISA to determine virus production, while stimulated ACH-2 cells were subjected to immunostaining with individual nAbs and non-nAbs as indicated to determine reactivity of these MAbs to HIV-1 Env. (A) HIV-1 p24 ELISA measurement of virus production in the supernatants of stimulated ACH-2 cells. (B) Representative experiments of flow cytometry analysis show the results of ACH-2 cells expressing Env on the cell surface 24 h after stimulation. (C) Representative experiments of Western blot show the reactive intensity of nAbs and non-nAbs with HIV-1 Env in the cellular lysates of ACH-2 cells 24 h after stimulation. PMA, PMA (10 ng/ml) plus ionomycin (1 μg/ml). (D) Colocalization of Env, CD59 and lipid rafts on the surface of ACH-2 cells upon provirus activation. ACH-2 cells were treated with PMA (10 ng/ml) plus ionomycin (1 μg/ml) or DMSO for 24 h. After treatment, cells are subjected to immunostaining for confocal microscopy analysis of Env, CD59, and lipid raft colocalization on the cell surface. Cells were subjected to surface staining with CTB conjugated with Alexa Fluor 488 (for lipid rafts), an individual nAbs (2G12, 2F5, and 4E10) or non-nAbs (2.2C, A32, N5-i5, and N12-i15) at 1 μg/ml (for HIV-1 Env), and BRIC229 (for hCD59). The white color in the merged panel indicates the colocalization of HIV-1 Env (red) with lipid rafts (green) and CD59 (magenta) on the surfaces of provirus-activated ACH-2 cells. Cellular DNA content and nuclei were stained with DAPI (blue). Scale bars, 5 μm. The data showed the results of 2G12 staining, which represents the staining results of all nAbs and non-nAbs, except N12-i15 that did not detect Env expression on the surface of provirus-activated ACH-2 cells. The results were obtained from at least three independent experiments. CTB, cholera toxin subunit B as a marker for lipid rafts. PMA, PMA (10 ng/ml) plus ionomycin (1 μg/ml).
We used the Western blot assay to confirm our flow cytometry data described above. We found that all MAbs that reacted with the native form of Env on the surface of provirus-activated ACH-2 cells in the flow cytometry assay were also able to react with Env in the cell lysates of provirus-activated ACH-2 cells (Fig. 4C). Similar to flow cytometry analysis of the native form of Env on the surface of provirus-activated ACH-2 cells, 2G12 strongly reacted with Env in the cell lysates of provirus-activated ACH-2 cells (Fig. 4C). However, the reactive intensities of 2F5 and 4E10 were different from those observed in flow cytometry analysis. As shown in Fig. 4C, 4E10 detected Env in the cell lysates of provirus-activated ACH-2 cells to the highest degree, whereas 2F5 detected a moderate degree of Env expression. These results are opposite of those observed in flow cytometry analysis, indicating that 4E10 may bind better to the denatured form of Env. Two non-nAbs (A32 and N5-i5) strongly reacted with Env in the cell lysates of provirus-activated ACH-2 cells, whereas 2.2C weakly reacted with the soluble Env and N12-i15 did not detect Env expression. Thus, the non-nAbs show a similar pattern of reactivation with Env in the cellular lysates and its native form on the surface of provirus-activated ACH-2 cells.
Productively HIV-1-infected cells use their surface RCA molecules, mainly CD59, to resist ADCML, allowing them to escape the vigorous and sustained anti-HIV-1 Ab responses observed in almost all infected individuals (34). This suggests that CD59 is located in close proximity to viral proteins such as Env on the cell surface, facilitating direct interaction between them. We used a confocal microscopy assay to analyze CD59 and Env colocalization on the surface of provirus-activated latently infected cells. The confocal microscopy analysis confirmed our flow cytometry and Western blot data that PMA/ionomycin stimulated provirus activation, leading to Env expression on the cell surface (Fig. 4D, Env panel). It is well known that Env on the surface of productively infected cells is made of a trimer consisting of three gp120/gp41 and that this trimeric Env is strongly associated with lipid rafts (50–52). We found a similar association between Env and lipid rafts on the surface of provirus-activated cells. A multiclustered Env distribution was observed on the surface of provirus-activated cells (Fig. 4D, Env panel), which was exclusively colocalized with cell membrane lipid rafts (Fig. 4D, CTB panel). Importantly, CD59 was also strongly associated with cell membrane lipid rafts (Fig. 4D, CD59 panel), and Env and CD59 were colocalized in lipid rafts on the surface of provirus-activated cells (Fig. 4D, white in merge panel), suggesting that Env may directly interact with RCA molecules on the cell surface. Figure 4D showed the results of 2G12 staining, which represents the staining results of all nAbs and non-nAbs, except N12-i15 that did not detect Env expression on the surface of provirus-activated ACH-2 cells.
Anti-Env nAbs and non-nAbs triggered ADCML of latently HIV-1-infected cells upon provirus activation.
Next, we tested whether blockage of CD59 function would render provirus-activated ACH-2 cells sensitive to ADCML by nAbs, non-nAbs, or both. We used BRIC229 to block hCD59 on the surface of provirus-activated cells to analyze the ADCML of latently HIV-1-infected cells. We found that blockage of hCD59 with BRIC229 markedly increased cytolysis of provirus-activated ACH-2 cells by nAbs/non-nAbs that reacted with Env on the surface of ACH-2 cells upon provirus activation (Fig. 5, upper panel). The lysis resulted from specific cell killing by anti-HIV-1 Env Ab-mediated ADCML, since lysis did not occur in both N12-i15 non-nAb that had no reaction with Env and the controls including the following: (i) no BRIC229, (ii) irrelevant human IgG1 in place of anti-HIV-1 Env nAbs or non-nAbs, (iii) heat-inactivated complement, (iv) ACH-2 cells treated with DMSO, and (v) A3.01 cells that are ACH-2 parental cells prior to HIV-1-infection (Fig. 5, lower panel).
FIG 5.
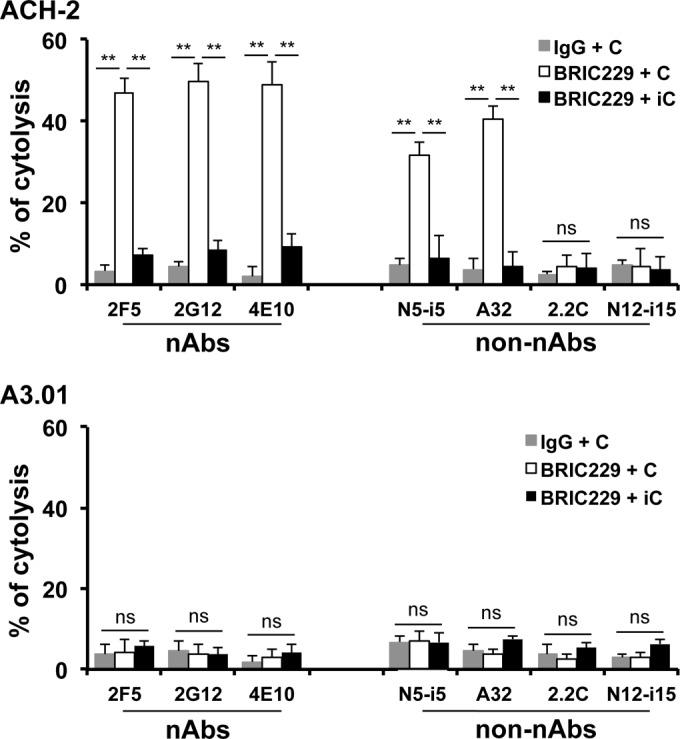
Anti-Env nAbs and non-nAbs triggered ADCML of latently HIV-1-infected cells upon provirus activation. Specific lysis of provirus-activated ACH-2 cells by anti-Env nAbs and non-nAbs. The calcein release assay was used to quantitate ADCML of latently HIV-1-infected cells by anti-HIV-1 nAbs and non-nAbs. ACH-2 or A3.01 cells treated or untreated with PMA/ionomycycin or DMSO. Cells were preincubated with BRIC229 (20 μg/ml) to block hCD59 function. An irrelevant IgG control Ab (20 μg/ml) was used in parallel as a negative control of hCD59 blockage. After preincubation, nAbs or non-nAbs were added, followed by the exposure to either complement-competent or heat-inactivated sera from three healthy blood donors. Triton X-100 was used for determining the total cytolysis. IgG + C, irrelevant Ab IgG plus complement; BRIC229 + C, BRIC229 Ab plus complement; BRIC229 + iC, BRIC229 Ab plus inactivated complement. Statistical significance (**, P < 0.01 versus IgG plus complement or BRIC229 plus inactivated complement) is indicated by asterisks. ns, not significant.
DISCUSSION
When a host is infected with a virus, Abs are produced against many epitopes on viral proteins. These Abs consist of a mixture of nAbs and non-nAbs, which can contribute to antiviral immunity in four ways. First, nAbs directly neutralize free virus particles by preventing receptor engagement, an essential step in the life cycle of all viruses, or by interfering with the fusion process of the viral surface proteins with host plasma membrane (10). Second, both nAbs and non-nAbs trigger specific ADCML of free virus particles and infected cells. Third, both nAbs and non-nAbs bind to and coat virus to mediate opsonization and phagocytosis by macrophages and other cells. Finally, both nAbs and non-nAbs trigger cytolysis of virus-infected cells by stimulating other immune responses such as ADCC. In the case of HIV-1 infection, both nAbs and non-nAbs against Env (gp160 or gp120 plus gp41) are simultaneously elicited, and the levels of these Abs rise in response to the gradual increase in viral load and are maintained at high levels throughout the infection (7, 8). Although anti-HIV-1 nAbs have been extensively studied in vitro and in vivo, non-nAbs have received little attention (44, 53). Studies have demonstrated that almost all HIV-1-infected individuals develop Abs capable of neutralizing autologous viruses (16). Indeed, Abs with potent neutralizing activity against a broad range of HIV-1 strains across HIV-1 clades have been found in HIV-1-infected individuals (17–19, 41, 54–57). However, these nAbs generated in HIV-1-infected patients fail to play their expected antiviral role. In addition, passive immunization with a cocktail of the potent first generation nAbs confers little to no clinical benefits to HIV-1-infected subjects (23). The new generation of nAbs uncovered by recently developed single-cell-based Ab cloning methods have demonstrated potent antivirus activities in reducing HIV-1 viremia (58). However, the reemergence of plasma viremia after decay of the nAb used in vivo has been observed and resistant strains of HIV-1 occur rapidly (58). Similarly, non-nAbs are commonly induced in both the acute and chronic phases of HIV-1 or SIV (simian immunodeficiency virus) infection (44, 59, 60). These non-nAbs have demonstrated potent ADCC activity in vitro (44, 59, 60) but fail to alter the pathogenic course of established HIV-1 or SIV infection in vivo (44, 59–62). These results indicate that HIV-1 virions and infected cells are able to escape Ab-mediated immunity, regardless of nAb-mediated neutralization and nAb/non-nAb-mediated complement attack, opsonization and phagocytosis, and ADCC. We found that the three well-characterized anti-HIV-1 nAbs (2G12, 2F5, and 4E10) and two non-nAbs (A32 and N5-i5) strongly reacted to laboratory-adapted T-tropic, M-tropic, and dualtropic strains of HIV-1, six primary isolates examined and provirus-activated ACH-2 cells, as determined by ELISA (Fig. 1), flow cytometry (Fig. 4B), Western blotting (Fig. 4C), and confocal microscopy analysis (Fig. 4D). 2.2C and N12-i15 are two non-nAbs that weakly reacted or did not react to these targets, respectively (Fig. 1 and Fig. 4B, C, and D). Regardless of their reactivity to Env, these nAbs and non-nAbs are not able to trigger ADCML of HIV-1 virions (Fig. 2D and Fig. 3) and infected cells that expressed Env on the cell surface (Fig. 5). The resistance of HIV-1 virions and infected cells to ADCML depends on RCA members, particularly CD59, on their surface. Blockage of CD59 function restores activities of these reactive nAbs and non-nAbs in triggering ADCML of HIV-1 virions and provirus-activated latently infected cells (Fig. 2D and E, Fig. 3, and Fig. 5). The ADMCL efficacy is positively correlated with the reactive intensity of these Abs with their targets, regardless of their neutralizing activities (Fig. 2D and E, Fig. 3, and Fig. 5). As a consequence of ADCML, HIV-1 infectivity was significantly reduced (Fig. 2F). Thus, blockage of RCA function restores activities of both nAbs and non-nAbs in triggering ADCML of HIV-1 virions and infected cells, including provirus-activated latently infected cells.
The native HIV-1 Env spike consists of a non-covalently linked trimer of gp120/gp41 heterodimers, each heterodimer composed of one surface gp120 subunit and one transmembrane gp41 subunit (63). Although gp120 (∼480 amino acids) carries the CD4 and chemokine receptor binding sites, gp41 (∼345 amino acids) is crucial for fusion between viral particle and the cell membrane. Both gp120 and gp41 have neutralizing and non-neutralizing epitopes (either linear or discontinuous) that evoke nAbs and non-nAbs, respectively (64–68). Most nAbs can be placed into four categories on the basis of their binding epitope locations: (i) CD4-binding site-directed nAbs such as b12, VRC01, VRC02, and VRC03 (69), (ii) gp120 variable regions 1 and 2 (V1/V2)-directed nAbs such as PG9 and PGT145 (69), (iii) glycan V3-directed nAbs such as 2G12 and PGT121 (69), and (iv) gp41 MPER-directed nAbs such as 2F5 and 4E10 (69). All of the three nAbs (2G12, 2F5, and 4E10) tested in our study are able to recognize denatured or native HIV-1 Env, albeit to different degrees (Fig. 1 and 4). 2G12 is superior to 2F5 and 4E10 in binding to native Env since this nAb reacts to provirus-activated ACH-2 cells at the highest degree (Fig. 4B). In addition, 2G12 coated to microplates captures free HIV-1 particles of either HIV-1 laboratory strains or primary isolates more efficiently than 2F5 and 4E10 (data not shown). Given that 2G12 is unique among the broadly nAbs because it targets the Env glycan structures that usually function to shield HIV-1 virions from Ab-mediated neutralization, this nAb is effective in neutralizing HIV-1 at low concentrations compared to other nAbs (69, 70). For example, 2G12 at 4.85 μg/ml can neutralize 80% of HIV-1 infection (IC80), whereas 2F5 and 4E10 require 9.42 μg/ml and 8.98 μg/ml to achieve an IC80 (69). In our study, the reactions of all nAbs used at the highest concentration (5 μg/ml) have not reached the plateau (Fig. 1A), and the reaction profiles may change if the nAb concentrations are increased. Thus, the superior reactive activity of 2G12 observed in the present study may be related to the Ab concentration (5 μg/ml) used, rather than its binding epitope location.
In comparison to the three nAbs, the four non-nAbs tested exhibit moderate to weak degrees of Env recognition. Among the four non-Abs, A32 is a potent mediator of ADCC (71). A32 epitope is expressed on the surface of HIV-1-infected CD4+ T cells earlier than the epitopes recognized by other Abs such as 2G12 (71). In addition, A32 at 0.25 μg/ml can start to react to Env on the surface of HIV-1-infected cells and the reaction reaches a peak of mean fluorescence intensity at 10 μg/ml (71). In our study, we found that A32 at 5 μg/ml could recognize its epitope of Env, either purified soluble gp160 (Fig. 1A), protein extracts from HIV-1 virions of laboratory strains (Fig. 1B) or primary isolates (Fig. 1C), Env on the surface of provirus-activated cells (Fig. 4B), and denatured Env (Fig. 4C). These reactions triggered ADCML of HIV-1 virions (Fig. 2D and E and Fig. 3) and provirus-activated ACH-2 cells (Fig. 5) upon blockage of CD59 function. Interestingly, N5-i5, an anti-gp120 non-nAb that exhibits similar binding and ADCC effects as A32 (44), also showed very similar activity in recognition of Env and in triggering ADCML of HIV-1 virions (Fig. 2D and E and Fig. 3) and provirus-activated ACH-2 cells (Fig. 5) upon blockage of CD59 function. 2.2C showed a weak reaction to all types of Env probably because the MAb concentration used was not high enough. N12-i15 did not react with any type of these Env proteins because this MAb recognizes a conformational gp120 epitope that is exposed only after Env binding to CD4 (44). Our data demonstrate that non-nAbs that are able to react with Env can trigger ADCML of HIV-1 virions and provirus-activated latently infected cells if CD59 function is blocked.
Because CD59 is widely expressed on almost all human cells and protects self-cells from destruction by complement activation, potential side effects may be the main obstacle in clinical application of CD59 blockers. However, accumulating data suggest that short-term or transient blockage of CD59 may be feasible and tolerable to HIV-1 patients, since CD59 blockage, deficiency, or knockout does not cause severe off-target effects in vitro and in vivo (72–80). We and others have demonstrated that the use of CD59 blockers in vitro to block CD59 does not cause nonspecific cytolytic effect on cell lines, human RBCs, or PBMCs in the presence of sera/plasma from HIV-1-infected patients (33–35). Thus, CD59 blockers that are expected to be used short term, not lifelong, will be better tolerated in HIV-1 patients. However, unmodified full-length hCD59-blocking Abs such as BRIC229 may have limitations in future clinical trials due to their molecular size, immunogenicity, and Fc effect. In order to overcome these limitations, we generated F(ab′)2 fragments of BRIC229 (Fig. 2A). We found that BRIC229 F(ab′)2 fragments retained the functional activity of intact BRIC229 molecules in blocking the function of hCD59. Similar to intact BRIC229, BRIC229 F(ab′)2 fragments blocked hCD59 function in a dose-dependent manner (Fig. 2B and C). BRIC229 or BRIC229 F(ab′)2 fragments at 20 μg/ml completely blocked hCD59 function as ADCML of hRBCs reached almost 100%, whereas hRBCs were resistant to ADCML in the absence of BRIC229 or BRIC229 F(ab′)2 fragments (Fig. 2C). Importantly, BRIC229 F(ab′)2 fragments restored activities of both nAbs and non-nAbs in triggering ADCML of HIV-1 laboratory strains to a similar degree as intact BRIC229 molecules (Fig. 2D and E). Ab fragments of F(ab′)2 offer several advantages over intact Ab molecules for in vivo use, including (i) being more efficient in circulation and penetration of bloodstream and tissues because of their smaller size, (ii) elimination of Fc-associated effector functions (e.g., complement fixation or nonspecific binding that results from Fc interaction with Fc receptor carrying cells, and (iii) lower immunogenicity than unmodified full-length Ab. Thus, a BRIC229 F(ab′)2 fragment has advantages over its full size Ab in future clinical applications. However, BRIC229 F(ab′)2 fragments of mouse MAb are still highly immunogenic in humans, especially when repeated administration is required. As a means of circumventing this problem, humanization strategies have been developed to preserve the specificity and affinity of the rodent MAbs, whereas significantly or completely eliminate their immunogenicity in humans. Humanized MAbs such as rituximab (a murine anti-human CD20 MAb) have been successfully administered to cancer patients for prolonged treatment duration without causing severe side effects (81, 82). The excellent safety and efficacy of therapeutic humanized MAbs against cancer provide the rationale to generate humanized CD59 MAbs as a therapeutic agent in combination with anti-HIV-1 Abs for purging HIV-1 virions and infected cells.
We have recently reported that blockage of CD59 renders provirus-activated ACH-2 cells and primary human CD4+ T cells that were latently infected with HIV-1 sensitive to ADCML by anti-HIV-1 polyclonal Abs or plasma from HIV-1-infected patients (35). In the present study, we used the well-characterized anti-HIV-1 nAbs and non-nAbs to clarify whether blockage of hCD59 function could enable nAbs, non-nAbs, or both to trigger ADCML of latently HIV-1-infected cells upon provirus activation. Our confocal microscopy data showed that the binding epitopes of these Abs were colocalized with CD59 in lipid rafts on the surface of provirus-activated ACH-2 cells (Fig. 4D, white in Merge panel). This suggests that Env may directly interact with RCA molecules on the infected cell surface to escape Ab immunity. Blockage of CD59 function can restore the functions of both nAbs and non-nAbs to trigger ADCML of provirus-activated latently HIV-1-infected cells (Fig. 5). The lysis resulted from specific cell killing by anti-HIV-1 Env Ab-mediated ADCML, since lysis did not occur in both N12-i15 non-nAb that had no reaction with Env and the controls, including (i) no BRIC229, (ii) irrelevant human IgG1 in place of anti-HIV-1 Env nAbs or non-nAbs, (iii) heat-inactivated complement, (v) ACH-2 cells treated with DMSO, and (v) A3.01 cells that are ACH-2 parental cells prior to HIV-1-infection (Fig. 5, lower panel). Since latently infected cells constitute the major reservoir of HIV-1 in vivo, these cells represent a major obstacle for HIV-1 eradication (83–87). A combination of provirus stimulants with RCA blockers represents a novel approach to purge latently HIV-1-infected cells, thereby has the potential to eradicate HIV-1 infection.
ACKNOWLEDGMENTS
This study was supported in part by Grand Challenges Explorations Phase II through the Bill and Melinda Gates Foundation (grant OPP1035237 to Y.Q.), the NIH (grants R21AI104268 and R01AI117835 to Y.Q.), the Showalter Research Trust Fund (Y.Q.), the National Natural Science Foundation of Zhejiang Province (grant Y2110608 to H.N.), and Research Facilities Improvement program grant C06 RR015481-01 from the National Center for Research Resources (NIH) to the Indiana University School of Medicine.
The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH: ACH-2 cells and A3.01 cells from Thomas Folks (88, 89), nAbs (2F5, 2G12, and 4E10) and HIV-1 strains, including two R5 (JR-CSF and BAL), three X4 (LAI, IIIB, and MN) and one dual R5X4 (RF) strains. We thank Yongjun Guan at University of Maryland (Baltimore, MD) and James Robinson at Tulane University (New Orleans, LA) for the non-nAbs (2.2C, A32, N5-i5, and N12-i15).
REFERENCES
- 1.Pincus SH, Messer KG, Nara PL, Blattner WA, Colclough G, Reitz M. 1994. Temporal analysis of the antibody response to HIV envelope protein in HIV-infected laboratory workers. J Clin Invest 93:2505–2513. doi: 10.1172/JCI117260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belec L, Dupre T, Prazuck T, Tevi-Benissan C, Kanga JM, Pathey O, Lu XS, Pillot J. 1995. Cervicovaginal overproduction of specific IgG to human immunodeficiency virus (HIV) contrasts with normal or impaired IgA local response in HIV infection. J Infect Dis 172:691–697. doi: 10.1093/infdis/172.3.691. [DOI] [PubMed] [Google Scholar]
- 3.Binley JM, Klasse PJ, Cao Y, Jones I, Markowitz M, Ho DD, Moore JP. 1997. Differential regulation of the antibody responses to Gag and Env proteins of human immunodeficiency virus type 1. J Virol 71:2799–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pellegrin I, Legrand E, Neau D, Bonot P, Masquelier B, Pellegrin JL, Ragnaud JM, Bernard N, Fleury HJ. 1996. Kinetics of appearance of neutralizing antibodies in 12 patients with primary or recent HIV-1 infection and relationship with plasma and cellular viral loads. J AIDS Hum Retrovirol 11:438–447. doi: 10.1097/00042560-199604150-00003. [DOI] [PubMed] [Google Scholar]
- 5.Richman DD, Wrin T, Little SJ, Petropoulos CJ. 2003. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc Natl Acad Sci U S A 100:4144–4149. doi: 10.1073/pnas.0630530100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aasa-Chapman MM, Hayman A, Newton P, Cornforth D, Williams I, Borrow P, Balfe P, McKnight A. 2004. Development of the antibody response in acute HIV-1 infection. AIDS 18:371–381. doi: 10.1097/00002030-200402200-00002. [DOI] [PubMed] [Google Scholar]
- 7.Humbert M, Dietrich U. 2006. The role of neutralizing antibodies in HIV infection. AIDS Rev 8:51–59. [PubMed] [Google Scholar]
- 8.Bonsignori M, Moody MA, Parks RJ, Holl TM, Kelsoe G, Hicks CB, Vandergrift N, Tomaras GD, Haynes BF. 2009. HIV-1 envelope induces memory B cell responses that correlate with plasma antibody levels after envelope gp120 protein vaccination or HIV-1 infection. J Immunol 183:2708–2717. doi: 10.4049/jimmunol.0901068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stamatatos L, Morris L, Burton DR, Mascola JR. 2009. Neutralizing antibodies generated during natural HIV-1 infection: good news for an HIV-1 vaccine? Nat Med 15:866–870. doi: 10.1038/nm.1949. [DOI] [PubMed] [Google Scholar]
- 10.Huber M, Trkola A. 2007. Humoral immunity to HIV-1: neutralization and beyond. J Intern Med 262:5–25. doi: 10.1111/j.1365-2796.2007.01819.x. [DOI] [PubMed] [Google Scholar]
- 11.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Q, Yu R, Qin X. 2010. The good and evil of complement activation in HIV-1 infection. Cell Mol Immunol 7:334–340. doi: 10.1038/cmi.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLinden R, Paris R, Polonis V, Close N, Su Z, Shikuma C, Margolis D, Kim J. 2012. Association of HIV neutralizing antibody with lower viral load after treatment interruption in a prospective trial (A5170). AIDS 26:1452. doi: 10.1097/QAD.0b013e3283550b8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bar KJ, Tsao CY, Iyer SS, Decker JM, Yang Y, Bonsignori M, Chen X, Hwang KK, Montefiori DC, Liao HX, Hraber P, Fischer W, Li H, Wang S, Sterrett S, Keele BF, Ganusov VV, Perelson AS, Korber BT, Georgiev I, McLellan JS, Pavlicek JW, Gao F, Haynes BF, Hahn BH, Kwong PD, Shaw GM. 2012. Early low-titer neutralizing antibodies impede HIV-1 replication and select for virus escape. PLoS Pathog 8:e1002721. doi: 10.1371/journal.ppat.1002721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barouch DH, Whitney JB, Moldt B, Klein F, Oliveira TY, Liu J, Stephenson KE, Chang HW, Shekhar K, Gupta S, Nkolola JP, Seaman MS, Smith KM, Borducchi EN, Cabral C, Smith JY, Blackmore S, Sanisetty S, Perry JR, Beck M, Lewis MG, Rinaldi W, Chakraborty AK, Poignard P, Nussenzweig MC, Burton DR. 2013. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature 503:224–228. doi: 10.1038/nature12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris L. 2002. Neutralizing antibody responses to HIV-1 infection. IUBMB Life 53:197–199. doi: 10.1080/15216540212656. [DOI] [PubMed] [Google Scholar]
- 17.Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PW, Sawyer LS, Hendry RM, Dunlop N, Nara PL, Lamacchia M, Garratty E, Stiehm ER, Bryson YJ, Cao Y, Moore JP, Ho DD, Barbas CF III. 1994. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024–1027. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 18.Zwick MB, Labrijn AF, Wang M, Spenlehauer C, Saphire EO, Binley JM, Moore JP, Stiegler G, Katinger H, Burton DR, Parren PW. 2001. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol 75:10892–10905. doi: 10.1128/JVI.75.22.10892-10905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stiegler G, Kunert R, Purtscher M, Wolbank S, Voglauer R, Steindl F, Katinger H. 2001. A potent cross-clade neutralizing human monoclonal antibody against a novel epitope on gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses 17:1757–1765. doi: 10.1089/08892220152741450. [DOI] [PubMed] [Google Scholar]
- 20.Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Srinivasan K, Sodroski J, Moore JP, Katinger H. 1996. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J Virol 70:1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manrique A, Rusert P, Joos B, Fischer M, Kuster H, Leemann C, Niederost B, Weber R, Stiegler G, Katinger H, Gunthard HF, Trkola A. 2007. In vivo and in vitro escape from neutralizing antibodies 2G12, 2F5, and 4E10. J Virol 81:8793–8808. doi: 10.1128/JVI.00598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. 2005. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22:163–173. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 23.Trkola A, Kuster H, Rusert P, Joos B, Fischer M, Leemann C, Manrique A, Huber M, Rehr M, Oxenius A, Weber R, Stiegler G, Vcelar B, Katinger H, Aceto L, Gunthard HF. 2005. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med 11:615–622. doi: 10.1038/nm1244. [DOI] [PubMed] [Google Scholar]
- 24.Hangartner L, Zellweger RM, Giobbi M, Weber J, Eschli B, McCoy KD, Harris N, Recher M, Zinkernagel RM, Hengartner H. 2006. Nonneutralizing antibodies binding to the surface glycoprotein of lymphocytic choriomeningitis virus reduce early virus spread. J Exp Med 203:2033–2042. doi: 10.1084/jem.20051557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward R. 2009. Mechanisms of protection against rotavirus infection and disease. Pediatr Infect Dis J 28:S57–S59. doi: 10.1097/INF.0b013e3181967c16. [DOI] [PubMed] [Google Scholar]
- 26.Taylor G, Stott EJ, Bew M, Fernie BF, Cote PJ, Collins AP, Hughes M, Jebbett J. 1984. Monoclonal antibodies protect against respiratory syncytial virus infection in mice. Immunology 52:137–142. [PMC free article] [PubMed] [Google Scholar]
- 27.Walsh EE, Hall CB, Schlesinger JJ, Brandriss MW, Hildreth S, Paradiso P. 1989. Comparison of antigenic sites of subtype-specific respiratory syncytial virus attachment proteins. J Gen Virol 70(Pt 11):2953–2961. [DOI] [PubMed] [Google Scholar]
- 28.Hraber P, Seaman MS, Bailer RT, Mascola JR, Montefiori DC, Korber BT. 2014. Prevalence of broadly neutralizing antibody responses during chronic HIV-1 infection. AIDS 28:163–169. doi: 10.1097/QAD.0000000000000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin BG, Yun MR, Kim SS, Kim GJ. 2011. Neutralizing antibody responses and evolution of the viral envelope in the course of HIV-1 Korean clade B infection. Osong Public Health Res Perspect 2:151–157. doi: 10.1016/j.phrp.2011.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saifuddin M, Hedayati T, Atkinson JP, Holguin MH, Parker CJ, Spear GT. 1997. Human immunodeficiency virus type 1 incorporates both glycosyl phosphatidylinositol-anchored CD55 and CD59 and integral membrane CD46 at levels that protect from complement-mediated destruction. J Gen Virol 78(Pt 8):1907–1911. [DOI] [PubMed] [Google Scholar]
- 31.Stoiber H, Pinter C, Siccardi AG, Clivio A, Dierich MP. 1996. Efficient destruction of human immunodeficiency virus in human serum by inhibiting the protective action of complement factor H and decay accelerating factor (DAF, CD55) J Exp Med 183:307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saifuddin M, Parker CJ, Peeples ME, Gorny MK, Zolla-Pazner S, Ghassemi M, Rooney IA, Atkinson JP, Spear GT. 1995. Role of virion-associated glycosylphosphatidylinositol-linked proteins CD55 and CD59 in complement resistance of cell line-derived and primary isolates of HIV-1. J Exp Med 182:501–509. doi: 10.1084/jem.182.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu W, Yu Q, Hu N, Byrd D, Amet T, Shikuma C, Shiramizu B, Halperin JA, Qin X. 2010. A high-affinity inhibitor of human CD59 enhances complement-mediated virolysis of HIV-1: implications for treatment of HIV-1/AIDS. J Immunol 184:359–368. doi: 10.4049/jimmunol.0902278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitz J, Zimmer JP, Kluxen B, Aries S, Bogel M, Gigli I, Schmitz H. 1995. Antibody-dependent complement-mediated cytotoxicity in sera from patients with HIV-1 infection is controlled by CD55 and CD59. J Clin Invest 96:1520–1526. doi: 10.1172/JCI118190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan J, Yang K, Byrd D, Hu N, Amet T, Shepherd N, Desai M, Gao J, Gupta S, Sun Y, Yu Q. 2014. Provirus activation plus CD59 blockage triggers antibody-dependent complement-mediated lysis of latently HIV-1-infected cells. J Immunol 193:3577–3589. doi: 10.4049/jimmunol.1303030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchacher A, Predl R, Strutzenberger K, Steinfellner W, Trkola A, Purtscher M, Gruber G, Tauer C, Steindl F, Jungbauer A, Katinger H. 1994. Generation of human monoclonal antibodies against HIV-1 proteins: electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res Hum Retroviruses 10:359–369. doi: 10.1089/aid.1994.10.359. [DOI] [PubMed] [Google Scholar]
- 37.Mascola JR, Lewis MG, Stiegler G, Harris D, VanCott TC, Hayes D, Louder MK, Brown CR, Sapan CV, Frankel SS, Lu Y, Robb ML, Katinger H, Birx DL. 1999. Protection of Macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J Virol 73:4009–4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Etemad-Moghadam B, Sun Y, Nicholson EK, Karlsson GB, Schenten D, Sodroski J. 1999. Determinants of neutralization resistance in the envelope glycoproteins of a simian-human immunodeficiency virus passaged in vivo. J Virol 73:8873–8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crawford JM, Earl PL, Moss B, Reimann KA, Wyand MS, Manson KH, Bilska M, Zhou JT, Pauza CD, Parren PW, Burton DR, Sodroski JG, Letvin NL, Montefiori DC. 1999. Characterization of primary isolate-like variants of simian-human immunodeficiency virus. J Virol 73:10199–10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Purtscher M, Trkola A, Grassauer A, Schulz PM, Klima A, Dopper S, Gruber G, Buchacher A, Muster T, Katinger H. 1996. Restricted antigenic variability of the epitope recognized by the neutralizing gp41 antibody 2F5. AIDS 10:587–593. doi: 10.1097/00002030-199606000-00003. [DOI] [PubMed] [Google Scholar]
- 41.Purtscher M, Trkola A, Gruber G, Buchacher A, Predl R, Steindl F, Tauer C, Berger R, Barrett N, Jungbauer A, Katinger H. 1994. A broadly neutralizing human monoclonal antibody against gp41 of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses 10:1651–1658. doi: 10.1089/aid.1994.10.1651. [DOI] [PubMed] [Google Scholar]
- 42.Thali M, Moore JP, Furman C, Charles M, Ho DD, Robinson J, Sodroski J. 1993. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J Virol 67:3978–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiang SH, Doka N, Choudhary RK, Sodroski J, Robinson JE. 2002. Characterization of CD4-induced epitopes on the HIV type 1 gp120 envelope glycoprotein recognized by neutralizing human monoclonal antibodies. AIDS Res Hum Retroviruses 18:1207–1217. doi: 10.1089/08892220260387959. [DOI] [PubMed] [Google Scholar]
- 44.Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, Fouts TR, Gallo RC, DeVico AL, Lewis GK. 2013. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc Natl Acad Sci U S A 110:E69–E78. doi: 10.1073/pnas.1217609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore JP, Thali M, Jameson BA, Vignaux F, Lewis GK, Poon SW, Charles M, Fung MS, Sun B, Durda PJ, Akerblom L, Wahren B, Ho DD, Sattentau QJ, Sodroski J. 1993. Immunochemical analysis of the gp120 surface glycoprotein of human immunodeficiency virus type 1: probing the structure of the C4 and V4 domains and the interaction of the C4 domain with the V3 loop. J Virol 67:4785–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wyatt R, Moore J, Accola M, Desjardin E, Robinson J, Sodroski J. 1995. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J Virol 69:5723–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smalls-Mantey A, Doria-Rose N, Klein R, Patamawenu A, Migueles SA, Ko SY, Hallahan CW, Wong H, Liu B, You L, Scheid J, Kappes JC, Ochsenbauer C, Nabel GJ, Mascola JR, Connors M. 2012. Antibody-dependent cellular cytotoxicity against primary HIV-infected CD4+ T cells is directly associated with the magnitude of surface IgG binding. J Virol 86:8672–8680. doi: 10.1128/JVI.00287-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silbermann B, Tod M, Desaint C, Pialoux G, Petitprez K, Slama L, Poncelet H, Moreau C, Mazarin V, Heshmati F, Salmon-Ceron D, Guillet JG, Launay O. 2008. Short communication: long-term persistence of vaccine-induced HIV seropositivity among healthy volunteers. AIDS Res Hum Retroviruses 24:1445–1448. doi: 10.1089/aid.2008.0107. [DOI] [PubMed] [Google Scholar]
- 49.Byrd D, Amet T, Hu N, Lan J, Hu S, Yu Q. 2013. Primary human leukocyte subsets differentially express vaccinia virus receptors enriched in lipid rafts. J Virol 87:9301–9312. doi: 10.1128/JVI.01545-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ono A. 2010. Relationships between plasma membrane microdomains and HIV-1 assembly. Biol Cell 102:335–350. doi: 10.1042/BC20090165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muranyi W, Malkusch S, Muller B, Heilemann M, Krausslich HG. 2013. Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal tail. PLoS Pathog 9:e1003198. doi: 10.1371/journal.ppat.1003198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malkusch S, Muranyi W, Muller B, Krausslich HG, Heilemann M. 2013. Single-molecule coordinate-based analysis of the morphology of HIV-1 assembly sites with near-molecular spatial resolution. Histochem Cell Biol 139:173–179. doi: 10.1007/s00418-012-1014-4. [DOI] [PubMed] [Google Scholar]
- 53.Ray K, Mengistu M, Yu L, Lewis GK, Lakowicz JR, DeVico AL. 2014. Antigenic properties of the HIV envelope on virions in solution. J Virol 88:1795–1808. doi: 10.1128/JVI.03048-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buratti E, McLain L, Tisminetzky S, Cleveland SM, Dimmock NJ, Baralle FE. 1998. The neutralizing antibody response against a conserved region of human immunodeficiency virus type 1 gp41 (amino acid residues 731-752) is uniquely directed against a conformational epitope. J Gen Virol 79(Pt 11):2709–2716. [DOI] [PubMed] [Google Scholar]
- 55.Muster T, Steindl F, Purtscher M, Trkola A, Klima A, Himmler G, Ruker F, Katinger H. 1993. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J Virol 67:6642–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Binley JM, Wrin T, Korber B, Zwick MB, Wang M, Chappey C, Stiegler G, Kunert R, Zolla-Pazner S, Katinger H, Petropoulos CJ, Burton DR. 2004. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J Virol 78:13232–13252. doi: 10.1128/JVI.78.23.13232-13252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muster T, Guinea R, Trkola A, Purtscher M, Klima A, Steindl F, Palese P, Katinger H. 1994. Cross-neutralizing activity against divergent human immunodeficiency virus type 1 isolates induced by the gp41 sequence ELDKWAS. J Virol 68:4031–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caskey M, Klein F, Lorenzi JC, Seaman MS, West AP Jr, Buckley N, Kremer G, Nogueira L, Braunschweig M, Scheid JF, Horwitz JA, Shimeliovich I, Ben-Avraham S, Witmer-Pack M, Platten M, Lehmann C, Burke LA, Hawthorne T, Gorelick RJ, Walker BD, Keler T, Gulick RM, Fatkenheuer G, Schlesinger SJ, Nussenzweig MC. 2015. Viremia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 522:487–491. doi: 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Forthal DN, Landucci G, Daar ES. 2001. Antibody from patients with acute human immunodeficiency virus (HIV) infection inhibits primary strains of HIV type 1 in the presence of natural-killer effector cells. J Virol 75:6953–6961. doi: 10.1128/JVI.75.15.6953-6961.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Forthal DN, Landucci G, Keenan B. 2001. Relationship between antibody-dependent cellular cytotoxicity, plasma HIV type 1 RNA, and CD4+ lymphocyte count. AIDS Res Hum Retroviruses 17:553–561. doi: 10.1089/08892220151126661. [DOI] [PubMed] [Google Scholar]
- 61.Nakane T, Nomura T, Shi S, Nakamura M, Naruse TK, Kimura A, Matano T, Yamamoto H. 2013. Limited impact of passive non-neutralizing antibody immunization in acute SIV infection on viremia control in rhesus macaques. PLoS One 8:e73453. doi: 10.1371/journal.pone.0073453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burton DR, Hessell AJ, Keele BF, Klasse PJ, Ketas TA, Moldt B, Dunlop DC, Poignard P, Doyle LA, Cavacini L, Veazey RS, Moore JP. 2011. Limited or no protection by weakly or nonneutralizing antibodies against vaginal SHIV challenge of macaques compared with a strongly neutralizing antibody. Proc Natl Acad Sci U S A 108:11181–11186. doi: 10.1073/pnas.1103012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu P, Liu J, Bess J Jr, Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH. 2006. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 64.Dong XN, Chen YH. 2006. Neutralizing epitopes in the membrane-proximal region of HIV-1 gp41: genetic variability and co-variation. Immunol Lett 106:180–186. doi: 10.1016/j.imlet.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 65.Montero M, Gulzar N, Klaric KA, Donald JE, Lepik C, Wu S, Tsai S, Julien JP, Hessell AJ, Wang S, Lu S, Burton DR, Pai EF, Degrado WF, Scott JK. 2012. Neutralizing epitopes in the membrane-proximal external region of HIV-1 gp41 are influenced by the transmembrane domain and the plasma membrane. J Virol 86:2930–2941. doi: 10.1128/JVI.06349-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sattentau QJ. 1995. Conservation of HIV-1 gp120 neutralizing epitopes after formalin inactivation. AIDS 9:1383–1385. doi: 10.1097/00002030-199512000-00017. [DOI] [PubMed] [Google Scholar]
- 67.Braibant M, Brunet S, Costagliola D, Rouzioux C, Agut H, Katinger H, Autran B, Barin F. 2006. Antibodies to conserved epitopes of the HIV-1 envelope in sera from long-term non-progressors: prevalence and association with neutralizing activity. AIDS 20:1923–1930. doi: 10.1097/01.aids.0000247113.43714.5e. [DOI] [PubMed] [Google Scholar]
- 68.Kwong PD, Mascola JR, Nabel GJ. 2011. Rational design of vaccines to elicit broadly neutralizing antibodies to HIV-1. Cold Spring Harb Perspect Med 1:a007278. doi: 10.1101/cshperspect.a007278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kwong PD, Mascola JR, Nabel GJ. 2013. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat Rev Immunol 13:693–701. doi: 10.1038/nri3516. [DOI] [PubMed] [Google Scholar]
- 70.Hessell AJ, Rakasz EG, Poignard P, Hangartner L, Landucci G, Forthal DN, Koff WC, Watkins DI, Burton DR. 2009. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog 5:e1000433. doi: 10.1371/journal.ppat.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferrari G, Pollara J, Kozink D, Harms T, Drinker M, Freel S, Moody MA, Alam SM, Tomaras GD, Ochsenbauer C, Kappes JC, Shaw GM, Hoxie JA, Robinson JE, Haynes BF. 2011. An HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent antibody-dependent cellular cytotoxicity (ADCC) activity and defines a common ADCC epitope in human HIV-1 serum. J Virol 85:7029–7036. doi: 10.1128/JVI.00171-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsuo S, Ichida S, Takizawa H, Okada N, Baranyi L, Iguchi A, Morgan BP, Okada H. 1994. In vivo effects of monoclonal antibodies that functionally inhibit complement regulatory proteins in rats. J Exp Med 180:1619–1627. doi: 10.1084/jem.180.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Holt DS, Botto M, Bygrave AE, Hanna SM, Walport MJ, Morgan BP. 2001. Targeted deletion of the CD59 gene causes spontaneous intravascular hemolysis and hemoglobinuria. Blood 98:442–449. doi: 10.1182/blood.V98.2.442. [DOI] [PubMed] [Google Scholar]
- 74.Macor P, Tripodo C, Zorzet S, Piovan E, Bossi F, Marzari R, Amadori A, Tedesco F. 2007. In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increases the antitumor activity of rituximab. Cancer Res 67:10556–10563. doi: 10.1158/0008-5472.CAN-07-1811. [DOI] [PubMed] [Google Scholar]
- 75.Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, Hillmen P, Luzzatto L, Young N, Kinoshita T, Rosse W, Socie G, International PNHIG . 2005. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 106:3699–3709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hall C, Richards S, Hillmen P. 2003. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH). Blood 102:3587–3591. doi: 10.1182/blood-2003-01-0009. [DOI] [PubMed] [Google Scholar]
- 77.Richards SJ, Hill A, Hillmen P. 2007. Recent advances in the diagnosis, monitoring, and management of patients with paroxysmal nocturnal hemoglobinuria. Cytometry B Clin Cytom 72:291–298. [DOI] [PubMed] [Google Scholar]
- 78.Azenishi Y, Ueda E, Machii T, Nishimura J, Hirota T, Shibano M, Nakao S, Kinoshita T, Mizoguchi H, Kitani T. 1999. CD59-deficient blood cells and PIG-A gene abnormalities in Japanese patients with aplastic anaemia. Br J Haematol 104:523–529. doi: 10.1046/j.1365-2141.1999.01214.x. [DOI] [PubMed] [Google Scholar]
- 79.Brodsky RA. 2006. New insights into paroxysmal nocturnal hemoglobinuria. Hematol Am Soc Hematol Educ Program 24-28:516. [DOI] [PubMed] [Google Scholar]
- 80.Brodsky RA, Hu R. 2006. PIG-A mutations in paroxysmal nocturnal hemoglobinuria and in normal hematopoiesis. Leukemia Lymphoma 47:1215–1221. doi: 10.1080/10428190600555520. [DOI] [PubMed] [Google Scholar]
- 81.Trail PA, King HD, Dubowchik GM. 2003. Monoclonal antibody drug immunoconjugates for targeted treatment of cancer. Cancer Immunol Immunother 52:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lim SH, Beers SA, French RR, Johnson PW, Glennie MJ, Cragg MS. 2010. Anti-CD20 monoclonal antibodies: historical and future perspectives. Haematologicae 95:135–143. doi: 10.3324/haematol.2008.001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sedaghat AR, Siliciano RF, Wilke CO. 2008. Low-level HIV-1 replication and the dynamics of the resting CD4+ T cell reservoir for HIV-1 in the setting of HAART. BMC Infect Dis 8:2. doi: 10.1186/1471-2334-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shen L, Siliciano RF. 2008. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J Allergy Clin Immunol 122:22–28. doi: 10.1016/j.jaci.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dahl V, Palmer S. 2009. Establishment of drug-resistant HIV-1 in latent reservoirs. J Infect Dis 199:1258–1260. doi: 10.1086/597760. [DOI] [PubMed] [Google Scholar]
- 86.Pace MJ, Agosto L, Graf EH, O'Doherty U. 2011. HIV reservoirs and latency models. Virology 411:344–354. doi: 10.1016/j.virol.2010.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lewin SR, Evans VA, Elliott JH, Spire B, Chomont N. 2011. Finding a cure for HIV: will it ever be achievable? J Int AIDS Soc 14:4. doi: 10.1186/1758-2652-14-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Clouse KA, Powell D, Washington I, Poli G, Strebel K, Farrar W, Barstad P, Kovacs J, Fauci AS, Folks TM. 1989. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J Immunol 142:431–438. [PubMed] [Google Scholar]
- 89.Folks TM, Clouse KA, Justement J, Rabson A, Duh E, Kehrl JH, Fauci AS. 1989. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc Natl Acad Sci U S A 86:2365–2368. doi: 10.1073/pnas.86.7.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]