

Abstract
The gut bile acid pool is millimolar in concentration, varies widely in composition among individuals, and is linked to metabolic disease and cancer. Although these molecules derive almost exclusively from the microbiota, remarkably little is known about which bacterial species and genes are responsible for their biosynthesis. Here, we report a biosynthetic pathway for the second most abundant class in the gut, iso (3β-hydroxy) bile acids, whose levels exceed 300 µM in some humans and are absent in others. We show, for the first time, that iso bile acids are produced by Ruminococcus gnavus, a far more abundant commensal than previously known producers; and that the iso bile acid pathway detoxifies deoxycholic acid, favoring the growth of the keystone genus Bacteroides. By revealing the biosynthetic genes for an abundant class of bile acids, our work sets the stage for predicting and rationally altering the composition of the bile acid pool.
Introduction
In the molecular milieu of the mammalian gut, bile acids are exemplars of collaborative metabolism between the host and its microbiota. The primary bile acids, cholic acid (CA) and chenodeoxycholic acid (CDCA), are synthesized from cholesterol by a series of oxidative transformations in the host’s liver; following a meal, they are secreted into the small intestine where they play an important role in digestion by acting as detergents.1 While the majority (∼97%) of bile acids are reabsorbed in the ileum and return to the liver, the remaining ∼3% enter the large intestine at a rate of 400–800 mg/day, where they exist at remarkably high concentrations (∼200 to ∼1000 µM)2. Importantly, nearly 100% of the bile acid pool in the large intestine consists of bacterially modified bile acids.
The principal actions of gut bacteria on bile acids are C24 amide hydrolysis and 7α-dehydroxylation, transforming the glycine and taurine conjugates of CA and CDCA into the secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA), respectively (Fig. 1a).3,4 Although DCA and LCA are the most abundant secondary bile acids, ∼50 different secondary bile acids have been detected in human feces.5 Notably, the relative and absolute amount of each bile acid varies widely among individuals in a manner that is influenced both by diet and the microbiota (for example, DCA: 30–700 µM, LCA: 1–450 µM).2 Because bile acids are present in high micromolar concentrations, a compound that makes up even 1% of this pool is present at a biologically relevant concentration.
Figure 1. Chemical transformations of bile acids by gut bacteria.

(a) Conversion of the primary bile acids CA and CDCA to the secondary bile acids DCA and LCA, respectively, involves the net reductive removal of the 7–OH group. (b) IsoDCA and isoLCA are 3β–OH epimers of DCA and LCA.
In addition to their role in digestion, bacterially produced bile acids affect host physiology in other ways. Various secondary bile acids are ligands for the farnesoid X receptor (FXR), the liver X receptor (LXR), the G-protein coupled receptor TGR5, and the vitamin D receptor, initiating a variety of signaling cascades relevant to metabolic and hepatic diseases.6,7,8 DCA production has been shown to limit the outgrowth of the enteric pathogen Clostridium difficile,9 and DCA and LCA have been implicated as causative agents in colon carcinogenesis.10,11 A recent study linked the development of obesity-associated hepatocellular carcinoma with DCA12; while the mode of action remains unclear, the consumption of a high-fat diet results in a larger quantity of 7α-dehydroxylating bacteria in the large intestine and higher fecal concentrations of DCA,13,14 suggesting a potential link between diet and cancer mediated by a microbiota-derived bile acid. In contrast, ursodeoxycholic acid (UDCA), the 7β isomer of CDCA, protects against the development of chemically induced colon cancer in animal studies15 and is used to treat gallstones.16
Given the biological importance of DCA, LCA, and UDCA and the fact that bile acids are among the most abundant and variable microbiota-derived metabolites, it is remarkable how little is known about the dozens of other secondary bile acids: their biological activities, how their levels vary among individuals, which bacterial strains produce them, and what genes are responsible. Several individual 7-hydroxysteroid dehydrogenase-encoding genes have been cloned,17,18,19,20 including an enzyme capable of transforming 7-oxoCDCA into UDCA.21 There is one example of a well-characterized gene cluster, the bai operon,1 which encodes enzymes responsible for the 7α-dehydroxylation of primary bile acids. However, this pathway is incomplete; the genes that comprise the reductive portion of the pathway have not yet been discovered. Identifying the bacterial genes responsible for bile acid transformations will enable the bile acid metabolic potential of a community to be predicted from metagenomic sequence data and will facilitate the use of synthetic ecology and genetic engineering to reprogram the bile acid metabolic capabilities of a gut community.
Here, we describe the delineation of a biosynthetic pathway for isoDCA (Fig. 1b), the 3β–OH epimer of DCA. We chose to focus on iso bile acids for three reasons. First, after DCA and LCA, isoDCA and isoLCA are the most abundant bile acids in the healthy human gut, with mean concentrations of approximately 50 µM (vs. 150 µM for LCA and 200 µM for DCA).2 Second, the iso bile acids vary widely in concentration among individuals, with isoDCA present at 0–390 µM and isoLCA at 0–260 µM.2 Third, iso bile acids are thought to undergo unusually rapid absorption by enterohepatic recirculation, raising the possibility that they are more readily sensed by the host than other secondary bile acids.2 We demonstrated that isoDCA has less detergent activity and causes less cell wall damage than DCA, and that the conversion of DCA to isoDCA favors the growth of the abundant genus Bacteroides.
Results
Screening candidate iso bile acid producers
Only two bacterial species, Eubacterium lentum22 (later reclassified as Eggerthella lenta) and Clostridium perfringens,23,24 had been previously shown to convert primary and secondary bile acids into their 3β isomers in whole cell culture; since these studies were performed before the advent of modern genetic tools, the genes responsible for these transformations were not identified. Furthermore, none of the strains assayed in these studies were deposited in culture collections, making them inaccessible for reanalysis. However, these studies provided valuable insight into the mechanism of this transformation ,in particular suggesting that bacterial epimerization of the 3–OH group occurs through 3-oxo-intermediates.25 Subsequent efforts identified nicotinamide-dependent 3α-hydroxysteroid dehydrogenase (HSDH) activities in the cell lysates of E. lenta26 and C. perfringens27, 28 strains capable of transforming bile acids into their 3-oxo-bile acid congeners, while 3β-HSDH activity was found in the cell lysates of Peptostreptococcus productus29 (later reclassified as Blautia producta) and Clostridium sp. 25.11.c; however, the genes responsible for these activities were never cloned or otherwise identified.30
Based on these early results, we conjectured that the biosynthetic pathway for iso bile acid formation is composed of one 3α- and one 3β-HSDH, and we set out to elucidate this pathway and uncover novel iso bile acid producers. We performed a small screen of gut commensal Firmicutes from the order Clostridiales, as this bacterial taxon possesses the vast majority of the known bile acid-metabolizing capabilities, including 7α-dehydroxylation and sterol alcohol oxidation and reduction.1 We also tested Eggerthella lenta DSM 2243, the type strain of a species from the genetically distant phylum Actinobacteria (see Supplementary Results, Supplementary Table 1 for strains screened). Identification of iso bile acid producing genes from this species and from species of Clostridiales would allow us to determine the degree of genetic conservation of this pathway across bacterial phyla.
Based on GC-MS analysis of purified culture supernatant, we determined that E. lenta DSM 2243 and two unrelated organisms, Ruminococcus gnavus ATCC 29149 and Lachnospiraceae sp. 2_1_58FAA, converted CA, CDCA, and DCA to isoCA, isoCDCA, and isoDCA, respectively (Supplementary Fig. 1). To our knowledge, this is the first report of either a Ruminococcus or Lachnospiraceae species producing iso-bile acids31, a notable finding given that R. gnavus is a more prominent member of the gut community than any previously known iso bile acid producer.32R. gnavus ATCC 29149 and Lachnospiraceae sp. 2_1_58FAA are highly similar on a genome-wide scale (average protein ortholog pair % identity = 98.8%), indicating that they are closely related strains of the same species. We therefore decided to focus our efforts on finding the genes responsible for iso bile acid production in R. gnavus ATCC 29149 and E. lenta DSM 2243.
Computational identification of candidate HSDHs
Given that the genes for the oxidative arm of the 7α-dehydroxylation pathway (baiA-I) are physically clustered, we performed a computational search for biosynthetic gene clusters that harbor at least two putative hydroxysteroid dehydrogenases. One candidate cluster harboring homologs of the bai genes was identified in E. lenta DSM 2243 (Supplementary Fig. 2), but, to our surprise, no clear candidates were found in R. gnavus ATCC 29149. As a result, we reconsidered our assumption that the genes for iso bile acid biosynthesis were contiguous, prompting us to broaden our search to consider unclustered gene candidates. Supporting this change in hypothesis, the UDCA-producing 7β-HSDH recently identified in R. gnavus is a solitary gene.21
To identify 3α- and 3β-HSDH candidates independent of genomic context, we performed BLASTP searches using a panel of 3α-HSDHs that are part of the bai operon and had been previously characterized from Clostridium hiranonis TO-931,33Clostridium scindens VPI 12708,34 and Clostridium hylemonae DSM 15053.35 From this search, 18 genes were identified: 11 from E. lenta and 7 from R. gnavus (Supplementary Table 2). Since no 3β-HSDH genes have been identified or characterized from gut bacterial species, a 3β-HSDH from the soil bacterium C. testosteroni (Accession # KGH18088) and the 7β-HSDH from R. gnavus21 were used as additional queries; no additional hits were obtained, validating the completeness of the results. Of the 18 genes, one (Rumgna_01650) was eliminated as a potential candidate because it resides within a putative fatty acid synthase-encoding cluster that is also present in Lachnospiraceae sp. 1_4_56FAA, one of the iso bile acid non-producers from the initial screen. The remaining 17 genes were cloned into pET28b and expressed as N-terminal hexahistidine fusion proteins in E. coli. Remarkably, 16 of the 17 proteins expressed in soluble form (Supplementary Fig. 3a); one protein (Elen_0666) was insoluble and was not characterized further (Supplementary Fig. 3b).
Biochemical characterization of candidate HSDH enzymes
To assess the ability of the heterologously expressed proteins to convert DCA to 3-oxoDCA or 3-oxoDCA to isoDCA, we first performed a qualitative cell lysate assay, the results of which we analyzed using TLC (Supplementary Fig. 4a, b) and confirmed by GC-MS (Supplementary Fig. 4c, d). From the 17 enzymes assayed, we identified two candidate 3α-HSDHs (Elen_0690 and Rumgna_02133) and three candidate 3β-HSDHs (Elen_0198, Elen_1325, and Rumgna_00694) (Fig. 2a).
Figure 2. Elucidation of the biosynthetic pathway for isoDCA formation inEggerthella lentaand Ruminococcus gnavus.
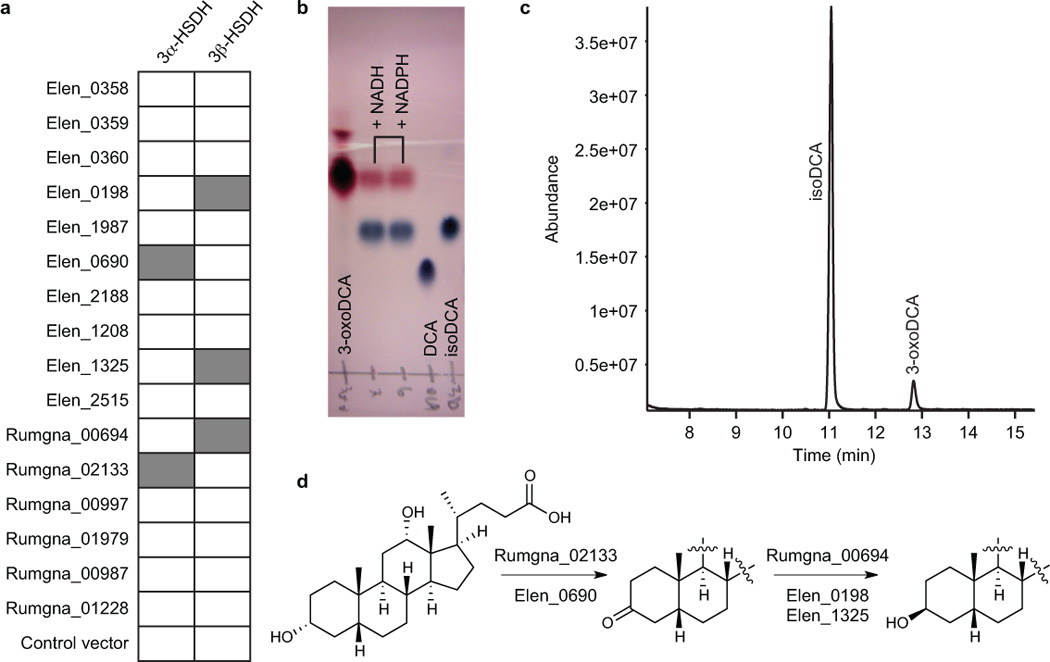
(a) Results of cell lysate assay in which candidate HSDH enzymes were expressed in E. coli and their ability to convert DCA to 3-oxoDCA (column 1) and 3-oxoDCA to isoDCA (column 2) was analyzed by TLC. Gray box: Product spot was present; white box: no product was observed. (b-c) Representative example showing enzymatic conversion by purified Rumgna_00694 of 3-oxoDCA into isoDCA by (b) TLC and (c) GC-MS. Retention times: isoDCA, 11.1 min; 3-oxoDCA, 12.9 min. In (c), NADPH was used as cofactor in the enzymatic reaction. (d) The biosynthetic pathway we elucidated for conversion of bile acids to iso bile acids involves the consecutive action of 3α- and 3β-hydroxysteroid dehydrogenases (HSDHs).
In order to confirm the results of the cell lysate assay, these five enzymes were overexpressed in E. coli and purified using Ni-NTA affinity chromatography (Supplementary Fig. 5). The activities of all five enzymes were confirmed using an assay in which purified enzyme (1 µM) was incubated in 10 mM MOPS buffer (pH 7.0) at 37 °C with substrate (DCA or 3-oxoDCA) and cofactor (NAD(P)+ or NAD(P)H) and conversion to product (3-oxoDCA or isoDCA) was confirmed by TLC (Fig. 2b, Supplementary Fig. 6) and GC-MS analysis (Fig. 2c, Supplementary Fig. 7). Although we were primarily concerned with the production of isoDCA, we also qualitatively confirmed these enzymes’ abilities to act as 3α- or 3β-HSDHs for the other bile acid substrates LCA, CDCA, and CA (Supplementary Fig. 8). The apparent lack of substrate selectivity exhibited by these enzymes suggests that they may act as general bile acid 3-epimerases. This result is consistent with our observation that E. lenta and R. gnavus convert CDCA and CA along with DCA to their iso congeners, and also with previous literature precedent that iso bile acid producers biosynthesize multiple iso variants.22,23
Next, we determined the kinetic parameters of these enzymes by measuring the change in absorbance of the nicotinamide cofactor at 340 nm.34 All of the enzymes displayed robust kinetic parameters comparable to previously characterized HSDHs (Table 1, Supplementary Fig. 9, 10, supporting the 3α- and 3β-HSDH functions proposed above. Notably, the Km values for the E. lenta HSDHs are in the low millimolar range, while those of the R. gnavus HSDHs are in 100 µM range. The reason for this difference is unclear, but may be related to the ambient bile acid concentrations to which these strains are exposed in the distinct niches they occupy in the gut. Overall, these data imply that we have elucidated the pathway for iso bile acid formation in R. gnavus and E. lenta (Figure 2d), a finding of notable importance given the abundance of iso bile acids in the human gut.
Table 1.
Kinetic parameters for 3α- and 3β-HSDHs.
| Rumgna_02133 | Elen_0690 | Rumgna_00694 | Elen_0198 | Elen_1325 | |
|---|---|---|---|---|---|
| Substrate | DCA | DCA | 3-oxoDCA | 3-oxoDCA | 3-oxoDCA |
| Cofactor | NADP+ | NAD+ | NADPH | NADH | NADH |
| Km (µM) | 369 ± 30 | 1280 ± 88 | 56.0 ± 5.6 | 2660 ± 539 | 2630 ± 365 |
| kcat (min−1) | 99.3 ± 2.0 | 291 ± 7 | 9430 ± 272 | 584 ± 35 | 1750 ± 66 |
Values represent mean ± s.e.m. from at least three independent experiments.
HSDHs are unclustered and evolutionarily diverse
Importantly, four of the five genes encoding the 3α- or 3β-HSDH enzymes are unclustered, and the fifth gene (Rumgna_00694) is found in a three-gene cluster that does not include any additional putative bile acid metabolic genes. These results suggest that bile acid metabolic genes, in contrast to nearly all other secondary metabolite biosynthetic genes, may be often unclustered, and they suggest that the oxidative and reductive steps of the iso bile acid pathway might be regulated independently of each other.
A dendrogram resulting from a multiple sequence alignment of the 18 HSDH candidates, the 5 query genes, and 42 genes belonging to the same enzyme family (PF00106, short chain dehydrogenases) reveals a surprising lack of homology among the 3α- and 3β-HSDH enzymes from E. lenta and R. gnavus (Supplementary Fig. 11). Notably, the putative 3β-HSDHs – including two enzymes from the same genome, Elen_0198 and Elen_1325, are found in separate clades and have relatively low pairwise sequence identity (36–38%). Furthermore, these three genes reside in clades with other enzymes with different functions; while Elen_1325 is found in the same clade as 3α−HSDHs, Rumgna_00694 is located alongside ketoreductases, and Elen_0198 resides in a clade with glucose dehydrogenases. Taken together, these findings suggest that 3β−HSDH activity has evolved multiple times independently from distantly related members of the short chain dehydrogenase superfamily.
Iso bile acids have less detergent activity and toxicity
The biological activities of iso bile acids have not yet been explored; this represents a notable gap in knowledge given that they can be present at high micromolar concentrations in the gut lumen. In light of the well-characterized cytotoxicity of DCA, we hypothesized that its conversion to isoDCA may decrease its growth inhibitory activity, favoring gut commensals sensitive to DCA. Deoxycholic acid acts as a detergent, solubilizing membrane lipids and causing dissociation of membrane proteins; at high concentrations (5–10 mM), membrane damage leads to cell lysis.36 Inversion of the C3–hydroxyl stereocenter to form isoDCA would disrupt the hydrophobic/hydrophilic facial nature of this molecule (Fig. 1b), likely decreasing its detergent properties and rendering it less toxic to bacteria. In order to evaluate the relative detergent capabilities of these compounds, we determined their critical micelle concentrations (CMCs). In support of our hypothesis, isoDCA has a higher CMC (12 mM) than DCA (5 mM), indicating that its detergent properties are greatly diminshed at physiological concentrations (Supplementary Fig. 12). Moreover, the aforementioned chemoprotective agent UDCA, which like isoDCA is a bile acid whose hydrophobic face is disrupted by a β-hydroxyl group, has a similar reported CMC to isoDCA (12 mM),37 further suggesting a link between diminished detergent capability and decreased toxicity.
In order to assess their relative antimicrobial activities, DCA and isoDCA were assayed against a panel of common gut commensals. The screen included Gram-negative Bacteroides species as well as Gram-positive Firmicutes and Actinobacteria, including the iso bile acid producers E. lenta and R. gnavus. While the growth of E. lenta was unaffected by the addition of compound at the concentrations assayed, R. gnavus displayed more robust growth in the presence of isoDCA than DCA, suggesting that R. gnavus benefits from its production of isoDCA (Table 2). Importantly, the growth of the four Bacteroides species assayed also differed in the presence of the two compounds, with minimal inhibitory concentrations of isoDCA that were 3- to 4-fold higher than those of DCA (Table 2). The growth of C. sporogenes and E. faecalis was similarly affected, although to a lesser degree. In order to investigate in more detail the growth of Bacteroides species in the presence of physiological, sub-inhibitory concentrations2 of compound, B. ovatus was grown in the presence of 250 µM DCA, isoDCA, or solvent (DMSO) control. A representative growth curve shows a lag in the growth of DCA-treated cultures in comparison to the DMSO control, while growth of isoDCA-treated cultures mirrors that of the control (Fig. 3a). In addition, the saturated cell density of DCA-treated cultures is lower than that of isoDCA- or DMSO-treated cultures (Fig. 3b). These observations are consistent with the possibility that at physiological bile acid concentrations, the conversion of DCA to isoDCA by R. gnavus or E. lenta favors the growth of Bacteroides.
Table 2.
Minimal inhibitory concentrations for isoDCA and DCA for selected Gram-negative and Gram-positive gut commensals
| B. theta | B. vulgatus | B. uniformis | B. ovatus | C. sporogenes | E. faecalis | R. gnavus | |
|---|---|---|---|---|---|---|---|
| DCA | 350 µM | 400 µM | 350 µM | 400 µM | 500 µM | 800 µM | 600 µM |
| isoDCA | 1.5 mM | 1.0 mM | 1.0 mM | 1.5 mM | 1.5 mM | 1.5 mM | 1.5 mM |
Data are representative of at least three independent experiments.
Figure 3. IsoDCA is less bacteriostatic than DCA.

(a) B. ovatus growth rate in the presence of 250 µM DCA, 250 µM isoDCA, or DMSO control. Data shown are from one experiment with three technical replicates. Error bars represent standard deviation. (b) Saturated culture density of B. ovatus was lower in DCA-treated versus isoDCA-treated (P = 0.0002) or DMSO-treated cultures (P < 0.0001) (asterisks represent significant differences between groups, Welch’s t test, n = 3 per group). (c) Co-culture experiment showing the growth of B. ovatus in the presence of either the isoDCA producer E. lenta or the common gut commensal and non-producer Eubacterium eligens with or without 300 µM DCA (starting concentration). Data shown are from four biological replicates. Error bars represent standard deviation. (d) Cell membrane damage caused by exposure of actively dividing B. ovatus cells to either DCA or isoDCA was assessed using a propidium iodide (Pi) fluorescence assay. IsoDCA caused less membrane damage than DCA at both 200 µM (½ DCA MIC, P = 0.0158) and 400 µM (DCA MIC, P = 0.0367). Data shown are from three biological replicates. Error bars represent standard deviation. Asterisks represent significant differences between groups, Welch’s t test, n = 3 per group. (DMSO vs. 200 µM DCA, P = 0.0068; DMSO vs. 200 µM isoDCA, P <0.0001; DMSO vs. 400 µM DCA, P = 0.0004; DMSO vs. 400 µM isoDCA, P = 0.0435.)
IsoDCA benefits the non-producer Bacteroides
To test this hypothesis, we performed a co-culture experiment in which we cultivated B. ovatus in the presence of a physiologically relevant, sub-minimal inhibitory concentration of DCA (300 µM) along with either the isoDCA producer E. lenta or the common gut commensal and non-isoDCA producer Eubacterium eligens. E. eligens was chosen as a control strain for E. lenta because it is a common gut commensal that does not metabolize DCA, and unlike the majority of strains assayed in this study, its growth is not significantly inhibited in the presence of 300 µM DCA. Following co-inoculation, growth was monitored by measuring colony-forming units (CFUs) at 0, 5, and 21 h. In the absence of DCA, B. ovatus grew robustly in binary culture with either E. lenta or E. eligens. However, in the presence of DCA and the non-DCA-metabolizer E. eligens, B. ovatus did not divide and its cell numbers drop by t = 21 h, consistent with the growth inhibitory effect of DCA on Bacteroides. In contrast, in the presence of DCA and the isoDCA producer E. lenta, B. ovatus continued to grow robustly (Fig. 3c, Supplementary Fig. 13). In combination with the MIC and B. ovatus growth data above, these results strongly suggest that the conversion of DCA to isoDCA by E. lenta favors the growth of Bacteroides.
Effect of bile acids on cell membrane integrity
In order to further investigate the mechanism by which isoDCA and DCA exert their differential effect on bacterial growth, we sought to determine whether these bile acids have a direct effect on bacterial membrane integrity at submicellar concentrations. Exposure of both bacterial and mammalian cells to super-micellar concentrations of bile acids leads is known to lead to membrane damage and lysis.36 However, at low/submicellar concentrations that are more physiologically relevant, the source of bile salt toxicity is less clear. It has been suggested that at lower concentrations, these compounds may disrupt membrane integrity by alternative mechanisms that include changing the activity of membrane-bound enzymes,36 affecting the physical properties of cell surfaces,38 and increasing cellular permeability.39 We incubated B. ovatus and R. gnavus cultures during log phase growth in the presence of DCA at its minimal inhibitory concentration (MIC) or ½ MIC. We also cultivated B. ovatus and R. gnavus in the presence of the same concentration of isoDCA. Each culture was then treated with the nucleic acid stain propidium iodide (Pi), which cannot pass through intact cellular membranes and is commonly used to evaluate bacterial viability. Only in severely damaged cells can the large, hydrophilic compound pass through the hydrophobic membrane, bind to DNA, and emit strong red (FL3) fluorescence.40 Our results indicate that DCA compromises membrane integrity at physiologically relevant concentrations (200 µM to 400 µM) in both species in a dose-dependent manner (Fig. 3d, Supplementary Figure 14). In addition, exposure to isoDCA results in less membrane damage than DCA, suggesting that its diminished bacterial toxicity can be explained at least in part by its diminished effect on cell membrane integrity.
Discussion
We have uncovered a biosynthetic pathway for an abundant, microbiota-derived bile acid, isoDCA, and revealed that this pathway is harbored not only by Eggerthella lenta but also by the prominent gut commensal Ruminococcus gnavus. We also show that isoDCA is less cytotoxic than its parent compound, DCA, for the abundant commensal genus Bacteroides through a mechanism that involves diminished cell membrane damage, uncovering a potential role for iso bile acids in modulating gut community composition. Furthermore, isoDCA production is an example of a collaborative biosynthetic pathway in which bacteria possessing the bai operon (e.g. Clostridium scindens, C. hiranonis) 7α-dehydroxylate CA to form DCA, which is then transformed into isoDCA by E. lenta and R. gnavus. It has become standard practice to use the bai genes to predict the level of DCA in a gut community. Our results suggest that the levels of the isoDCA biosynthetic genes must also be taken into account, given their ability to divert DCA to isoDCA. These results underscore the impact that metagenomics can have on advancing a mechanistic understanding of molecular transformations carried out by the microbiota, and they highlight the importance of further efforts to characterize the metabolism of bile acids by gut bacteria.
Online Methods
Materials, general methods, and instrumentation
Bacterial strains were obtained from ATCC, DSMZ, Japan Collection of Microorganisms (JCM), or BEI Resources. Strains were grown under anaerobic conditions (75% N2, 20% CO2, 5% H2) at 37 °C in an anaerobic chamber (Coy Laboratory Products). Growth medium was kept in the chamber overnight to ensure that it was anaerobic prior to use. Bacteria were grown in reinforced clostridial medium (RCM), Eggerth-Gagnon medium (EG), Difco Brain Heart Infusion medium supplemented with 0.02% fructose w/v (BHI-F)35, peptone yeast extract fructose medium (PYF),41 or tryptone-yeast extract glucose medium (TYG). Escherichia coli strains were grown aerobically at 37 °C (unless otherwise noted) in Luria Broth (LB) medium supplemented with antibiotic as indicated. E. coli TOP10 and BL21(DE3) pLysS chemically competent cells were purchased from Invitrogen. Plasmid pET28b was purchased from Novagen. Oligonucleotide primers were synthesized by Elim Biopharm. Phusion High Fidelity DNA Polymerase, restriction enzymes, and T4 DNA ligase were purchased from New England Biolabs. IsoDCA used in preliminary whole cell growth assays was a generous gift from Professor Alan Hofmann. IsoDCA used for MIC and kinetic experiments and oxo-bile acids were purchased from Steraloids. All other chemicals were purchased from Sigma-Aldrich. Thin layer chromatography was performed on silica gel PLC 60 F254 plates (0.5 mm) (EMD Chemicals Inc.). Pipet column chromatography was performed using Purasil silica gel 60 (38–63 mm) (GE Healthcare Whatman). Gas chromatography (GC)-MS analyses were performed on an Agilent G2570A 6850 GC/MSD System using an Agilent J&W HP-5ms GC Column (30 m × 0.25 mm × 0.25 µm, 5 inch cage) at 1 mL/min from 280 °C to 290 °C. Kinetic and critical micelle concentration experiments were performed using a Varian Cary 50 UV-Vis (Agilent Technologies). Growth data at OD600 were obtained using an Ultrospec 10 (GE).
Whole cell screen for bile acid metabolism by gut commensal bacteria
Selected strains (Supplementary Results, Supplementary Table 1) were grown to saturation overnight. Cells were diluted to a starting OD600 of 0.15 into medium containing bile acid (CA, CDCA, DCA, or LCA) at a final concentration of 100 µM. 2 mL aliquots were removed at early-log, mid-log, and stationary phase and quenched with 200 µL of 6N HCl. The acidified medium was extracted with 2 × 2 mL of EtOAc, dried by passing through a plug of Na2SO4, and concentrated to dryness. The residue was purified using a 1.5 cm SiO2 pipet column. The column was washed with 1.5 mL of 5% MeOH/CH2Cl2 and eluted with 4 mL of 9% MeOH/EtOAc, and the eluent was concentrated to dryness. Prior to GC-MS analysis, the purified bile acids were converted to their methyl ester – trimethylsilyl ether derivatives.42 Briefly, to a solution of the bile acids in 50 µL of MeOH was added 30 µL of a 2.0 M solution of (trimethylsilyl)diazomethane in diethyl ether. Once gas evolution had ceased, the solution was concentrated to dryness, and 40 µL of a 3:1 solution of N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) and chlorotrimethylsilane was added. GC-MS analysis was performed on 3 µL of the resulting solution. Bile acids were identified based either on retention times and MS fragmentation patterns of standards or on published MS fragmentation patterns.42
Bioinformatic searches for candidate 3α- and 3β-HSDHs in E. lenta and R. gnavus
BLASTP searches were performed on IMG (JGI) using 3α−HSDHs from C. scindens (AAB61155), C. hiranonis (AAF22845), and C. hylemonae (ACF20977), a 7β-HSDH from R. gnavus (RUMGNA_02585, EDN76974), and a 3β-HSDH from C. testosteroni (WP_003063214) as query sequences, with a cutoff expectation value of 1e−5.
Phylogenetic analysis of candidate and query HSDHs
A multiple sequence alignment was calculated using MUSCLE.43 A phylogenetic tree was then computed from this alignment using PhyML,44 choosing the LG substitution model, the SPR & NNI tree improvement method, 10 random starting trees, and bootstrap with 500 replicas. The phylogenetic tree was visualized using iTOL.45
Cloning and expression of candidate 3α- and 3β-HSDH genes
Candidate 3α- and 3β-HSDH genes were cloned from genomic DNA prepared using gene-specific primers (Supplementary Table 2). PCR reactions were performed with Phusion High Fidelity Polymerase, and PCR products were purified (MinElute, Qiagen). The amplified gene sequences were digested with the appropriate restriction enzymes, ligated into pET28b, and transformed into E. coli TOP10 cells. The identities of the resulting pET28b–HSDH constructs were confirmed by DNA sequencing. Expression constructs were transformed into E. coli BL21 (DE3) pLysS cells, grown to saturation in LB medium supplemented with kanamycin (30 µg/mL) and chloramphenicol (25 µg/mL) at 37 °C, and diluted 1:100 into LB medium and supplemented with kanamycin (30 µg/mL) and chloramphenicol (25 µg/mL). The expression of N-terminal His6 fusion proteins was induced at OD600 0.5–0.6 with 400 µM isopropyl β-D-1-thiogalactopyranoside, and the induced cells were incubated at 20 °C for 20 h. Cells from 200 mL of culture were pelleted by centrifugation (20 min at 7,000 X g and 4 °C), resuspended in 15 mL of ice-cold phosphate-buffered saline containing ½ of a cOmplete Protease Inhibitor cocktail tablet (Roche Diagnostics), and lysed by sonication. Cell debris was removed by ultracentrifugation (20 min at 20,000 X g and 4 °C). Protein expression was confirmed by SDS/Page analysis using Mini-PROTEAN TGX Gels, 4–20% (Bio-Rad Laboratories). Gels were stained with Coomassie Blue for visualization. The clarified supernatant was used directly in the cell lysate assay described below.
Cell lysate assay to identify functional 3α- and 3β-HSDHs among candidate genes
Bile acid substrate (2 µL of a 100 mM solution of CA, CDCA, DCA, or LCA in MeOH) was added to 2 mL of clarified supernatant. After incubation at 37 °C for 7 h, the reactions were quenched with 200 µL of 6N HCl and extracted with 1 × 2 mL of EtOAc. The organic extracts were passed through a plug of Na2SO4 and concentrated to dryness. The resulting residues were resuspended in 50 µL of EtOAc by vortexing and spotted on TLC plates, which were resolved using 70:20:2 benzene/dioxane/acetic acid and visualized using p-anisaldehyde stain. 3α- and 3β-HSDH activities were determined by the appearance of new spots that co-migrated with 3-oxoDCA and isoDCA standards, respectively. Following sample purification, these results were confirmed by GC-MS analysis as described above.
Expression and purification of Elen_0690, Rumgna_02133, Elen_0198, Elen_1325, and Rumgna_00694
Proteins were overproduced using the procedure described above. Cells from 500 mL of culture were pelleted by centrifugation, resuspended in 40 mL of ice-cold lysis buffer (300 mM NaCl, 10 mM imidazole, 50 mM NaH2PO4, pH 8.0) containing one cOmplete Protease Inhibitor cocktail tablet, and lysed by 8 min of continuous passage through a cell disruptor (EmulsiFlex-C3; Avestin, Inc.) at 15,000 lbs per square inch. Cell debris was removed by ultracentrifugation (20 min at 20,000 X g and 4 °C), and the cell-free extract was applied to 0.5 mL of HisPur Ni-NTA Resin (Thermo Scientific) pre-equilibrated with lysis buffer by gentle rocking at 4 °C for 2 h. Non-absorbed materials and weakly bound proteins were removed by washing the column with 2 × 25 mL of wash buffer (300 mM NaCl, 20 mM imidazole, 50 mM NaH2PO4, pH 8.0). His6-tagged protein was eluted with 5 mL of elution buffer (300 mM NaCl, 200 mM imidazole, 50 mM NaH2PO4, pH 8.0). After SDS-PAGE analysis, eluent containing pure protein was dialyzed (Spectra/Por Dialysis Membrane, 6 – 8 kDa molecular weight cutoff; Spectrum Labs) against 500 mL of extraction buffer (10 mM 3-(N-morpholino)propanesulfonic acid (MOPS) and 300 mM NaCl, adjusted to pH 7.0 with NaOH) for 3 h at 4 °C and again with 1 L of extraction buffer for 12 h at 4 °C. The proteins were flash-frozen in liquid nitrogen as solutions in 10% glycerol and stored at –80 °C. The concentrations of purified proteins were determined spectrophotometrically by the Bradford method using the Coomassie (Bradford) Protein Assay Kit (Thermo Scientific).
Purified enzyme assays to confirm activity
Oxidation
Purified protein (1 µM final concentration or protein-free control) was added to a solution of 2 mL of 10 mM MOPS buffer (pH 7.0) containing substrate (DCA, 400 µM final concentration, or DMSO control) and cofactor (NAD(P)+, 100 µM final concentration) at 37 °C.
Reduction
As above, with the following exceptions: 10 mM MOPS buffer (pH 7.0) or 0.2 N glycine (adjusted to pH 10.0 with NaOH), 3-oxoDCA substrate NAD(P)H cofactor.
After 30 min of incubation at 37 °C, all reactions were quenched by the addition of 200 µL of 6N HCl. 3α- and 3β-HSDH activities were confirmed by TLC and GC-MS analysis.
Determination of kinetic parameters
3α- and 3β-HSDH activity was determined spectrophotometrically by measuring the change in NAD(P)H concentration. Change in absorbance at 340 nm was monitored, and enzyme activity was calculated using the Beer-Lambert law using a molar extinction coefficient of 6.22 mM−1 × cm−1 for NAD(P)H33 and a path length of 1 cm.
Determination of preferred cofactor for each enzyme
TLC analysis purified enzyme reactions had previously determined the following preferred cofactors: Elen_0690 and Elen_0198: NAD+/H; Rumgna_02133: NADP+/H. The preferred cofactors for Elen_1325 (NADH) and Rumgna_00694 (NADPH) were determined by qualitatively assessing rate of absorbance decay for 120 seconds under the following conditions: purified protein (Rumgna_00694, 5 nM, or Elen_1325, 50 nM) was added to a solution of 1000 µL (final volume) of 10 mM MOPS buffer (pH 7.0) containing substrate (3-oxoDCA; Rumgna_00694, 10 µM, or Elen_1325, 100 µM) and cofactor (NADH or NADPH, 100 µM) at 37 °C.
General procedure
Substrate was added to a solution of 100 µL of 10 mM MOPS buffer (pH 7.0) and cofactor at 37 °C. In order to obtain absorbance measurements within the range of the spectrophotometer, the solution was diluted to a volume of 1000 µL using 10 mM MOPS buffer (pH 7.0) at 37 °C, and purified protein was added to start the reaction. Change in absorbance was monitored every 5 s for 100 s at 37 °C for all enzymes except Rumgna_00694, for which absorbance was monitored every 3 s for 60 s. Absorbance was converted to substrate concentration based on the 1:1 reaction stoichiometry of cofactor to substrate. Initial velocity conditions were maintained by limiting substrate consumption to 10% or less. The kinetic parameters kcat and Km were determined by varying the concentration of substrate (Supplementary Table 3). Note that all concentrations and initial velocities refer to the original 100 µL reaction volume. Measurements were made from a single batch of purified enzyme. Initial velocity data were fit to the Michaelis-Menten equation by using the program GraphPad.
Determination of minimal inhibitory concentrations (MICs) for DCA and isoDCA
B. thetaiotaomicron, B. ovatus, B. uniformis, B. vulgatus, E. faecalis, C. sporogenes, and R. gnavus were grown to saturation overnight in BHI-F. E. lenta was grown in BHI-F + 1% Arg. The next morning, 50 µL of saturated culture was added to a solution of 930 µL medium and 20 µL of either DMSO containing bile acid (Supplementary Table 4) or DMSO only (control) at 37 °C in 14 mm diameter glass tubes. To compensate for diminished growth in BHI-F, for B. uniformis, B. vulgatus and R. gnavus, 100 µL of saturated culture was added to a solution of 880 µL medium and 20 µL of DMSO. Cultures were incubated for 12 h, at which time the control cultures had reached stationary phase. MICs were determined by the concentration of either isoDCA or DCA at which no opacity (and therefore no growth) was observed in the culture tube for a given strain.
Growth curves of B. ovatus at sub-minimal inhibitory concentrations of DCA and isoDCA
B. ovatus was grown to saturation overnight at 37 °C. The next morning, the strain was diluted into three triplicate sets of 4 mL of TYG medium containing (set 1) DCA (10 µL of a 100 mM solution in DMSO, 250 µM final concentration), (set 2) isoDCA (10 µL of a 100 mM solution in DMSO, 250 µM final concentration), or (set 3) 10 µL of DMSO only (control). The starting OD600 for each culture was 0.1. The growth of all samples was monitored until stationary phase was reached. Time points were taken every 15 minutes during log phase growth. Differences in final carrying capacities were determined using Welch’s t tests with GraphPad software.
Determination of critical micelle concentrations (CMCs) for DCA and isoDCA
Aqueous solutions of sodium isodeoxycholate and sodium deoxycholate were prepared by the addition of a stoichiometric amount of NaOH to suspensions of isoDCA and DCA in water, respectively. To a series of nine 1.5 mL Eppendorf tubes was added 0.25 mL of isodeoxycholate solution (range: 7 – 15 mM) or deoxycholate solution (range: 1 – 10 mM) and two Optimizer-blueBALLS™ (G Biosciences). The tubes were vigorously vortexed and incubated without shaking at room temperature for 20 h. The tubes were again vigorously vortexed and then centrifuged at 6,000 x g for 5 min. The supernatants (0.2 mL) were removed, transferred to a 96-well plate, and centrifuged again at 6,000 x g for 5 min to pellet any residual blue particles suspended in the solution. The supernatant (0.115 mL) was transferred to UV-micro cuvettes, taking care not to disturb the pelleted particles, and dye absorbance was read at 640 nm. Critical micelle concentration (CMC) was determined graphically (Supplementary Figure 12). The quantity of dye in solution is proportional to the number of detergent micelles. The CMC was determined by plotting optical density of solubilized dye against detergent concentration and fitting a standard five-parameter logistic growth curve to the data using Graphpad. The computed inflection point of each curve corresponds to the CMC for each compound.
Effect of DCA and isoDCA on cell membrane integrity
The entire experiment aside from the final fluorescence measurement was performed in an anaerobic chamber. B. ovatus and R. gnavus were grown to saturation overnight at 37 °C in BHI-F. The next morning, the strain was diluted into six triplicate cultures of 6 mL (B. ovatus) or 5.2 mL (R. gnavus) BHI-F medium. The starting OD600 for each culture was 0.25. For B. ovatus: The growth of all samples was monitored until an OD600 of 0.8 was reached for each culture. The sample sets were then treated as follows: (set 1) + DCA (12 µL of a 100 mM solution in DMSO, 200 µM final concentration), (set 2) + isoDCA (200 µM), (set 3) + DCA (24 µL of a 100 mM solution in DMSO, 400 µM), (set 4) + isoDCA (400 µM), (set 5) + 24 µL of DMSO only (control), (set 6) no additive, to be used for heat-killed cell control. After 1.5 hours, the OD600 of each culture was recorded. For R. gnavus: The growth of all samples was monitored until an OD600 of 0.55 was reached for each culture. The sample sets were then treated as follows: (set 1) + DCA (15.2 µL of a 100 mM solution in DMSO, 300 µM final concentration), (set 2) + isoDCA (300 µM), (set 3) + DCA (31.2 µL of a 100 mM solution in DMSO, 600 µM), (set 4) + isoDCA (600 µM), (set 5) + 31.2 µL of DMSO only (control), (set 6) no additive, to be used for heat-killed cell control. After 3.5 hours, the OD600 of each culture was recorded. For both strains: A modified version of a previously published propidium iodide (Pi)-based procedure was used to evaluate cell membrane integrity.46 1 mL aliquots from set 6 were heat killed (80 °C for 10 min). Aliquots from all cultures (1 mL each) were then centrifuged at 2,000 x g for 4 min. The supernatant was carefully removed and the cell pellet was resuspended in 1 mL of anaerobic PBS pre-warmed to 37 °C. To these aqueous resuspensions was then added 2 µL of a freshly made 2 mg/mL solution of Pi in water (20 µg/mL final Pi concentration). Samples were incubated in the dark at 37 °C for 15 min, at which time 100 µL aliquots were transferred to a black 96-well plate (Corning Costar). The plate was then removed from the anaerobic chamber, and red fluorescence was measured using a Tecan Safire2 plate reader (excitation: 540 nm, emission 610 nm). In order to correct for variations in cell number, fluorescence was normalized by dividing by the OD600 of the corresponding culture measured immediately prior to centrifugation. In the event that the OD600 of a given culture had dropped following addition of compound, fluorescence was normalized by diving by the maximal OD600 recorded, i.e., the OD600 of the culture immediately prior to compound addition. Differences in normalized fluorescence were determined using Welch’s t tests with GraphPad software.
Co-culture of B. ovatus with E. lenta or E. eligens with or without DCA
B. ovatus, E. lenta, and E. eligens were grown to saturation overnight at 37 °C in BHI-F + 1% Arg. The next morning, experimental cultures were co-inoculated in a 5 mL final volume of BHI-F + 1% Arg as follows: condition 1: B. ovatus + E. lenta + 300 µM (final concentration) DCA; condition 2: B. ovatus + E. eligens + 300 µM DCA; condition 3: B. ovatus + E. lenta; condition 4: B. ovatus + E. eligens. The starting OD600 for each strain in each co-culture was 0.15. Time points were taken by plating serial dilutions on BHI + 10% horse blood + 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal, 2 mL of 100 mg/mL solution in DMF per 1000 mL of media) at t=0, 5, and 21 h. Note: B. ovatus harbors a β-galactosidase that enables it to metabolize X-gal, while E. lenta and E. eligens do not, allowing blue B. ovatus colonies to be distinguished from translucent E. lenta and white E. eligens colonies. Plates were incubated anaerobically at 37 °C for 48 h, removed from the chamber, and allowed to rest for 6 hours aerobically at 23 °C to allow for full blue color to develop prior to colony counting. Growth curves were plotted using GraphPad software.
Supplementary Material
Acknowledgements
We are grateful to Prof. Alan Hofmann for helpful discussions and for providing an initial sample of isoDCA, and to Prof. Takashi Iida for providing us with an authentic sample of 12-epi-isoDCA. We thank Paul Ortiz de Montellano for use of his GC-MS, Yug Varma for background research and experimentation on bile acids, Peter Cimermancic for help with the phylogenetic analysis, and members of the Fischbach Group for helpful discussions. This research was supported by an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund (M.A.F.), a Glenn Award for Research in Biological Mechanisms of Aging (M.A.F.), a Medical Research Program Grant from the W.M. Keck Foundation (M.A.F.), a Fellowship for Science and Engineering from the David and Lucile Packard Foundation (M.A.F.), and NIH grants OD007290 and GM081879 (M.A.F.).
Footnotes
Author Contributions
A.S.D. and M.A.F. conceived of the project, designed the experiments, and wrote the manuscript. A.S.D. performed the experiments.
Competing Financial Interests
M.A.F. is on the scientific advisory board of NGM Biopharmaceuticals.
Reference
- 1.Ridlon J, Kang D, Hylemon P. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Hamilton JP, et al. Human cecal bile acids: concentration spectrum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;293:G256–G263. doi: 10.1152/ajpgi.00027.2007. [DOI] [PubMed] [Google Scholar]
- 3.Macdonald I, Bokkenheuser VD, Winter J, McLernon AM, Mosbach EH. Degradation of steroids in the human gut. J. Lipid Res. 1983;24:675–700. [PubMed] [Google Scholar]
- 4.Hofmann AF, et al. A proposed nomenclature for bile acids. J. Lipid Res. 1992;33:599–604. [PubMed] [Google Scholar]
- 5.Setchell KDR, Lawson AM, Tanida N, Sjovall J. General methods for the analysis of metabolic profiles of bile acids and related compounds in feces. J. Lipid Res. 1983;24:1085–1100. [PubMed] [Google Scholar]
- 6.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bileacid signalling for metabolic diseases. Nat. Rev. Drug Disc. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 7.Makishima M, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 8.Talukdar S, Bhatnagar S, Dridi S, Hillgartner FB. Chenodeoxycholic acid suppresses the activation of acetyl-coenzyme A carboxylase-alpha gene transcription by the liver X receptor agonist T0-901317. J. Lipid Res. 2007;48:2647–2663. doi: 10.1194/jlr.M700189-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Buffie CG, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile . Nature. 2015;517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy BS, Narisawa T, Weisenburger JH, Wynder EL. Promoting effect of sodium deoxycholate on colon adenocarcinomas in germfree rats. J. Natl. Cancer Inst. 1976;56:441–442. doi: 10.1093/jnci/56.2.441. [DOI] [PubMed] [Google Scholar]
- 11.Narisawa T, Magadia NE, Weisburger JH, Wynder EL. Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N’-nitro-Nnitrosoguanidine in rats. J. Natl. Cancer Inst. 1974;53:1093–1097. doi: 10.1093/jnci/53.4.1093. [DOI] [PubMed] [Google Scholar]
- 12.Yoshimoto S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 13.Rafter JJ, et al. Cellular toxicity of fecal water depends on diet. Am. J. Clin. Nutr. 1987;45:559–563. doi: 10.1093/ajcn/45.3.559. [DOI] [PubMed] [Google Scholar]
- 14.Reddy BS, Weisenburger JH, Wynder EL. Effects of high risk, low risk diets for colon carcinogenesis on fecal microflora, steroids in man. J. Nutr. 1975;105:878–884. doi: 10.1093/jn/105.7.878. [DOI] [PubMed] [Google Scholar]
- 15.Im E, Martinez JD. Ursodeoxycholic acid(UDCA) can inhibit deoxycholic acid (DCA)-induced apoptosis via modulation of EGFR/Raf-1/ERK signaling in human colon cancer cells. J. Nutr. 2004;134:483–486. doi: 10.1093/jn/134.2.483. [DOI] [PubMed] [Google Scholar]
- 16.Bachrach WH, Hofmann AF. Ursodeoxycholic acid in the treatment of cholesterol cholelithiasis. Part I. Dig. Dis. Sci. 1982;27:737–761. doi: 10.1007/BF01393771. [DOI] [PubMed] [Google Scholar]
- 17.Bennett MJ, McKnight SL, Coleman JP. Cloning and characterization of he NAD-dependent 7α-hydroxysteroid dehydrogenase from Bacteroides fragilis. Curr. Microbiol. 2003;47:475–484. doi: 10.1007/s00284-003-4079-4. [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Aigner A, Schmid RD. Identification, cloning, heterologous expression characterization of a NADPH-dependent 7β-hydroxysteroid dehydrogenase from Collinsella aerofaciens. Appl. Microbiol. Biotechnol. 2011;90:127–135. doi: 10.1007/s00253-010-3052-y. [DOI] [PubMed] [Google Scholar]
- 19.Baron SF, Franklund CV, Hylemon PB. Cloning, sequencing, expression of the gene encoding for bile acid 7α-hydroxysteroid dehydrogenase from Eubacterium sp. strain VPI12708J. Bacteriol. 1991;173:4558–4569. doi: 10.1128/jb.173.15.4558-4569.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleman JP, Hudson LL, Adams MJ. Characterization and regulation of the NADP-linked 7α-hydroxysteroid dehydrogenase gene from Clostridium sordellii . J. Bacteriol. 1994;176:4865–4874. doi: 10.1128/jb.176.16.4865-4874.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JY, et al. Contribution of the 7β-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J. Lipid Res. 2013;11:3062–3069. doi: 10.1194/jlr.M039834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirano S, Masuda N. Transformation of bile acids by Eubacterium lentum . Environ. Appl. Microbiol. 1981;42:912–915. doi: 10.1128/aem.42.5.912-915.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirano S, Masuda N, Oda H, Mukai H. Transformation of bile acids by Clostridium perfringens . Appl. Environ. Microbiol. 1981;42:394–399. doi: 10.1128/aem.42.3.394-399.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macdonald IA, et al. Metabolism of primary bile acids byClostridium perfringens . J. Steroid Biochem. 1983;18:97–104. doi: 10.1016/0022-4731(83)90336-9. [DOI] [PubMed] [Google Scholar]
- 25.Hayakawa S. Microbiological transformation of bile acids. Adv. Lipid Res. 1973;11:143–192. doi: 10.1016/b978-0-12-024911-4.50011-8. [DOI] [PubMed] [Google Scholar]
- 26.Macdonald IA, Meier EC, Mahony DE, Costain GA. 3α-, 7α-, and 12α- hydroxysteroid dehydrogenase activity from Clostridium perfringens . Biochim. Biophys. Acta. 1976;450:142–153. doi: 10.1016/0005-2760(76)90086-2. [DOI] [PubMed] [Google Scholar]
- 27.Macdonald IA, Jellett JF, Mahony DE, Holdeman LV. Bile salt 3 α- and 12 α- hydroxysteroid dehydrogenases from Eubacterium lentum and related organisms. Appl. Environ. Microbiol. 1979;37:992–1000. doi: 10.1128/aem.37.5.992-1000.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macdonald IA, Mahony DE, Jellet JF, Meier CE. NAD-dependent 3α- and 12α-hydroxysteroid dehydrogenase activities from Eubacterium lentum ATCC no. 25559. Biochim. Biophys. Acta. 1977;489:466–476. doi: 10.1016/0005-2760(77)90167-9. [DOI] [PubMed] [Google Scholar]
- 29.Edenharder R, Pfutzner A, Hammann R. Characterization of NAD-dependent 3α- and 3β−hydroxysteroid dehydrogenase and of NADP-dependent 7β−hydroxysteroid dehydrogenase from Peptostreptococcus productus . Biochim. Biophys. Acta. 1989;1004:230–238. doi: 10.1016/0005-2760(89)90272-5. [DOI] [PubMed] [Google Scholar]
- 30.Edenharder R, Pfutzner A, Hammann R. NADP-dependent 3β−, 7α- and 7β-hydroxysteroid dehydrogenase activities from a lecithinase-lipase-negative Clostridium species 25.11.c. Biochim. Biophys. Acta. 1989;1002:37–44. doi: 10.1016/0005-2760(89)90061-1. [DOI] [PubMed] [Google Scholar]
- 31.Akao T, Akao T, Hattori M, Namba T, Kobashi K. 3β-Hydroxysteroid dehydrogenase of Ruminococcus sp. from human intestinal bacteria. J. Biochem. 1986;99:1425–1431. doi: 10.1093/oxfordjournals.jbchem.a135612. [DOI] [PubMed] [Google Scholar]
- 32.Kraal L, Abubucker S, Kota K, Fischbach MA. & Mitreva, M. The prevalence of species and strains in the human microbiome: a resource for experimental efforts. PLoS ONE. 2014;9:e97279. doi: 10.1371/journal.pone.0097279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wells JE, Hylemon PB. Identification characterization of a bile acid 7α-dehydroxylation operon in Clostridium sp. strain TO-931 a highly active 7α-dehydroxylating strain isolated from human feces. Appl. Environ. Microbiol. 2000;66:1107–1113. doi: 10.1128/aem.66.3.1107-1113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mallonee DH, Lijewski MA, Hylemon PB. Expression in Escherichia coli, characterization of a bile acid-inducible 3α-hydroxysteroid dehydrogenase from Eubacterium sp. strain VPI12708. Curr. Microbiol. 1995;30:259–263. doi: 10.1007/BF00295498. [DOI] [PubMed] [Google Scholar]
- 35.Ridlon JM, Kang D, Hylemon PB. Isolation and characterization of a bile acid inducible 7α-dehydroxylating operon in Clostridium hylemonae TN271. Anaerobe. 2010;16:137–146. doi: 10.1016/j.anaerobe.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Begley M, Gahan CGM, Hill C. The interaction bacteria, bile. FEMS Microbiol. Rev. 2005;29:625–651. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Matsuoka K, Moroi Y. Micelle formation of sodium deoxycholate and sodium ursodeoxycholate (part 1) Biochim. Biophys. Acta. 2002;1580:189–199. doi: 10.1016/s1388-1981(01)00203-7. [DOI] [PubMed] [Google Scholar]
- 38.Gomez-Zavaglia A, Kociubinski G, Perez P, Disalvo E, De Antonio G. Effect of bile on the lipid composition, surface properties of bifidobacteria. J. Appl. Microbiol. 2002;93:794–799. doi: 10.1046/j.1365-2672.2002.01747.x. [DOI] [PubMed] [Google Scholar]
- 39.Noh DO, Gilliland SE. Influence of bile on cellular integrity, β-galactosidase activity of Lactobacillus acidophilus. J Dairy Sci. 1993;76:1253–1259. doi: 10.3168/jds.S0022-0302(93)77454-8. [DOI] [PubMed] [Google Scholar]
- 40.Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holdeman LV, Moore WEC. New genus,Coprococcus, twelve new species emended descriptions of four previously described species of bacteria from human feces. Int. J. Syst. Bacteriol. 1974;24:260–277. [Google Scholar]
- 42.Lawson AM, Setchell KDR. In: Mass spectrometry of bile acids. In The Bile Acids: Chemistry, Physiology, and Metabolism. Setchell KDR, Kritchevsky D, Nair PP, editors. Vol. 4. New York, USA: Plenum Press; 1988. pp. 167–267. [Google Scholar]
- 43.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy, high throughput Nucl. Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guindon S, et al. New algorithms methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 45.Letunic I, Bork P. Interactive Tree of Life v2: online annotation display of phylogenetic trees made easy. Nucl. Acids Res. 2011;39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stiefel P, Schmidt-Emrich S, Maniura-Weber K, Ren Q. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Micriobiol. 2015;15 doi: 10.1186/s12866-015-0376-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.