

Abstract
Background and Purpose
Interstitial lung disease accounts for a group of chronic and progressive disorders associated with severe pulmonary vascular remodelling, peripheral vascular rarefaction and fibrosis, thus limiting lung function. We have previously shown that Akt is necessary for myofibroblast differentiation, a critical event in organ fibrosis. However, the contributory role of the Akt-mTOR pathway in interstitial lung disease and the therapeutic benefits of targeting Akt and mTOR remain unclear.
Experimental Approach
We investigated the role of the Akt-mTOR pathway and its downstream molecular mechanisms in chronic hypoxia- and TGFβ-induced pulmonary vascular pruning and fibrosis in mice. We also determined the therapeutic benefits of the Akt inhibitor triciribine and the mTOR inhibitor rapamycin for the treatment of pulmonary fibrosis in mice.
Key Results
Akt1−/− mice were protected from chronic hypoxia-induced peripheral vascular pruning. In contrast, hyperactivation of Akt1 induced focal fibrosis similar to TGFβ-induced fibrosis. Pharmacological inhibition of Akt, but not the Akt substrate mTOR, inhibited hypoxia- and TGFβ-induced pulmonary vascular rarefaction and fibrosis. Mechanistically, we found that Akt1 modulates pulmonary remodelling via regulation of thrombospondin1 (TSP1) expression. Hypoxic Akt1−/− mice lungs expressed less TSP1. Moreover, TSP1−/− mice were resistant to adMyrAkt1-induced pulmonary fibrosis.
Conclusions and Implications
Our study identified Akt1 as a novel target for the treatment of interstitial lung disease and provides preclinical data on the potential benefits of the Akt inhibitor triciribine for the treatment of interstitial lung disease.
Table of Links
| TARGETS |
|---|
| Enzymes |
| Akt1 |
| mTOR |
| PI3 kinase |
| LIGANDS | |
|---|---|
| Bosentan | Pirfenidone |
| Everolimus | Rapamycin (sirolimus) |
| LY294002 | SU5416 (semaxanib) |
| Nintedanib | Triciribine (Akt inhibitor V) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Interstitial lung disease (ILD) is characterized by reduced lung volume due, in part, to progressive hypertrophic scar formation leading to fibrosis and vascular remodelling (McLaughlin et al., 2009; King et al., 2011; Raghu et al., 2011; Hinz et al., 2012; Meyer, 2014). Although its pathogenesis is multifactorial, persistent myofibroblast accumulation and excess deposition of extracellular matrix (ECM) proteins are critical events in this fibro-proliferative process. Thus, identifying novel pathways is important in the hopes of devising new targeted therapeutics for pulmonary remodelling.
Research from our laboratory has established the central role of Akt1, a serine-threonine protein kinase Bα, in ECM assembly and secretion (Somanath et al., 2007; Somanath and Byzova, 2009; Goc et al., 2011). We previously reported that genetic ablation of Akt1 in mice resulted in impaired secretion and assembly of ECM proteins and enhanced angiogenesis leading to vascular leakage and skin abnormalities (Chen et al., 2005a). Genetic knock-down of Akt1, not Akt2, resulted in impaired angiogenesis in cutaneous wound healing (Somanath et al., 2008). We also demonstrated that sustained hyperactivation of Akt1 resulted in increased myofibroblast differentiation via increased α-smooth muscle cell actin (αSMA) synthesis involving serum response transcription factor (SRF) and myocardin (Abdalla et al., 2013). Furthermore, pharmacological inhibition of Akt or genetic ablation of Akt1 impaired myofibroblast differentiation and ED-A fibronectin (ED-A-FN) synthesis in vitro (Abdalla et al., 2013).
Myofibroblasts are the hallmark of pulmonary fibrosis (Raghu et al., 2011; Hinz et al., 2012). Pulmonary vascular remodelling in pulmonary hypertension (PH) has also been demonstrated to be mediated by adventitial myofibroblasts (Barman et al., 2014). Our previous findings on the role of Akt-mTOR pathway in extracellular remodelling and myofibroblast differentiation suggest that Akt and/or mTOR may be potential targets for therapeutic interventions in ILD. Other studies have also reported a possible correlation between Akt hyperactivation and fibrotic diseases. Mice deficient in the protein PTEN (phosphatase and tensin homologue), an endogenous inhibitor of Akt pathway, developed a fibroproliferative response consistent with fibrosis (Xia et al., 2008). Interestingly, Akt expression has been found to be elevated in fibrotic lung tissues from patients with idiopathic pulmonary fibrosis (IPF) (Xia et al., 2008). However, the precise role of the Akt-mTOR pathway in the pathogenesis of pulmonary fibrosis and the therapeutic benefits of targeting Akt and mTOR in ILD remain unknown.
In this study, we investigated the contributory role of Akt in general, and Akt1 in particular, in pulmonary tissue remodelling. We identified that Akt1−/− mice are protected from hypoxia-induced pulmonary fibrosis and vascular remodelling and that adenovirus-mediated overexpression of hyperactive Akt1 (adMyrAkt1) in mouse lungs resulted in increased fibrosis. Pharmacological inhibition of Akt using triciribine (TCBN) significantly reversed TGFβ- and chronic hypoxia-induced pulmonary fibrosis. However, targeting mTOR using rapamycin did not yield therapeutic benefits on chronic hypoxia- or TGFβ-induced pulmonary vascular rarefaction and fibrosis. Mechanistically, we found that genetic ablation of Akt1 and pharmacological inhibition of Akt were associated with impaired thrombospondin1 (TSP1) expression by the fibroblasts in vitro and in the lung tissues in vivo. Furthermore, TSP1−/− mice were protected from adMyrAkt1-induced fibrosis. Collectively, we identified Akt1 as a novel target in ILD as it is a critical mediator of hypoxia- and TGFβ-induced pulmonary remodelling, in part through TSP1 regulation.
Methods
Antibodies
Anti-αSMA, anti-TSP1 and anti-fibronectin antibodies were purchased from Sigma (St Louis, MO, USA). GAPDH, phosphoAkt-S473, phosphoT37/46-4E-BP1 and anti-SRF antibodies were purchased from Cell Signaling (Boston, MA, USA). Anti-myocardin antibodies were purchased from R&D Systems (Minneapolis, MN, USA). Collagens type I and III antibodies were purchased from Rockland (Gilbertsville, PA, USA). Antibodies for ED-A-FN, collagen VI and αSMA were purchased from Abcam (Cambridge, MA, USA).
Cell culture
Normal and fibrotic human lung fibroblasts (FHLFs) and FGM™-2 fibroblast growth medium-2 were purchased from Lonza (Walkersville, MD, USA). Normal human lung fibroblasts were cultured on a 6-well plate. After reaching 70% of confluence, cells were subjected to serum starvation in the presence or absence of 100 pM TGFβ (R&D Systems) for 48 h (Goc et al., 2011). This was followed by co-treatment for 24 h (total 72 h) with inhibitors of PI3 kinase (25 μM LY294002), Akt (10 nM TCBN; 1,5-dihydro-5-methyl-1-β-D-ribofuranosyl-1,2,5,6,8-pentaazaacenaphthylen-3-amine) or mTOR (25 nM rapamycin), all of which are obtained from Calbiochem (Millipore, Billerica, MA, USA). Cells were subjected to Western analyses as described below. For FHLFs, cells were cultured in serum starvation in a 6-well plate and treated with inhibitors of Akt (10 nM TCBN) or mTOR (25 nM rapamycin) for 24 h.
Western blot analysis
Cell lysates were prepared using lysis buffer [20 mM Tris-HCl, pH 7.4; 1% Triton X-100, 3 mM EGTA, 5 mM EDTA, phosphatase inhibitors (10 mM sodium pyrophosphate, 5 mM sodium orthovanadate, 5 mM sodium fluoride and 10 μM okadaic acid), protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland) and 1 mM PMSF]. SDS-PAGE and Western blotting were performed as described previously (Abdalla et al., 2013).
Animals
All animal care and experimental procedures were approved by the Charlie Norwood VAMC Institutional Animal Care and Use Committee. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 230 animals were used in the experiments described here.
Akt1−/− mice were generated as previously described (Chen et al., 2005b) and were maintained in the C57BL/6 background. Age-matched male Akt1+/+ and Akt1−/− (8–12 weeks old) mice were utilized for the chronic hypoxia and TGFβ-induced pulmonary fibrosis models. Eight-week-old male TSP1−/− mice were purchased from Jackson mice (Bar Harbor, ME, USA).
Chronic hypoxia model
Akt1+/+ and Akt1−/− mice were subjected to normoxia or hypoxia (10% O2) (Biospherix, New York, NY, USA) for 7 and 14 days (n = 2–6 mice per group). Noteworthy, high mortality was observed in Akt1−/− mice exposed to hypoxia longer than 14–16 days. For pharmacological inhibition studies, Akt1+/+ mice, subjected to normoxia or chronic hypoxia for 14 days, received daily i.p. injection of saline, TCBN (0.5 mg·kg−1·day−1) or rapamycin (1.5 mg·kg−1·day−1) for 7 days, and the total continuous exposure to hypoxia or normoxia was 21 days (n = 6–8 mice per group). Pharmacological inhibitors were administered daily while the mice were maintained in the hypoxia chamber to minimize exposure to air and spontaneous reversal of pulmonary remodelling. After lung isolation, the left lung was subjected to histology for haematoxylin and eosin (H&E), Masson's trichrome and immunofluorescence staining against αSMA (to elucidate vascular remodelling) and fibronectin (to elucidate vascular and peripheral fibrosis). The right lung was subjected to Western analyses against various proteins including TGFβ, αSMA, myocardin, SRF, fibronectin, specialized fibronectin (ED-A), collagen (I, III, VI), phosphorylated Akt, mTOR, 4EBP, TSP1 and GAPDH.
Adenovirus administration
Mice were treated with intratracheal (i.t.) control adenovirus (adControl) or adenovirus TGFβ (adTGFβ) at a concentration of 1 × 106 pfu. Saline, TCBN or rapamycin was administered i.p. on days 7–10 (n = 6–8 mice per group). For the Akt-induced fibrosis experiment, TSP1+/+ and TSP1−/− mice were subjected to i.t. administration of control vector or adenovirus expressing constitutively active-Akt1 (adMyrAkt1) two times on days 0 and 7. Mice were killed and lungs were collected on day 14.
Vascular Microfil® casting
Following completion of adenovirus or hypoxia studies, mice were anesthetized and given 50 μL of heparin (50 mg·mL−1) subcutaneously. Microfil (FlowTech, Inc., Carver, MA, USA) casting agent (1:2 dilution + 3.2% curing agent) was infused through the right ventricle at a perfusion rate of 1 mL·min−1. After curing for approximately 30 min, lungs were isolated, fixed in 4% paraformaldehyde overnight, and cleared with ethanol and methyl salicylate per the manufacturer's manual. After approximately 2 months of vascular clearing, the peripheral vasculature was imaged using confocal microscopy (n = 5–8 mice per group).
Histological and immunohistochemical assessments
Left lung lobe, heart and liver tissues were fixed in 4% paraformaldehyde, embedded in paraffin and sectioned at 5 μm thickness. Quantitative and qualitative evaluation of fibrotic changes was obtained as follows: Using ImageJ software (National Institutes of Health, Bethesda, MD, USA), the threshold percentage area for each field was averaged and presented as percentage of fibrosed area (3–6 fields per lung section per mouse). The severity of pulmonary fibrosis was scored (0–8 scale; 0 for normal and 8 for total fibrosis) in lung sections stained for collagen with Masson's trichrome stains using the grading system described previously (Ashcroft et al., 1988). Immunostaining for αSMA and fibronectin was performed using specific antibodies. For vascular remodelling studies, lung sections from each animal were stained with H&E and Masson's trichrome and examined by digital photomicroscopy at various magnifications to determine the severity of the disease. Peripheral pulmonary vessels were examined to determine the medial wall/lumen diameter ratio, and the right ventricular wall thickness was measured in heart sections (average of 8–10 measurements per animal per group) to determine right ventricular hypertrophy. Finally, liver morphology was assessed semi-quantitatively using the threshold in ImageJ software in 5 fields per liver section per mouse per group. All quantitative vascular remodelling assessments following Microfil casting were performed on binary images.
Quantitative RT-PCR arrays
Control pBabe (retroviral) and DN-Akt1 (Akt1 K179M) expressing NIH-3T3 fibroblasts and FHLFs were used for the qRT-PCR arrays. Briefly, cells were lysed and RNA was isolated using RNAase Mini plus Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Next, cDNA was generated by RT2 First Strand Kit (SABiosciences, Valencia, CA, USA), mixed with qPCR Sybr Green master mix and loaded into fibrosis RT2 Profiler PCR Array plates (SA Biosciences). Reading was completed in Eppendorf Mastercycler realplex-2 equipment (Hamburg, Germany).
Data analysis
Data are presented as means ± SD. Statistical analysis was performed using two-way anova on the ranks of the data that were used to compare control, insult (chronic hypoxia or adTGFβ or adMyrAkt1), and control or insult, plus TCBN or rapamycin. Student's t-tests with a two-tailed distribution were used to compare control and treated as well as Akt1+/+ and Akt1−/− mice on all variables. Statistical significance was determined at P < 0.05.
Results
Akt1−/− mice are resistant to chronic hypoxia-induced pulmonary remodelling
To examine whether Akt is required for pulmonary remodelling, Akt1+/+ and Akt1−/− mice were subjected to normoxia, short-term hypoxia (7 days) or chronic hypoxia (14 days), and morphological alteration due to hypoxia was assessed. The results showed that Akt1+/+ mice subjected to chronic hypoxia developed patterns consistent with the disease progression as evidenced by patchy lesions, mild vessel luminal narrowing and medial thickening on day 7 that progressed to diffuse interstitial lesions and significant vascular remodelling on day 14 (Figure 1A and B). In contrast, hypoxic Akt1−/− mice showed histopathological patterns similar to normoxic Akt1+/+ mice on days 7 and 14 (Figure 1A and B). Despite chronic hypoxia exposure, Akt1−/− mice had minimal fibronectin accumulation in the interstitial space (Figure 1C and E). Furthermore, 14d-hypoxic Akt1−/− mice had normal appearing pulmonary arterioles as evidenced by the absence of any medial thickening and luminal narrowing, decreased αSMA assembly and decreased fibronectin deposition compared with Akt1+/+ mice lungs (Figure 1D and F). Thus, results suggest that absence of Akt1 impedes hypoxia-induced pulmonary remodelling.
Figure 1.
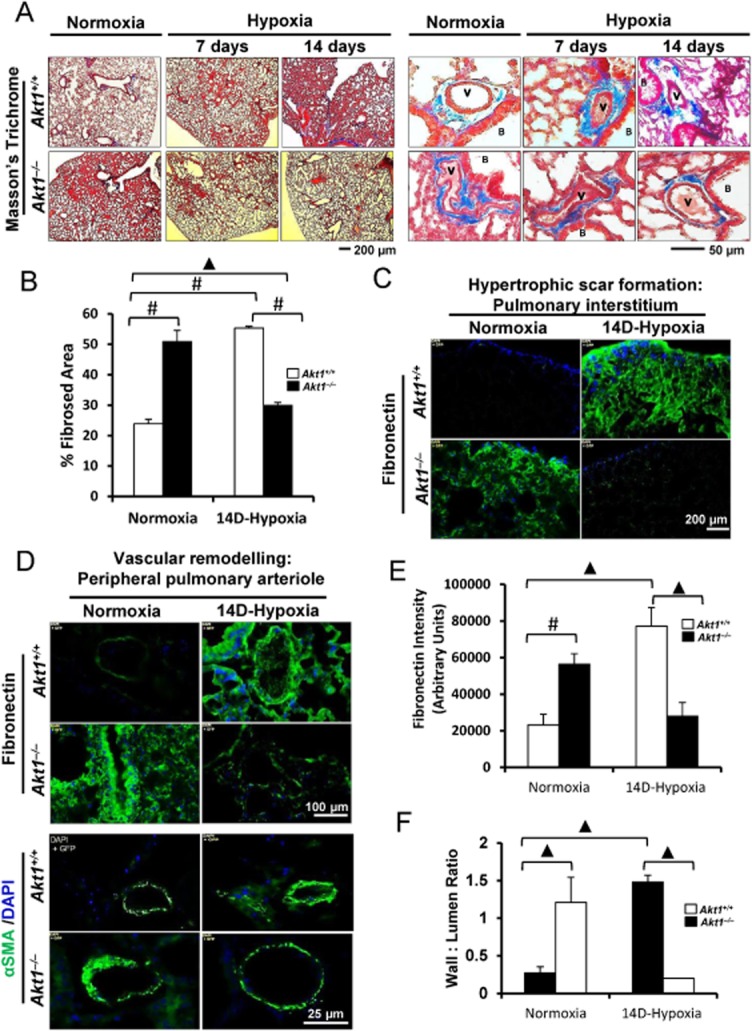
Akt1 deficiency protects against hypoxia-induced pulmonary remodelling. (A) Masson's trichrome stained section of the pulmonary interstitium (left) and peripheral pulmonary arterioles (right) of Akt1+/+ and Akt1−/− mice subjected to normoxia or chronic hypoxia for 7 and 14 days (n = 3–5 mice/group). (B) Histogram showing quantification of the fibrosed area in Akt1+/+ and Akt1−/− mice lungs after 14 day hypoxia compared with normoxia. (C) Immunostaining of frozen sections of 14 day hypoxia and normoxia Akt1+/+ and Akt1−/− mice lungs showing fibronectin expression in the interstitium. (D) Fibronectin and αSMA immunofluorescence staining in and around small pulmonary arteries of normoxic and 14d-hypoxic Akt1+/+ and Akt1−/− mice. (E) Histogram showing reduced fibronectin expression in Akt1−/− mice hypoxic lung sections compared with Akt1+/+ mice lungs (n = 4–5 mice/group). (F) Histogram showing vascular wall to lumen ratio in Akt1+/+ and Akt1−/− mice lung hypoxic sections measured from αSMA immunofluorescence (n = 3–5 mice/group). V, vasculature; B, bronchiole. #P < 0.01, ▲P < 0.001.
Interestingly, normoxic Akt1−/− mice exhibited morphological features similar to 14d-hypoxic Akt1+/+ mice on tissue remodelling (Figure 1A–C) and vascular wall thickening as well as αSMA expression (Figure 1D–F). Compared with normoxic Akt1+/+ mice, genetic ablation of Akt1 was associated with increased interstitial thickening, αSMA and fibronectin deposition.
Akt inhibition reverses chronic hypoxia-induced pulmonary remodelling in vivo
Hypoxic vasoconstriction and vascular remodelling are one of the contributing factors in the pathogenesis of pulmonary fibrosis (Strange and Highland, 2005). In our study, severe chronic hypoxia (21 days) induced medial thickness of peripheral pulmonary arterioles, which correlated with increased collagens, fibronectin and αSMA deposition as demonstrated by H&E, Masson's trichrome and immunofluorescence staining respectively (Figure 2A). This was also confirmed at the protein level where hypoxia increased the levels of αSMA and its transcription factors, SRF and myocardin, and various ECM proteins (Figure 2C). TCBN treatment, administered for 7 days after 14 days of hypoxia until 21 days of hypoxia is reached, reversed the vascular thickening as shown by immunohistochemistry and Western analyses (Figure 2A–C). On the other hand, rapamycin treatment did not prevent hypoxia-induced pulmonary alveolar haemorrhage and congestion (Figure 2A–C). Noteworthy, under normoxic conditions, rapamycin alone induced mild tissue and vascular remodelling in the lungs (Figure 2A).
Figure 2.
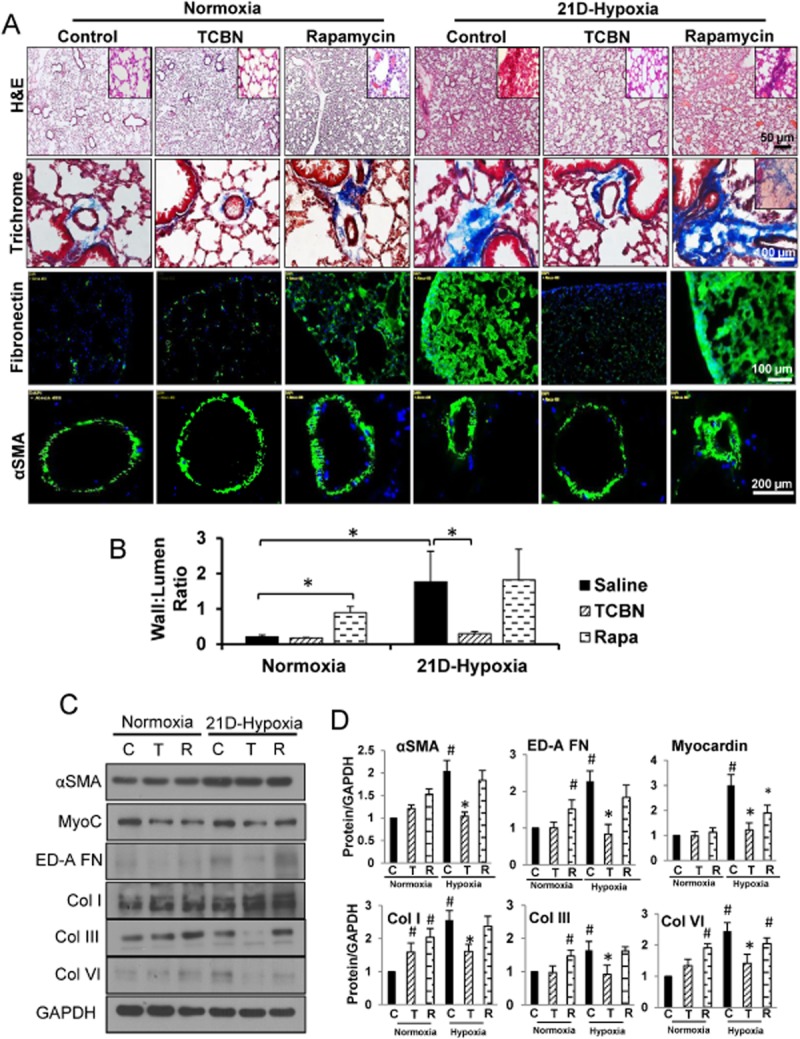
TCBN reverses hypoxia-induced pulmonary fibrosis and vascular remodelling in vivo. (A) H&E staining, Masson's trichrome staining, fibronectin and αSMA immunofluorescence staining of lung sections subjected for normoxia and 21 day chronic hypoxia. (B) Histogram showing vascular wall to lumen ratio (n = 6–8 mice/group). (C) Western analysis of αSMA and its transcription factors SRF and myocardin, ECM proteins including ED-A-FN, collagen types I, III and VI in TCBN, and rapamycin-treated lungs compared with vehicle-treated lungs after 21 days of hypoxia. (D) Histograms showing the densitometry analysis of Western protein bands showing changes in the expression of αSMA, SRF, myocardin, ED-A-FN, collagen types I, III and VI in control, TCBN and rapamycin-treated 21 day hypoxic lungs. *P < 0.05, #P < 0.01 (n = 6–8 mice/group).
Akt inhibition reverses adTGFβ-induced pulmonary fibrosis in vivo
Next, we sought to investigate the effects of Akt inhibition using TCBN and mTOR inhibition using rapamycin on TGFβ-induced model of pulmonary remodelling. Administration of adTGFβ (i.t.) to mouse lungs induced significant damage in the normal pulmonary architecture that correlated with an Ashcroft fibrosis score of ∼7.6 (categorized as severe fibrosis). Administration of adTGFβ resulted in a heterogeneous pattern consistent with advanced stage IPF (Figure 3A) with substantial interstitial fibrosis, ECM deposition, loss of alveolar parenchyma, microscopic honeycomb foci and an increase in the percentage of fibrosed area. This was also associated with marked increase in fibronectin and αSMA deposition and assembly as demonstrated by immunofluorescence staining (Figure 3A). Akt inhibition using TCBN blunted adTGFβ-induced fibrosis correlating to an Ashcroft fibrosis score of ∼1.2 (categorized as minimal fibrosis) (Figure 3A–C). This is evidenced by markedly decreased fibrotic patches, ECM deposition and αSMA expression (Figure 3A–C). In contrast, although rapamycin-treated animals had decreased fibrotic patches and improved Ashcroft fibrosis score of ∼5 (categorized as moderate fibrosis) (Figure 3A–C), they also demonstrated thickening of the alveolar space, diffuse alveolar micro-haemorrhage, capillary congestion and dense inflammatory infiltrates as evidenced by H&E and Masson's trichrome staining (Figure 3A). Notably, even in the absence of adTGFβ, control mice treated with rapamycin exhibited aberrant pulmonary morphological features (Figure 3A–C).
Figure 3.
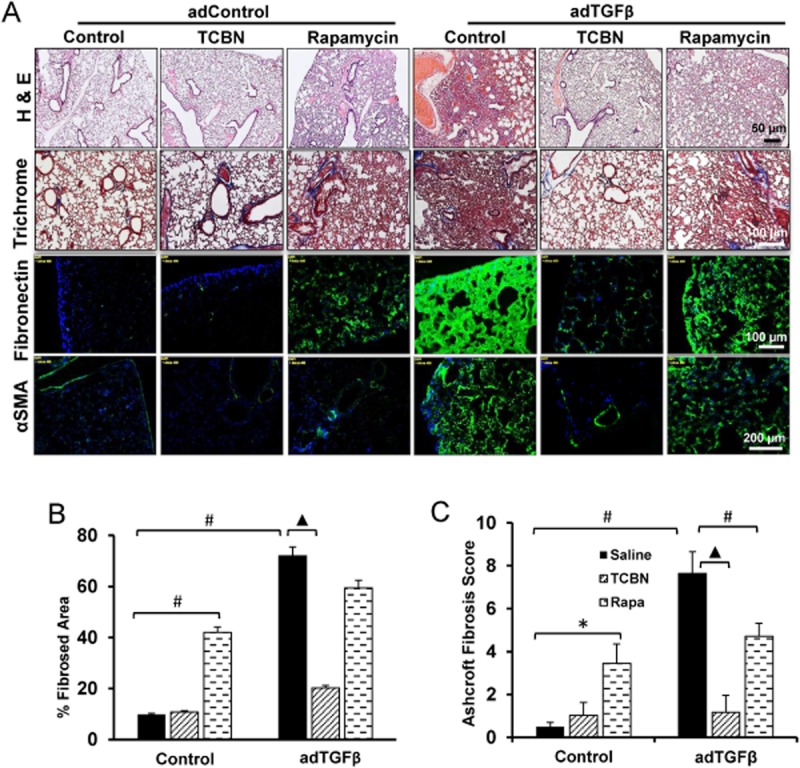
TCBN ameliorates adTGFβ-induced pulmonary fibrosis in vivo. (A) H&E staining, Masson's trichrome staining, fibronectin and αSMA immunofluorescence staining of lung sections subjected for adControl and adTGFβ treatments. (B) Histogram showing quantification of the fibrosed area in adControl and adTGFβ-expressing mice lungs (n = 6–8 mice/group). (C) Histogram showing Aschroft fibrosis score in adControl and adTGFβ-expressing mice lungs (n = 6–8 mice/group). *P < 0.05, #P < 0.01, ▲P < 0.001.
Akt inhibition ameliorates hypoxia- and adTGFβ-induced peripheral vascular rarefaction
The above findings prompted us to examine the effects of Akt inhibition on the aberrant peripheral vascular rarefaction that occurs during PH and pulmonary fibrosis (PF). To do this, we utilized arterial casting to visualize the vascular tree of mouse lungs subjected to 21d-chronic hypoxia and/or adTGFβ. First, compared with the diffuse vascular blush observed in the normoxic lung, chronic hypoxia resulted in significant vascular pruning as demonstrated by binary images of peripheral vessels (Figure 4A and B). TCBN partially inhibited progressive pruning of the vasculature (Figure 4A and B), which supports our previous finding that TCBN alleviates vessel occlusion in microcapillaries. In contrast, rapamycin treatment did not significantly reverse the reduced vascular density due to chronic hypoxia and had no significant effect on pruning of small vessels (Figure 4A and B). Noteworthy, while TCBN did not modulate vascular density in normoxic mice, rapamycin was associated with vascular filling and increased vessel pruning (Figure 4A and B).
Figure 4.
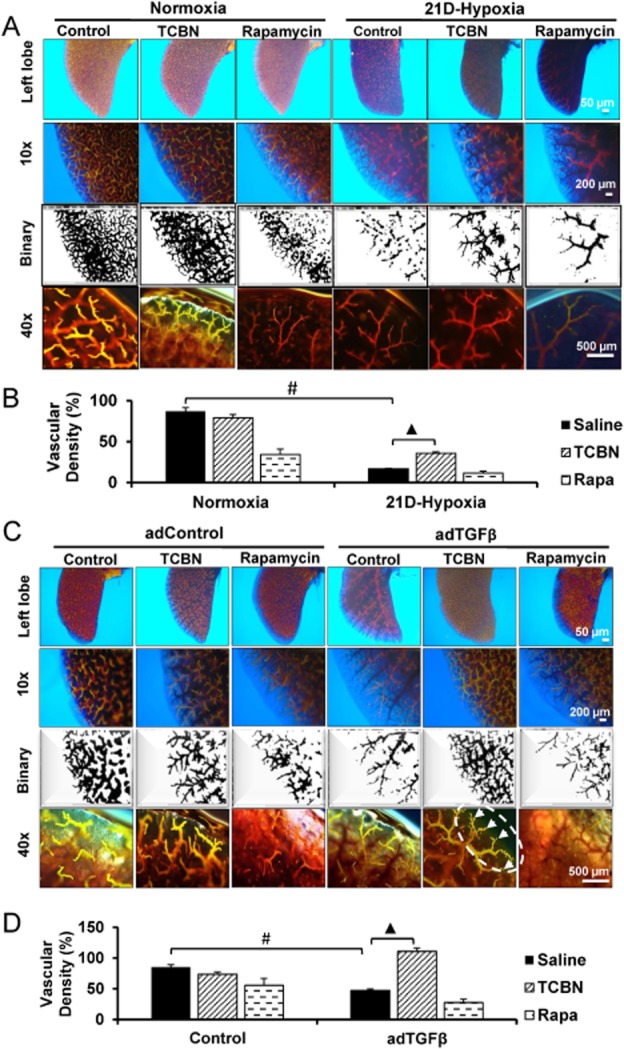
TCBN reverses hypoxia- and adTGFβ-induced vascular rarefaction. (A) Representative images showing vascular branching of the left lobe after Microfil casting of mice subjected to normoxia or chronic hypoxia and treated with saline, TCBN or rapamycin. (B) Histogram showing vascular density (%) in mouse lungs subjected to normoxia or chronic hypoxia and treated with saline, TCBN or rapamycin, and calculated using ImageJ software (n = 3–5 mice/group). (C) Representative images showing vascular branching of the left lobe after Microfil casting of mice subjected to adControl or adTGFβ, and treated with saline, TCBN or rapamycin. Arrows indicate increase in microvascular branching in TCBN-treated group. (D) Histogram showing vascular density (%) in mouse lungs subjected to adControl or adTGFβ treated with saline, TCBN or rapamycin, and calculated using ImageJ software (n = 3–5 mice/group). #P < 0.01, ▲P < 0.001. Scale bar in order 500, 200 and 50 μm.
Next, we subjected mice to the adTGFβ-induced pulmonary fibrosis and the lung tissues were analysed for vascular rarefaction. Expression with adTGFβ leads to significant pruning of small vessels on day 8 compared with adControl (Figure 4C and D). TCBN treatment maintained the vasculature even in the presence of adTGFβ whereas rapamycin- treated mice did not show a beneficial effect on peripheral vasculature protection (Figure 4C and D).
Akt inhibition ameliorates chronic hypoxia-induced right ventricular remodelling and hepatotoxicity
The hypoxia-induced pulmonary vasculopathies often lead to right ventricular hypertrophy (Voelkel et al., 2012). We determined the effects of inhibiting Akt or mTOR on right ventricular wall thickness in mice subjected to 21 day hypoxia. Under normoxic conditions, neither TCBN nor rapamycin induced any discernible effects on wall thickness (Figure 5A and B). The modest but significant increase in right ventricular wall thickness in hypoxic compared with normoxic mice was reversed by TCBN treatment (Figure 5A and B). However, rapamycin treatment did not induce a significant reduction in right ventricular wall thickness (Figure 5A and B).
Figure 5.
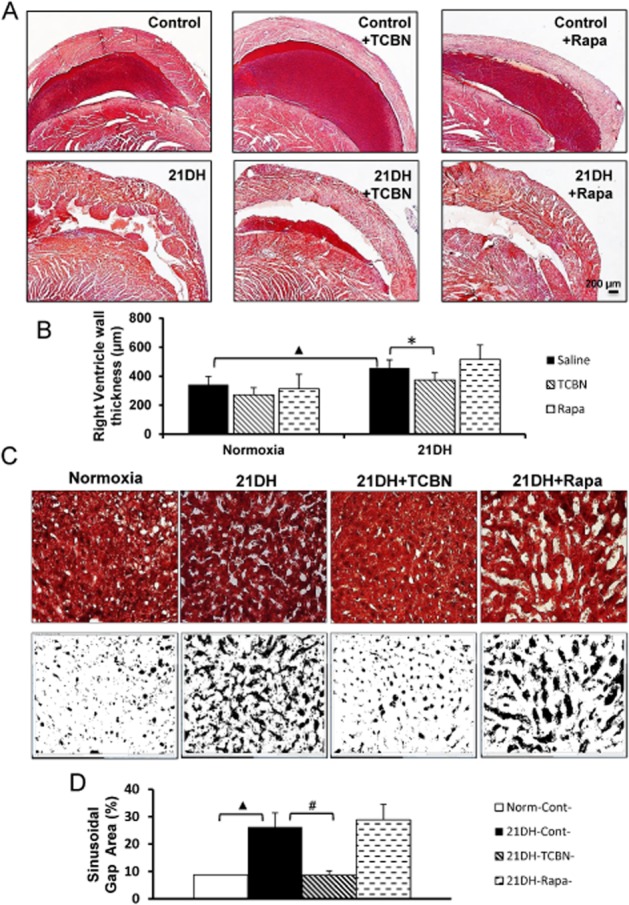
TCBN inhibits hypoxia-induced right ventricular remodelling and hepatic injury. (A) Images of Masson's trichrome stained cross sections of the right ventricle from control, chronic hypoxic, hypoxic treated with TCBN and hypoxic treated with rapamycin hearts. (B) Histogram showing quantification of right ventricular wall thickness showing the effect of TCBN and rapamycin on compensatory ventricular wall remodelling following hypoxia (n = 6–8 mice/group). (C) Pictures of Masson's trichrome stained liver sections and binary images (using ImageJ software). (D) Histogram showing quantification of per cent sinusoidal gap area as measured using ImageJ software from binary images (n = 6–8 mice/group). *P < 0.05, #P < 0.01, ▲P < 0.001.
Next, we assessed the safety profile of both drugs in the liver of mice subjected to chronic hypoxia. Compared with normoxia control, liver sections from 21 day chronic hypoxic mice exhibited significantly higher percentage of gap areas between the cells indicating significant liver damage due to hypoxia (Figure 5C and D). This is evident from the marked degeneration and atrophy of hepatic cords and the associated sinusoidal dilation. Liver histology also revealed that treatment with TCBN, but not rapamycin, blunted liver damage due to chronic hypoxia (Figure 5C and D). No significant difference in the ratio between lung, liver, heart or kidney weights and total body weight was observed, thus suggesting that both TCBN and rapamycin had no severe toxic effects on mice (Supporting Information Fig. S1).
Akt1 inhibition attenuates pulmonary remodelling through inhibition of TSP1 expression
To identify the signalling molecules downstream of Akt1 activation in pulmonary fibrosis, we performed gene arrays for fibrosis-related genes in both NIH-3T3 and FHLFs expressing inactive mutant of Akt1 (DN-Akt1; Akt1 K179M). A common target identified from gene arrays performed in both the cells after Akt1 inhibition was reduced expression of TSP1 (Figure 6A). Indeed, Western analyses revealed that the TSP1 expression in 14d-hypoxic Akt1+/+ mice was blunted in 14d-hypoxic Akt1−/− mice, which was also correlated with significantly reduced fibronectin expression (Figure 6B and C).
Figure 6.
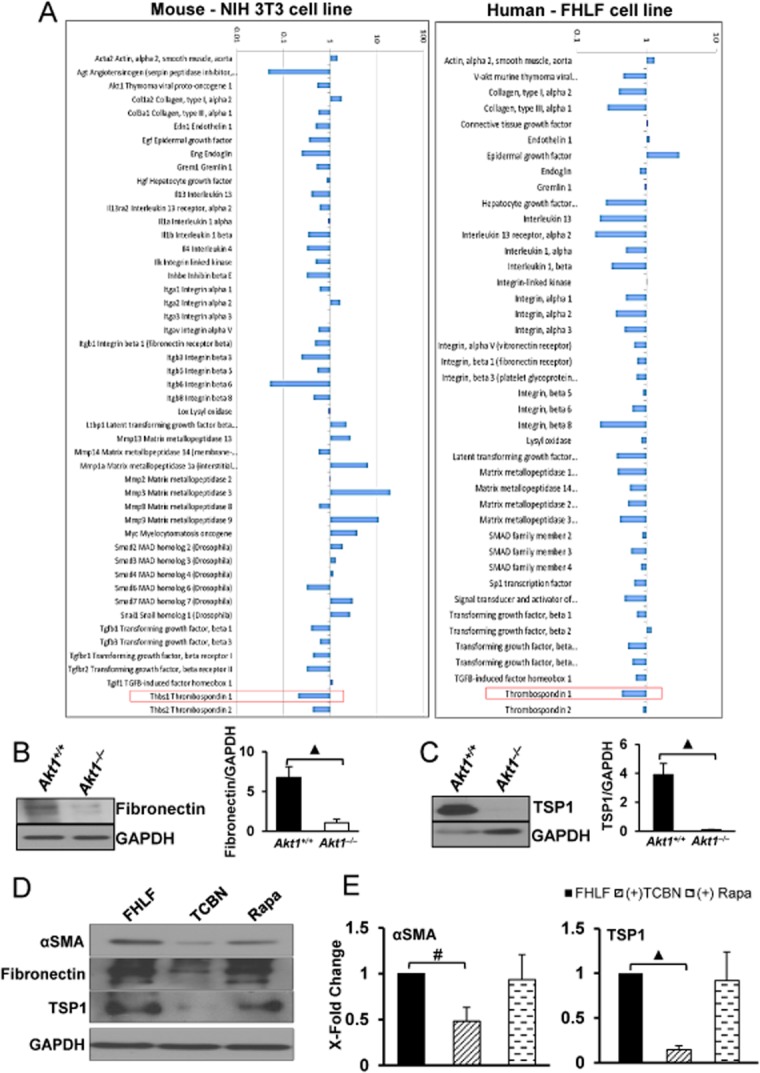
TSP1 expression is decreased in Akt1-deficient NIH 3T3, IPF fibroblasts and 14 day hypoxic mice. (A) Gene expression profiles of NIH 3T3 and IPF fibroblasts (FHLFs) expressing DN-Akt1 (dominant negative, inactive Akt1) (n = 3). (B and C) Western blot image and histogram showing total fibronectin and TSP1 expression levels normalized to GAPDH levels in 14 day hypoxic Akt1+/+ and Akt1−/− mice lungs (n = 3–5 per group). (D) Images of Western blots showing the effect of TCBN and rapamycin on the expression of αSMA and TSP1 expression in FHLFs. (E) Histogram showing densitometry analysis of the Western blot bands indicating changes in the expression levels of αSMA and TSP1, normalized to GAPDH after treatment with TCBN and rapamycin in FHLFs (n = 3). #P < 0.01, ▲P < 0.001.
To further confirm our previous in vivo and in vitro findings, we subjected FHLFs isolated from an IPF patient to serum starvation and treated with 10 nM TCBN or 25 nM rapamycin for 24 h, and analysed αSMA, fibronectin and TSP1 expression levels. Inhibiting Akt, but not mTOR, significantly reduced αSMA, fibronectin and TSP1 expressions in FHLFs (Figure 6D and E). The results were confirmed in vivo using Western analysis of the adControl and adTGFβ expressing mice lung tissues. Our analysis indicated that the lungs expressing adTGFβ expressed significantly higher concentrations of TSP1 along with the expression of increased expression of myofibroblast markers such as αSMA, ED-A-FN and ECM proteins, and that treatment with TCBN, but not rapamycin, significantly reversed their expression (Figure 7A and B).
Figure 7.
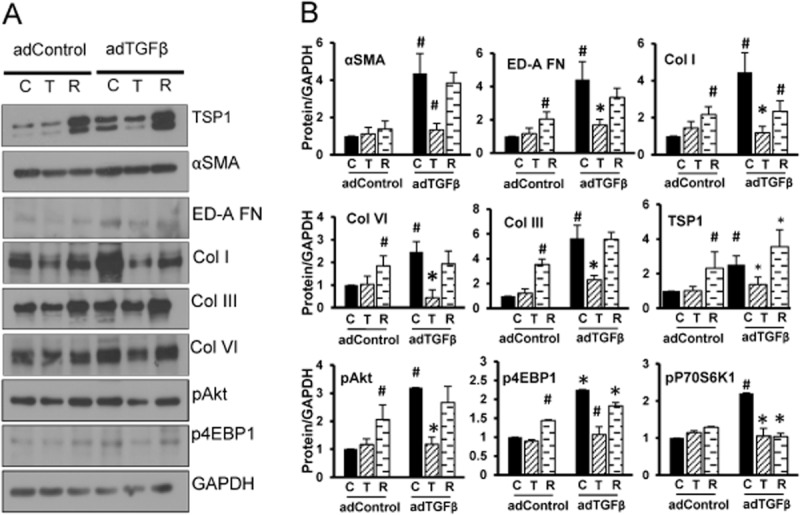
Targeting Akt, not mTOR, modulates expression of TSP1, αSMA and ECM proteins in the mice lungs with adTGFβ expression. (A) Western blot images of adControl and adTGFβ expressing lung tissue lysates showing the changes in expression of TSP1, αSMA, ED-A-FN, collagens (types I, III and VI), and phosphorylated Akt and 4E-BP1. (B) Densitometry analysis of Western blot bands of adControl and adTGFβ-expressing lung tissue lysates showing the changes in expression of TSP1, αSMA, ED-A-FN, collagens (types I, III and VI) and phosphorylated Akt and 4E-BP1 (n = 4). *P < 0.05, #P < 0.01.
TSP1−/− mice are protected from adMyrAkt1-induced pulmonary fibrosis
To further confirm the causal role of Akt1-TSP1 signalling axis in PF and lung tissue remodelling, we subjected TSP1+/+ and TSP1−/− mice to i.t. administration of adenovirus-mediated gene transfer of control vector or adenovirus-mediated expression of constitutively active Akt1 (adMyrAkt1). Administration of adMyrAkt1 in mice resulted in significant interstitial fibrosis. TSP1+/+ lung tissues expressing adMyrAkt1 exhibited significant increase in tissue remodelling correlating an Aschroft scale of ∼7 (Figure 8A–C). Although mild tissue remodelling was already present in TSP1−/− mice lungs, compared with control-TSP1+/+ mice, TSP1−/− mice did not exhibit significant histological alterations with adMyrAkt1 expression (Figure 8A–C), thus indicating that TSP1−/− mice are resistant to adMyrAkt1-induced pulmonary fibrosis (Figure 7A–C).
Figure 8.
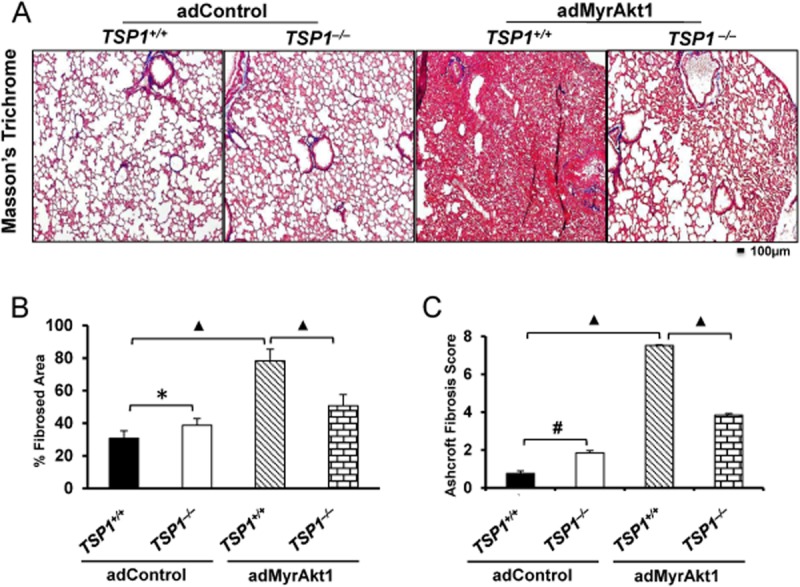
TSP1−/− mice are protected from adMyrAkt1-induced pulmonary fibrosis. (A) Masson's trichrome staining of WT and TSP1−/− mouse lung harvested 14 days after i.t. adenovirus gene transfer of control vector or adMyrAkt1 (constitutive active Akt1) (n = 3 mice/group). (B and C) Per cent fibrosed area quantified using ImageJ software, and Ashcroft fibrosis score of the degree of fibrosis in WT and TSP1−/− mice subjected to control vector or adMyr-Akt1 respectively. *P < 0.05, #P < 0.01, ▲P < 0.001.
Discussion and conclusions
Although Akt is hyperactivated in the fibroblastic foci in human IPF lungs (Xia et al., 2008), a causal link between Akt activation and the events leading to IPF has not been established. Further, the therapeutic benefits of targeting Akt for IPF have not yet been investigated. Our major findings from the current study are: (i) Akt1−/−mice are protected from chronic hypoxia-induced pulmonary vascular and tissue remodelling; (ii) Akt inhibitor TCBN ameliorates progressive chronic hypoxia- and adTGFβ-induced pulmonary fibrosis, peripheral vascular remodelling and rarefaction; (iii) targeting Akt substrate mTOR using rapamycin did not reverse chronic hypoxia- and adTGFβ-induced peripheral vascular remodelling and rarefaction, and pulmonary fibrosis, respectively; (iv) genetic ablation of Akt1 and pharmacological inhibition of Akt both resulted in significantly reduced expression of TSP1, a matricellular protein known to be involved in the hypoxia-induced pulmonary remodelling (Ochoa et al., 2010) as well as fibrosis progression (Bussolati et al., 2006; Xie et al., 2010); (v) sustained hyperactivation of Akt1 in the mice lungs through expression of adMyrAkt1-induced severe pulmonary fibrosis even in the absence of any TGFβ stimuli; and (vi) TSP1−/− mice are protected from adMyrAkt1-induced pulmonary fibrosis. Our study demonstrates the integral role of Akt1 in the disease progression of IPF through development of fibrotic foci and vascular rarefaction, partly through modulation of TSP1, ED-A-FN and collagen type I expression, in addition to its role in the expression of αSMA via a SRF/myocardin pathway. To our knowledge, these findings are novel and have not been reported previously.
The origin of the events leading to the development of IPF is still not clearly understood. This also became the bottleneck in the development of the appropriate treatment strategy for pulmonary fibrosis patients. A variety of theories such as ‘vascular’, ‘plasticity/growth factor receptor’, ‘inflammation’ and ‘matrix’ hypotheses have been proposed for the development of IPF (Bringardner et al., 2008). Others have argued that profound inflammation in the alveoli and the interstitium is the major cause of IPF (Gauldie, 2002; Strieter, 2002). However, clinical trials on anti-inflammatory agents for the treatment of IPF patients did not yield beneficial effects (Rafii et al., 2013). Even though thrombin and tissue factor expression was enhanced in IPF lungs suggesting a role for platelets in the disease progression, heparin or warfarin provided no therapeutic benefits (Kubo et al., 2005). Although vascular remodelling occurs in fibrosis (Barman et al., 2014), the VEGF inhibitor SU5416 (SUGEN) induces pulmonary vascular injury and remodelling leading to PH (Mendel et al., 2000). Although PH has been linked to the development of ILD, its management has proven ineffective in IPF patients. Clinical trials of the endothelin receptor antagonist bosentan used in the management of PH failed clinical trials in IPF patients (Rafii et al., 2013). In contrast, nintedanib, a broad spectrum receptor tyrosine kinase inhibitor, improved the quality of life for IPF patients (Richeldi et al., 2014). On the other hand, the pathological hallmark of fibrosis is the persistent myofibroblast differentiation and ECM deposition. TGFβ is the major mediator of tissue fibrosis through promotion of myofibroblast differentiation and ECM deposition (Tomasek et al., 2002; Hinz et al., 2012). A recent clinical trial on pirfenidone, an inhibitor of TGFβ, demonstrated very similar efficacy to that of nintedanib in IPF patients (Takeda et al., 2014). Although these two studies demonstrated improvement in the forced vital capacity in IPF patients leading to its approval for use in the USA by the Food and Drug Administration in 2014, unfortunately neither of these had any significant effect on the overall patient mortality. These clinical trials provided important clues that a multifactorial disease such as IPF cannot be treated by targeting just one arm of the disease.
Intracellular signalling molecules such as the PI3 kinase-Akt pathway, Ras-mitogen activated kinase (MAPK) pathway and Rho-GTPase signalling are activated by various receptor tyrosine kinases, GPCRs and TGFβ family of growth factors (Massagué, 2012). Among these, Akt is a potential target for IPF therapy due to its involvement in various processes leading to the development of IPF. We have previously demonstrated that Akt1, the predominant Akt isoform in endothelial cells and fibroblasts, is responsible for the promotion of angiogenesis and vascular protection (Chen et al., 2005a), wound healing (Somanath et al., 2008), and fibroblast-mediated ECM secretion (Goc et al., 2011) and assembly (Somanath et al., 2007; Somanath and Byzova, 2009). Akt1 has also been implicated in the regulation of pulmonary artery neointimal proliferation through smooth muscle cell activation (Tang et al., 2015). In pathological conditions, Akt1 also promoted myofibroblast differentiation in vitro (Abdalla et al., 2013) and post-ischaemic cardiac fibrosis in vivo (Ma et al., 2014). Furthermore, whereas Akt1 inhibition has been shown to induce vascular permeability, specific role for each of the Akt1, Akt2 and Akt3 isoforms has been implicated in platelet activation and thrombosis (Chen et al., 2004; Woulfe et al., 2004; O'Brien et al., 2011). Akt1 has also been shown to promote inflammation (Di Lorenzo et al., 2009). Several lines of evidence have reported that PI3K/Akt pathway is hyperactivated during fibrosis in general (Xia et al., 2008) and pulmonary fibrosis in particular (Xia et al., 2008). As events leading to the vascular complications and pulmonary tissue remodelling are consistent with deregulated Akt-mediated cellular and molecular processes, we hypothesized that hyperactivation of Akt would lead to pathological pulmonary tissue remodelling, and that pharmacological inhibition of Akt might be a logical solution for the treatment of IPF patients. Our data from Akt1−/− mice lungs showing impaired chronic hypoxia-induced pulmonary tissue and vascular remodelling evidenced by reduced interstitial lesions associated with decreased expression of fibronectin and αSMA further supported this hypothesis.
Currently, TCBN, an Akt inhibitor, is in clinical trials for the management of various types of cancers, and initial reports indicate no serious adverse events (Garrett et al., 2011). Its specificity and efficacy in cancer prompted us to test this drug in comparison with the anti-inflammatory mTOR inhibitor rapamycin. Our study identified anti-fibrotic and anti-remodelling properties of TCBN as evident by the reduced fibrotic lesions, αSMA and matrix deposition, decreased medial thickening of pulmonary arterioles, reduced vascular pruning and right ventricular thickening. In contrast, rapamycin exacerbated the conditions through increased alveolar congestion and micro-haemorrhage, severe pulmonary vascular rarefaction, right ventricular thickening and hepatotoxicity. Rapamycin did not induce anti-fibrotic and anti-remodelling response as previously reported in rodent models (Paddenberg et al., 2007; Korfhagen et al., 2009). Whereas one potential reason for this discrepancy could be due to the use of supra-optimal doses of rapamycin (4 and 3 mg·kg−1·day−1 respectively) in these studies, our results are consistent with clinical reports of pulmonary toxicities associated with two rapamycin derivatives, sirolimus and everolimus (Buhaescu et al., 2006; Damas et al., 2006; Khalife et al., 2007; Feagans et al., 2009; Depuydt et al., 2012). Importantly, this finding sheds light on the adverse effects of everolimus observed in IPF patients (Malouf et al., 2011). Mechanistically, the adverse effects of rapamycin could be attributed, at least in part, to the rebound Akt activation as reported by several laboratories (Sun et al., 2005; Breuleux et al., 2009; Zakikhani et al., 2010; Soares et al., 2013). On the other hand, TCBN exhibits dual role by targeting both myofibroblast differentiation and the ECM deposition, in addition to increasing vascular perfusion, platelet inactivation and reduced inflammation as reported in the literature. This suggests that targeting Akt, not mTOR, may serve as a favourable therapeutic strategy in IPF.
Our previous study has identified that Akt1 is directly involved in TSP1 expression by the vascular cells, and Akt1−/− mice skin and lung endothelial cells express reduced levels of TSP1 (Chen et al., 2005b). Interestingly, it has been shown that TSP1 deficiency protects against hypoxia-induced PH (Ochoa et al., 2010), but not in bleomycin-induced pulmonary fibrosis (Ezzie et al., 2012). Increased TSP1 has also been shown to promote vascular rarefaction (Gonzalez-Quesada et al., 2013). Incidentally, our gene array analysis in two different fibroblast cell lines, murine NIH 3T3 and human pulmonary fibrosis patient lung fibroblasts, also identified decreased TSP1 gene expression upon overexpression with DN-Akt1 (Akt1K179M inactive mutant). This was further confirmed at the protein level in Akt1−/− mice lungs and human fibrotic lung fibroblasts. As overexpression with adMyrAkt1 in lungs was sufficient to cause fibrosis, and because bleomycin was able to induce fibrosis TSP1−/− mice lungs, we determined whether TSP1−/− mice lungs will be resistant to pulmonary fibrosis induced by adMyrAkt1 overexpression. We found that TSP1-deficient mice are protected from adMyrAkt1-induced pulmonary fibrosis. The reason that TSP1−/− mice lungs were resistant to bleomycin-induced pulmonary fibrosis, but not to adMyrAkt1-induced pulmonary vascular remodelling along with our observation of pulmonary tissue remodelling in Akt1−/− mice lungs in normoxic conditions, suggests the existence of Akt-independent pathways in the regulation of TGFβ-induced pulmonary fibrosis. Collectively, our results provide direct evidence on the causal role of Akt1 in disease onset and progression in pulmonary fibrosis by promoting myofibroblast differentiation and ECM deposition, in part, through TSP1 regulation. Our study indicates potential therapeutic benefits of the Akt inhibitor TCBN for the treatment of ILD, particularly IPF.
Acknowledgments
Funds were provided by the National Institutes of Health grant (R01HL103952), the University of Georgia College of Pharmacy intramural grant and the Wilson Pharmacy Foundation to P. R. S., and the National Institutes of Health grant (R01 NS083559) to A. E. M. A. was supported by a pre-doctoral fellowship from the American Heart Association (13PRE17100070). This material is the result of work supported with resources and the use of facilities at the Charlie Norwood VAMC, Augusta, GA. The funders had no role in the study design, data collection, analysis and decision to publish. Preparation of the manuscript and the contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Glossary
- ECM
extracellular matrix
- FHLF
fibrotic human lung fibroblast
- FN
fibronectin
- ILD
interstitial lung disease
- IPF
idiopathic pulmonary fibrosis
- mTOR
mammalian target of rapamycin
- PH
pulmonary hypertension
- PTEN
phosphatase and tensin homologue
- SRF
serum response factor
- TCBN
triciribine
- TSP1
thrombospondin1
- αSMA
α-smooth muscle cell actin
Author contributions
M. A., H. S. and R. P. performed the research. M. A., A. E., S. C. F. and P. R. S. designed the research study. M. A., H. S., R. P. and P. R. S. analysed the data. M. A. and P. R. S. wrote the paper.
Conflict of interest
The authors have declared that no conflicts of interest exist.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Effect of triciribine and rapamycin on organ and body weight during hypoxia. The ratio of organ to body weight was quantified for the lung, heart, liver and kidney (n = 6–8 mice/group).
References
- Abdalla M, Goc A, Segar L, Somanath PR. Akt1 mediates α-smooth muscle actin expression and myofibroblast differentiation via myocardin and serum response factor. J Biol Chem. 2013;288:33483–33493. doi: 10.1074/jbc.M113.504290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman SA, Chen F, Su Y, Dimitropoulou C, Wang Y, Catravas JD, et al. Nadph oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler Thromb Vasc Biol. 2014;34:1704–1715. doi: 10.1161/ATVBAHA.114.303848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuleux M, Klopfenstein M, Stephan C, Doughty CA, Barys L, Maira SM, et al. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol Cancer Ther. 2009;8:742–753. doi: 10.1158/1535-7163.MCT-08-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringardner BD, Baran CP, Eubank TD, Marsh CB. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid Redox Signal. 2008;10:287–301. doi: 10.1089/ars.2007.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhaescu I, Izzedine H, Covic A. Sirolimus-challenging current perspectives. Ther Drug Monit. 2006;28:577–584. doi: 10.1097/01.ftd.0000245377.93401.39. [DOI] [PubMed] [Google Scholar]
- Bussolati B, Assenzio B, Deregibus MC, Camussi G. The proangiogenic phenotype of human tumor-derived endothelial cells depends on thrombospondin-1 downregulation via phosphatidylinositol 3-kinase/Akt pathway. J Mol Med (Berl) 2006;84:852–863. doi: 10.1007/s00109-006-0075-z. [DOI] [PubMed] [Google Scholar]
- Chen J, De S, Damron DS, Chen WS, Hay N, Byzova TV. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–1710. doi: 10.1182/blood-2003-10-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, et al. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med. 2005a;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, Law PY, Loh HH. Inhibition of PI3K/Akt signaling: an emerging paradigm for targeted cancer therapy. Curr Med Chem Anticancer Agents. 2005b;5:575–589. doi: 10.2174/156801105774574649. [DOI] [PubMed] [Google Scholar]
- Damas C, Oliveira A, Morais A. Lung toxicity induced by rapamycin. Rev Port Pneumol. 2006;12:715–724. doi: 10.1016/s0873-2159(15)30463-3. [DOI] [PubMed] [Google Scholar]
- Depuydt P, Nollet J, Benoit D, Praet M, Caes F. Fatal acute pulmonary injury associated with everolimus. Ann Pharmacother. 2012;46:e7. doi: 10.1345/aph.1Q623. [DOI] [PubMed] [Google Scholar]
- Di Lorenzo A, Fernández-Hernando C, Cirino G, Sessa WC. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc Natl Acad Sci U S A. 2009;106:14552–14557. doi: 10.1073/pnas.0904073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzie ME, Crawford M, Cho JH, Orellana R, Zhang S, Gelinas R, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. 2012;67:122–131. doi: 10.1136/thoraxjnl-2011-200089. [DOI] [PubMed] [Google Scholar]
- Feagans J, Victor D, Moehlen M, Florman SS, Regenstein F, Balart LA, et al. Interstitial pneumonitis in the transplant patient: consider sirolimus-associated pulmonary toxicity. J la State Med Soc. 2009;161:166. , 168–172. [PubMed] [Google Scholar]
- Garrett CR, Coppola D, Wenham RM, Cubitt CL, Neuger AM, Frost TJ, et al. Phase I pharmacokinetic and pharmacodynamic study of triciribine phosphate monohydrate, a small-molecule inhibitor of AKT phosphorylation, in adult subjects with solid tumors containing activated AKT. Invest New Drugs. 2011;29:1381–1389. doi: 10.1007/s10637-010-9479-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauldie J. Pro: inflammatory mechanisms are a minor component of the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2002;165:1205–1206. doi: 10.1164/rccm.2202054. [DOI] [PubMed] [Google Scholar]
- Goc A, Choudhary M, Byzova TV, Somanath PR. TGFβ- and bleomycin-induced extracellular matrix synthesis is mediated through Akt and mammalian target of rapamycin (mTOR) J Cell Physiol. 2011;226:3004–3013. doi: 10.1002/jcp.22648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Quesada C, Cavalera M, Biernacka A, Kong P, Lee DW, Saxena A, et al. Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ Res. 2013;113:1331–1344. doi: 10.1161/CIRCRESAHA.113.302593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalife WI, Kogoj P, Kar B. Sirolimus-induced alveolar hemorrhage. J Heart Lung Transplant. 2007;26:652–657. doi: 10.1016/j.healun.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King TE, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- Korfhagen TR, Le Cras TD, Davidson CR, Schmidt SM, Ikegami M, Whitsett JA, et al. Rapamycin prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2009;41:562–572. doi: 10.1165/rcmb.2008-0377OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo H, Nakayama K, Yanai M, Suzuki T, Yamaya M, Watanabe M, et al. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2005;128:1475–1482. doi: 10.1378/chest.128.3.1475. [DOI] [PubMed] [Google Scholar]
- Ma L, Kerr BA, Naga Prasad SV, Byzova TV, Somanath PR. Differential effects of Akt1 signaling on short- versus long-term consequences of myocardial infarction and reperfusion injury. Lab Invest. 2014;94:1083–1091. doi: 10.1038/labinvest.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malouf MA, Hopkins P, Snell G, Glanville AR Investigators EiIS. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology. 2011;16:776–783. doi: 10.1111/j.1440-1843.2011.01955.x. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573–1619. doi: 10.1016/j.jacc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Mendel DB, Schreck RE, West DC, Li G, Strawn LM, Tanciongco SS, et al. The angiogenesis inhibitor SU5416 has long-lasting effects on vascular endothelial growth factor receptor phosphorylation and function. Clin Cancer Res. 2000;6:4848–4858. [PubMed] [Google Scholar]
- Meyer KC. Diagnosis and management of interstitial lung disease. Transl Respir Med. 2014;2:4. doi: 10.1186/2213-0802-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien KA, Stojanovic-Terpo A, Hay N, Du X. An important role for Akt3 in platelet activation and thrombosis. Blood. 2011;118:4215–4223. doi: 10.1182/blood-2010-12-323204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa CD, Yu L, Al-Ansari E, Hales CA, Quinn DA. Thrombospondin-1 null mice are resistant to hypoxia-induced pulmonary hypertension. J Cardiothorac Surg. 2010;5:32. doi: 10.1186/1749-8090-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns HH, et al. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res. 2007;8:15. doi: 10.1186/1465-9921-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafii R, Juarez MM, Albertson TE, Chan AL. A review of current and novel therapies for idiopathic pulmonary fibrosis. J Thorac Dis. 2013;5:48–73. doi: 10.3978/j.issn.2072-1439.2012.12.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS ONE. 2013;8:e57289. doi: 10.1371/journal.pone.0057289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanath PR, Byzova TV. 14-3-3beta-Rac1-p21 activated kinase signaling regulates Akt1-mediated cytoskeletal organization, lamellipodia formation and fibronectin matrix assembly. J Cell Physiol. 2009;218:394–404. doi: 10.1002/jcp.21612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanath PR, Kandel ES, Hay N, Byzova TV. Akt1 signaling regulates integrin activation, matrix recognition, and fibronectin assembly. J Biol Chem. 2007;282:22964–22976. doi: 10.1074/jbc.M700241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanath PR, Chen J, Byzova TV. Akt1 is necessary for the vascular maturation and angiogenesis during cutaneous wound healing. Angiogenesis. 2008;11:277–288. doi: 10.1007/s10456-008-9111-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange C, Highland KB. Pulmonary hypertension in interstitial lung disease. Curr Opin Pulm Med. 2005;11:452–455. doi: 10.1097/01.mcp.0000174250.38188.6d. [DOI] [PubMed] [Google Scholar]
- Strieter RM. Con: inflammatory mechanisms are not a minor component of the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2002;165:1206–1207. doi: 10.1164/rccm.2202055. , discussion 1207–1208. [DOI] [PubMed] [Google Scholar]
- Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Tsujino K, Kijima T, Kumanogoh A. Efficacy and safety of pirfenidone for idiopathic pulmonary fibrosis. Patient Prefer Adherence. 2014;8:361–370. doi: 10.2147/PPA.S37233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, et al. Deficiency of Akt1, but Not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2015;308:L208–L220. doi: 10.1152/ajplung.00242.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ, Nicolls MR. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur Respir J. 2012;40:1555–1565. doi: 10.1183/09031936.00046612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woulfe D, Jiang H, Morgans A, Monks R, Birnbaum M, Brass LF. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Invest. 2004;113:441–450. doi: 10.1172/JCI20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med. 2008;205:1659–1672. doi: 10.1084/jem.20080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Khalil W, Kahm J, Jessurun J, Kleidon J, Henke CA. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am J Pathol. 2010;176:2626–2637. doi: 10.2353/ajpath.2010.091117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie XS, Liu HC, Wang FP, Zhang CL, Zuo C, Deng Y, et al. Ginsenoside Rg1 modulation on thrombospondin-1 and vascular endothelial growth factor expression in early renal fibrogenesis in unilateral obstruction. Phytother Res. 2010;24:1581–1587. doi: 10.1002/ptr.3190. [DOI] [PubMed] [Google Scholar]
- Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010;123:271–279. doi: 10.1007/s10549-010-0763-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of triciribine and rapamycin on organ and body weight during hypoxia. The ratio of organ to body weight was quantified for the lung, heart, liver and kidney (n = 6–8 mice/group).