

Abstract
Recent studies report that a history of antidepressant use is strongly correlated with the occurrence of Parkinson’s disease (PD). However, it remains unclear whether antidepressant use can be a causative factor for PD. In the present study, we examined whether tricyclic antidepressants amitriptyline and desipramine can induce dopaminergic cell damage, both in vitro and in vivo. We found that amitriptyline and desipramine induced mitochondria-mediated neurotoxicity and oxidative stress in SH-SY5Y cells. When injected into mice on a subchronic schedule, amitriptyline induced movement deficits in the pole test, which is known to detect nigrostriatal dysfunction. In addition, the number of tyrosine hydroxylase-positive neurons in the substantia nigra pars compacta was reduced in amitriptyline-injected mice. Our results suggest that amitriptyline and desipramine may induce PD-associated neurotoxicity.
Keywords: amitriptyline, desipramine, neurotoxicity, Parkinson’s disease, tricyclic antidepressant
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease. It is characterized by dopaminergic neuronal loss in the substantia nigra pars compacta (SNpc) of the midbrain and results in movement deficits. It is widely appreciated that environmental toxins, as well as genetic factors, can contribute to the pathogenesis of PD. Exposure to pesticides has been suggested as a risk factor for PD (Freire and Koifman, 2012). Many studies have shown that pesticides, including rotenone, paraquat, and maneb, cause dopaminergic neurotoxicity via mitochondrial damage and oxidative stress (Arif and Khan, 2010; Hatcher et al., 2008). When injected into mice, these pesticides can recapitulate some aspects of PD, such as dopaminergic neuronal loss in the SNpc and motor deficits (Hatcher et al., 2008; Meredith et al., 2008).
In addition to pesticides, certain psychoactive drugs have been associated with parkinsonian movement deficits (Mena and Yebenes, 2006). Many drugs, such as antipsychotics, calcium channel blockers, and antidepressants, can induce parkinsonism as an adverse side effect (Shin and Chung, 2012). Most of the drug-induced parkinsonism (DIP) subsides following discontinuation of the offending drug. However, the motor symptoms persist and develop into irreversible, progressive parkinsonism in some cases (Mena and Yebenes, 2006). This can be explained by the coincidence of PD patients at preclinical or asymptomatic stages taking the offending drug, revealing the progressive parkinsonian symptoms after terminating drug use. However, there have been studies suggesting that the persistent parkinsonism may result from irreversible damage in the affected brain area caused by the toxicity of the drug (Bishnoi et al., 2007a; Tsai and Lee, 1998). Some psychoactive drugs have been shown to leave signs of oxidative stress in vivo. Chronic administration of haloperidol, a typical antipsychotic, induced oxidative damage in all brain regions of rats (Bishnoi et al., 2007b). It was also reported that schizophrenic patients who received neuroleptic treatment and developed movement deficits exhibited signs of oxidative damage in brain tissues (Lohr et al, 2003). In addition, the direct toxicity of psychoactive drugs on nigrostriatal dopaminergic neurons has been reported (Mena et al., 1995; Rollema et al., 1994). Therefore, the possibility remains that psychoactive drugs can cause permanent damage to the nigrostriatal pathway.
Studies have reported that antidepressant use is associated with a high risk for developing PD (Alonso et al., 2009; Dick et al., 2007). In particular, tricyclic antidepressants (TCAs) have been reported to reduce the viability of various types of cells in vitro (Haller et al., 2007; Huang et al., 2007; Kitagawa et al., 2006; Lirk et al., 2006) and in vivo (Estebe and Myers, 2004). Amitriptyline, one of the TCAs, was shown to induce inhibition of cellular respiration in non-neuronal cells (Higgins and Pilkington, 2010; Hroudova and Fisar, 2012) and produce oxidative stress (Bautista-Ferrufino et al., 2011). Considering that mitochondrial dysfunction and oxidative stress have been suggested to be major mechanisms of neurotoxicity in drug-induced PD, it is plausible that the tricyclic antidepressants can cause neurotoxicity associated with PD.
In the present study, we investigated whether the TCAs amitriptyline and desipramine exhibit PD-associated neurotoxicity in a dopaminergic cell line. In addition, we examined whether amitriptyline causes dopaminergic neuronal loss in the SNpc and parkinsonian movement deficits in mice.
MATERIALS AND METHODS
Antibodies and reagents
Anti-active BCL2-associated X (Bax) protein and anti-apoptosis inducing factor (AIF) antibodies were purchased from Cell Signaling Technology (USA). Anti-tyrosine hydroxylase (TH) and anti-cytochrome c antibodies were from Millipore (USA). Amitriptyline, desipramine, Trolox, calpain inhibitor II (ALLM), and 3-methyladenine (3-MA) were purchased from Sigma-Aldrich (USA). Dichlorofluorescin diacetate (DCF-DA), JC-1, and MitoTracker Red 580 were from Invitrogen (USA). Carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylke-tone (zVAD-fmk) was from Bachem (USA). 3-aminobenzamide (3AB) was from Millipore. E64 was purchased from Tocris (UK). All other reagents were purchased from Sigma-Aldrich unless stated otherwise.
Cell culture
Human neuroblastoma SH-SY5Y cells were cultured in a medium consisting of MEM and Ham’s F12-K supplemented with 10% fetal bovine serum and 1% antibiotics plus antimycotics in a 37°C CO2 (5%) incubator. Medium was changed every 2 or 3 days and the cells were sub-cultured when the confluency reached approximately 70–80%. All of the medium, culture supplements, and dishes were from Jeevan Biosciences, Inc. (JBI, USA). For immunostaining, the cells were plated at a density of 2.0 × 104 cells per 12 mm coverslip (Marienfeld, Germany) pre-coated with poly-L-lysine (0.1 mg/ml) and laminin (2 μg/ml).
Cell viability assay
To monitor the cell viability, cells were incubated with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay reagent (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega, USA) for 1 h. Absorbance of the samples was read at 490 nm using a microplate reader (Bio-Rad, USA). The rate of cell death calculated from the MTS cell viability assay was the same as that measured by a lactate dehydrogenase release assay (data not shown). Therefore, we measured the cell viability using the MTS assay system throughout the study.
Cell staining
For immunocytochemistry, cells grown on coverslips were fixed with 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 100 mM Na2HPO4, 2 mM KH2PO4) for 10 min and permeabilized with 0.5% Triton X-100 in PBS (PBST) for 10 min. Cell immunostaining was performed as described previously (Kim et al., 2015). For JC-1 staining, cells in culture were loaded with JC-1 dye (5 μg/ml) for 30 min, washed with PBS three times, and then fixed with PBS containing PFA (4%). For MitoTracker staining, cells were incubated with MitoTracker Red 580 (100 nM) for 30 min. After washing, the cells were fixed with 4% paraformaldehyde.
For the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, the cells were fixed with 4% PFA and then processed according to the manufacturer’s manual. The stained cells were mounted with medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Slowfade Gold antifade reagent with DAPI, Invitrogen). The samples were examined under a fluorescence microscope (Axioplan 2, Zeiss) or a confocal microscope (Leica TCS SP5, Leica).
Measurement of ATP level
Cellular ATP levels were measured using the luminescence ATP detection assay system (PerkinElmer, USA). The cells were incubated with or without chemicals for various time points, 100 μl of the ATPlite 1step reagent was added to the wells, and the cell plates were orbitally shaken for 3 min. The plates were read for emitted luminescence using a microplate luminometer (GloMax 9100-100, Promega).
Measurement of reactive oxygen species
SH-SY5Y cells cultured in 24-well plates were treated with appropriate chemicals. After the treatment, cells were washed with Hank’s balanced salt solution (JBI) two times and incubated with DCF-DA (5 μM) for 30 min. After the medium was removed, cells were washed with PBS and solubilized in 200 μl lysis buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF). After incubation for 10 min on ice, the cells were harvested and transferred to a black 96-well plate. DCF fluorescence was used as an indicator of reactive oxygen species (ROS). The plates were read at λex=485 nm and λem=530 nm using a spectrofluorometer (SpectraMax Gemini EM, Molecular Devices, USA).
Animal care and amitriptyline injection
All animal use complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee at Sejong University. Mice were group housed under a 12:12 light-dark lighting schedule with free access to food and water. The temperature in the room was 22°C. Twelve- to sixteen-week-old male C57BL/6 mice were injected intraperitoneally with sterile PBS (100 μl each, n = 5) or amitriptyline (2 or 20 mg/kg in a volume of 100 μl PBS each, n = 5) twice a week for 4 weeks. Body weight was measured before every injection. The injected mice were subjected to behavioral tests one week after the last injection.
Pole test
A pole test (Ogawa et al., 1985) was performed to evaluate bradykinesia in the amitriptyline-injected mice. The mouse was positioned at the top of a rough-surfaced pole (10 mm diameter and 60 cm height), head up, and then the time to turn and reach the floor was measured. During a habituation period one day prior to testing, each mouse was allowed to attempt to descend the pole. On the test day, mice were allowed to practice three times and then tested. The time to turn was first measured from the beginning of movement until the mouse turned completely head down, and time to reach the floor was measured until the mouse arrived at the floor. Each testing session consisted of three trials, and each lasted for a maximum of 180 s. Each mouse was tested twice before the 4-week drug injection period and one week following the final injection.
Open field test
To monitor general activity levels, gross locomotor activity, and explorative behaviors, the mouse was placed in the center of the open field arena and allowed to freely move for 20 min while being tracked by an automated tracking system (Ethovision, The Netherlands). The time of stay in the center or corner area, rearing activity, total distance moved, and velocities were recorded for 20 min. Before each recording, the mouse was allowed to explore the arena to acclimate to the space. The recordings were analyzed by Ethovision.
Immunohistochemistry
One week after the last injection, the mice were anesthetized with isoflurane, and transcardially perfused, first with pre-chilled PBS and then with 4% PFA in PBS (pH 7.4). The brains were removed, dissected using mouse coronal brain matrices (Applied Scientific Instrumentation, USA), embedded in Optimum cutting temperature compound, and then stored at −80°C. Coronal sections (10 μm thick) corresponding to the substantia nigra (SN) level (bregma −2.70 to −3.70) were prepared using a cryotome (CM3000, Leica) and fixed with 4% PFA in PBS for 10 min. After rinsing and quenching endogenous peroxidase with 0.1% H2O2 in deionized water for 10 min, the samples were incubated in PBST with 10% NGS for 1 h. The sections were then incubated with anti-TH antibodies in 0.1% PBST containing 2% NGS overnight at 4°C. The bound antibodies were visualized using the ABC method according to the manufacturer’s protocol (ABC Elite kit, Vector Laboratories).
Statistics
For the statistical analysis, all experiments were repeated at least three times. The results were expressed as the mean ± SD of at least three independent experiments, unless stated otherwise. Statistical significance was evaluated by the student’s t-test.
RESULTS
Amitriptyline and desipramine induced SH-SY5Y cell death that was not suppressed by inhibitors of known cell death pathways
To examine whether amitriptyline and desipramine are cytotoxic to SH-SY5Y cells, the viability of the cultured cells was measured following amitriptyline or desipramine treatment for 24 h. As shown in Fig. 1A, amitriptyline and desipramine induced cell death in a dose-dependent manner. Amitriptyline induced approximately 40–50% cell death at a 60 μM concentration and desipramine at a 50 μM concentration. To examine if the TCA-induced cell death is caspase-dependent, the cells were preincubated with the pan-caspase inhibitor zVAD-fmk (50 μM, Fig. 1A) and then treated with the drugs. Notably, cell death induced by amitriptyline or desipramine was not suppressed by the pan-caspase inhibitor, suggesting that the observed cell death may not be caspase-dependent apoptosis. To examine the mode of cell death induced by these TCAs, cells were preincubated with poly(ADP-ribose) polymerase-1 (Parp-1) inhibitor 3AB (5 mM, Fig. 1B), ALLM, which inhibits calpain and cathepsin B/L (20 μM, Fig. 1C), or E64, which inhibits cathepsin K/L/S/B/H (10 μM, Fig. 1D). In addition, the cell death induced by amitriptyline or desipramine was not suppressed by an autophagy inhibitor 3-MA (3 mM, Fig. 1E). As shown in Fig. 1, none of the inhibitors of known death-inducing enzymes or pathways suppressed TCA-induced cell death.
Fig. 1.
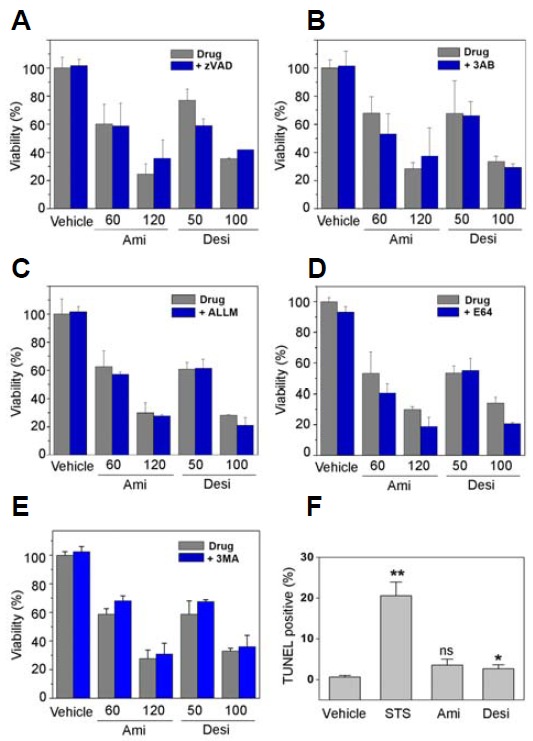
Amitriptyline- and desipramine-induced SH-SY5Y cell death was not suppressed by inhibitors of known cell death pathways. To examine the mode of cell death induced by amitriptyline or desipramine, SH-SY5Y cells were preincubated with inhibitors of caspases [40 μM zVAD-fmk, (A)], Parp-1 [5 mM 3AB, (B)], calpain and cathepsin B/L [20 μM ALLM, (C)], cathepsins [10 μM E64, (D)] and autophagy [3 mM 3-MA, (E)] for 30 min. Amitriptyline (Ami) and desipramine (Desi) were then added at the indicated concentrations (in μM) and the cells further incubated for 24 h. Cell viability was measured using MTS reagent (n = 3). (F) SH-SY5Y cells were treated with amitriptyline (Ami, 60 μM), desipramine (Desi, 50 μM), or staurosporine as a positive control (STS, 1 μM) for 24 h and then processed for TUNEL to monitor apoptosis (n = 3, *p < 0.05, **p < 0.01 vs. vehicle treated; ns, not significant).
To confirm whether the cell death induced by amitriptyline or desipramine is caspase-independent, a TUNEL assay was performed following drug treatment. As shown in Fig. 1F, the cells incubated with amitriptyline or desipramine exhibited only basal levels of apoptosis when examined by TUNEL assay. These results suggest that amitriptyline or desipramine may induce cell death independently of the known cell death pathways.
Amitriptyline and desipramine induced mitochondrial fragmentation without AIF or cytochrome c release
A previous study suggested that amitriptyline can induce apoptosis (Lirk et al., 2006), but we did not observe suppression of TCA-induced cell death with caspase inhibition. Thus, we examined whether there is a release or translocation of AIF following amitriptyline or desipramine treatment. As shown in Fig. 2A, immunostaining of AIF suggests that the drugs did not induce AIF translocation. Immunocytochemistry using anti-cytochrome c antibody revealed that cytochrome c was not released following amitriptyline or desipramine treatment (Fig. 2A, bottom panels). Interestingly, the drugs induced mitochondrial fragmentation, and both AIF and cytochrome c were confined in the fragmented mitochondria (Fig. 2B). These results suggest that amitriptyline and desipramine induce cell death through mitochondrial damage but in a different manner from the canonical cell death pathway.
Fig. 2.
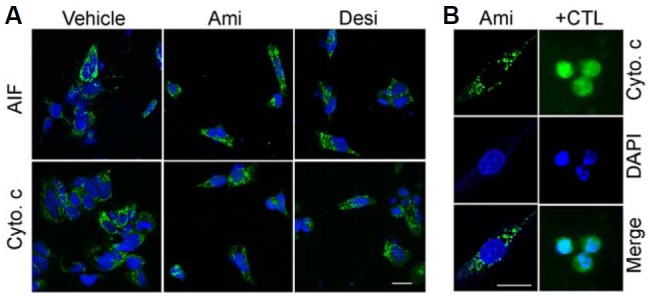
Amitriptyline and desipramine induced mitochondrial fragmentation without AIF or cytochrome c release. SH-SY5Y cells were treated with amitriptyline (Ami, 60 μM) or desipramine (Desi, 50 μM). At 24 h after the incubation, the cells were processed for immunostaining using anti-AIF (A) or anti-cytochrome c [Cyto. c, (A, B)] antibodies. As a positive control for the cytochrome c release, SH-SY5Y cells were incubated with 50 μM etoposide [+CTL, (B)] for 12 h. Note that the localization of cytochrome c was confined to the fragmented mitochondria in the amitriptyline-treated cells whereas the cytochrome c was released in etoposide-treated cells in (B). The cells were counterstained with DAPI to visualize nuclei. Scale bar = 10 μm.
Amitriptyline and desipramine induced ATP depletion and the activation of Bax
Because we observed signs of mitochondrial damage following amitriptyline and desipramine treatment, we then examined whether these drugs induce a decrease in mitochondrial membrane potential. To examine the changes in mitochondrial potential, the drug-treated cells were stained with JC-1 dye, and the changes in the dye fluorescence were monitored. As shown in Fig. 3A, the cells incubated with amitriptyline or desipramine exhibited a shift in JC-1 fluorescence from red to green, suggesting a decrease in mitochondrial membrane potential. In addition, MitoTracker staining confirmed that amitriptyline and desipramine induced a mitochondrial membrane potential drop because the MitoTracker fluorescence decreased following the drug treatment (Fig. 3B). Notably, MitoTracker staining also revealed fragmentation of mitochondria after the TCAs treatment (Fig. 3B). Consistent with these results, the level of cellular ATP decreased following the incubation with amitriptyline or desipramine (Fig. 3C).
Fig. 3.
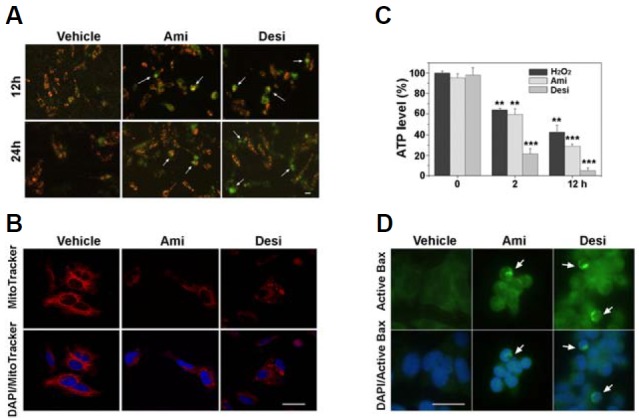
Amitriptyline and desipramine induced ATP depletion and the activation of Bax. (A, B) SH-SY5Y cells were incubated with amitriptyline (Ami, 60 μM) or desipramine (Desi, 50 μM) for 12 and 24 h and then incubated with JC-1 dye [5 μM, (A)] or MitoTracker [100 nM for 12 h, B)] for 30 min. Arrows indicate the cells with JC-1 green monomer fluorescence, suggesting decreased mitochondrial membrane potential (A). Note the decreased fluorescence of MitoTracker in the drug-treated cells in (B). (C) SH-SY5Y cells were incubated with amitriptyline or desipramine and the cellular ATP level was determined using ATPlite assay kit at indicated times. Hydrogen peroxide was used as a positive control for ATP depletion (n = 3, **p < 0.01 and ***p < 0.001 vs. 0 h). (D) To monitor the Bax activation, the cells were treated with amitriptyline or desipramine for 24 h and then processed for immunostaining using anti-active Bax antibodies (arrows). Scale bar = 10 μm.
We then examined whether these drugs activate Bax, because activation of Bax is involved in mitochondrial damage, including fragmentation and membrane potential decrease (Ghibelli and Diederich, 2010; Karbowski et al., 2006; Sheridan et al., 2008). SH-SY5Y cells were incubated with amitriptyline or desipramine and then immunostained with antibodies specific for active Bax. As shown in Fig. 3D, the cells incubated with TCAs were positive for active Bax, suggesting that amitriptyline or desipramine activated Bax.
Amitriptyline and desipramine promoted ROS generation, and the cell death was attenuated by an antioxidant
It is known that oxidative stress can induce mitochondrial damage and vice versa (Mammucari and Rizzuto, 2010; Peng and Jou, 2010; Van Houten et al., 2006). To examine whether amitriptyline and desipramine can induce oxidative stress, cellular ROS levels were monitored using DCF-DA after the drug treatment. As shown in Fig. 4A, both amitriptyline and desipramine promoted the generation of ROS. These results suggest that amitriptyline and desipramine can induce cytotoxicity via oxidative stress. To test this possibility, we examined whether an antioxidant can suppress cell death induced by amitriptyline or desipramine. The cells were preincubated with the antioxidant Trolox (400 μM) and then treated with amitriptyline or desipramine. As shown in Fig. 4B, preincubation with Trolox significantly suppressed the TCA-induced cell death, suggesting that the TCAs induced cell death via oxidative stress.
Fig. 4.
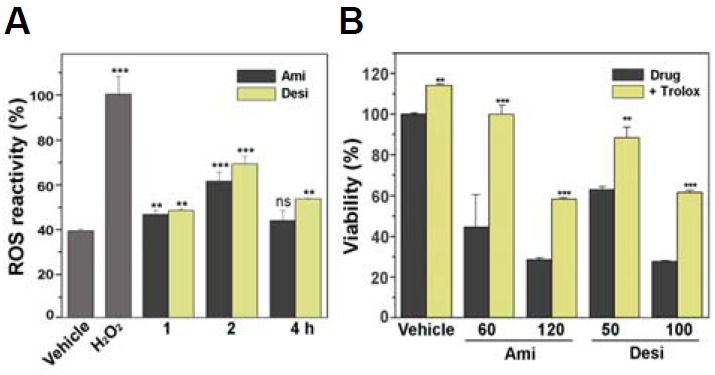
Amitriptyline and desipramine promoted ROS generation, and cell death was attenuated by an antioxidant. (A) SH-SY5Y cells were incubated with amitriptyline (Ami, 60 μM) or desipramine (Desi, 50 μM) for the indicated times and the cellular level of ROS was spectrofluorometrically measured using DCF-DA (5 μM). The reading from the hydrogen peroxide-incubated cells (400 μM for 30 min) served as 100% control (n = 4, **p < 0.01 and ***p < 0.001 vs. vehicle treated; ns, not significant). (B) SH-SY5Y cells were preincubated with Trolox (400 μM) for 30 min and then incubated with amitriptyline (60 μM) or desipramine (50 μM) for 24 h. The viability of the cells was then measured using MTS reagent (n = 4, **p < 0.01, ***p < 0.001 vs. drugs only).
Subchronically injected amitriptyline induced TH-positive neuronal loss in the SNpc
To examine whether TCA can induce neurotoxicity in dopaminergic neurons in vivo, mice were intraperitoneally injected with 20 mg/kg of amitriptyline on a subchronic schedule (twice a week for 4 weeks) and the number of TH-positive cells in the SNpc was counted. As can be seen in Fig. 5, the number of TH-positive cells in the SNpc of amitriptyline-injected mice was significantly reduced compared with vehicle-injected mice. About 35% of the TH-positive neurons were lost in the SNpc of the amitriptyline-injected mice, suggesting that the TCA can induce cytotoxicity of dopaminergic neurons in the brain area affected in PD.
Fig. 5.
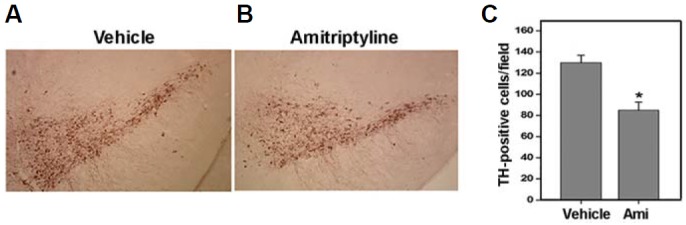
Subchronically injected amitriptyline induced TH-positive neuronal loss in the SNpc. To examine the in vivo effect of amitriptyline, mice were intraperitoneally injected with amitriptyline (20 mg/kg body weight) twice a week for 4 weeks. Brain tissue sections were prepared from the midbrain area of control vehicle- or amitriptyline-injected mice and immunostained using anti-TH antibodies. Representative immunohistochemistry photographs are shown in (A). (B) The number of TH-positive cells was counted from each SNpc area. At least ten sections from each mouse were counted (n = 5, *p < 0.5 vs. vehicle injected).
Subchronically injected amitriptyline induced movement deficits
To examine whether amitriptyline can induce parkinsonian behavioral changes, motor activity was assessed by open field test in two different groups of mice injected intraperitoneally with either 20 mg/kg or 2 mg/kg of amitriptyline twice a week for 4 weeks. Amitriptyline-injected mice exhibited slightly reduced motor activities in total distance moved (Fig. 6A) and mean velocity (Fig. 6B) compared with vehicle-injected control mice. To further assess the motor deficits induced by amitriptyline injection, a pole test was performed on the injected mice. The pole test is proposed to be the most sensitive behavioral test, because it can discriminate the subtle changes in the forepaw dexterity, reflecting motor dysfunction in PD (Matsuura et al., 1997; Rauch et al., 2010). As shown in Fig. 6C, amitriptyline-injected mice displayed a much longer time to turn on the pole, suggesting that they have motor dysfunction in the forepaws. These results suggest that subchronic injection of amitriptyline can induce parkinsonian motor deficits.
Fig. 6.
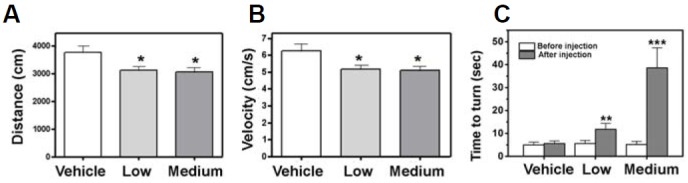
Subchronically injected amitriptyline induced movement deficits. Mice were injected with a low dose (2 mg/kg body weight) or a medium dose (20 mg/kg) of amitriptyline twice a week for 4 weeks. Behavioral tests were performed one week after the last injection (n = 5 each, *p < 0.05, **p < 0.01, and ***p < 0.001 vs. vehicle injected). (A, B) The drug-injected mice were tested in the open field for the total distance moved (A) and mean velocity of the movement (B). (C) To evaluate forepaw dexterity, the injected mice were subjected to a pole test. The time to turn downward on the test pole was measured.
DISCUSSION
PD is the second most prevalent neurodegenerative disorder and is influenced by various factors, including age, genetics, and environmental toxins (Foltynie et al., 2002; Hirtz et al., 2007). The use of pesticides and psychoactive drugs has been implicated in the pathogenesis of environmentally-induced PD. It has long been known that drugs, such as antipsychotics, can cause parkinsonian symptoms called drug-induced parkinsonism (DIP). In most cases, the DIP subsides upon the withdrawal of the causative drugs, but it can develop into PD in certain cases (Benito-León et al., 2009). In the present study, we investigated the neurotoxicity mechanism of the TCAs amitriptyline and desipramine in vitro and observed the in vivo effects of subchronic injection of amitriptyline in mice.
In SH-SY5Y cells, amitriptyline and desipramine induced cell death with accompanying mitochondrial damage and oxidative stress. Interestingly, the cell death induced by these TCAs was suppressed by an antioxidant but not by inhibitors of caspases, Parp-1, cathepsin, or calpains. Furthermore, despite signs of mitochondrial damage, such as fragmentation, activation of Bax and a decrease in membrane potential and ATP level, the release of cytochrome c or AIF was not detected. These results suggest that the cell death induced by amitriptyline or desipramine is different from conventional apoptosis or programmed necrosis. It has been suggested that features of apoptotic and necrotic cell death are shared and not discretely defined into independent death pathways (Han et al., 2008; Lemasters, 1999). Consistent with our observation of mitochondrial damage, a recent study reported that pharmacologically different groups of antidepressants, including amitriptyline, directly suppressed mitochondrial respiratory activity (Hroudova and Fisar, 2010). We observed that the drugs promoted the generation of ROS, and the cell death was suppressed by an antioxidant. Thus, it is plausible that mitochondrial damage is the main causative toxicity mechanism of amitriptyline and desipramine, i.e., inhibition of mitochondrial activity resulted in increased production of ROS and cell death. Another possibility is that amitriptyline or desipramine can induce autophagy-associated cell death. It has been reported that amitriptyline induced signs of autophagy with the appearance of LC3-II in rat primary neurons (Zschocke et al., 2011). However, we did not observe any protective or aggravating effects by the autophagy inhibitor 3-methyladenine on the TCA-induced cell death. Nonetheless, we cannot rule out the possibility of TCA inducing autophagic cell death, because ATP depletion and the induction of BH3-only proteins are also associated with autophagy (Mukhopadhyay et al., 2014). Furthermore, inhibitors of autophagy often exhibit dual functions in autophagic processes and cell death (Wu et al., 2010). A more detailed analysis is necessary to address this issue clearly.
When injected into mice on a subchronic schedule, amitriptyline induced a loss of dopaminergic neurons in the SNpc and behavioral deficits during the pole test. These results suggest that amitriptyline and desipramine can induce PD-associated neurotoxicity in vivo. TCAs such as amitriptyline and desipramine are used to treat depression and chronic pain (Chan et al., 2009). Similar to antipsychotics, the adverse effects of antidepressants include parkinsonism (Warner et al., 2006). In addition, it has been reported that amitriptyline and desipramine have toxicity on the brain, heart, and respiratory system (Crome, 1986). Importantly, it has recently been reported that the correlation between the use of antidepressants and the pathogenesis of PD is higher than that between exposure to pesticides and PD (Alonso et al., 2009; Priyadarshi et al., 2001). However, whether the antidepressants are causally related to the PD pathogenesis has not been addressed. We have shown in the present study that amitriptyline injection into mice resulted in the loss of dopaminergic neurons in the SNpc. Intriguingly, amitriptyline-injected mice exhibited motor deficits more prominently in the pole test than in the open field test. The decreased general locomotor activity in the drug-injected mice in the open field was the same level in low (2 mg/kg) and medium dose (20 mg/kg) groups. However, in the pole test, the medium dose group exhibited a strikingly poor performance when compared with the low dose group mice. The pole test is known to be one of the most sensitive behavioral tests that can measure fore-paw dexterity and detect nigrostriatal dysfunction (Meredith et al., 2008; Ogawa et al., 1985). Thus, it is highly possible that the loss of TH-positive neurons in the SNpc (Fig. 5) is responsible for the decreased forepaw dexterity (Fig. 6C), possibly reflecting parkinsonian motor dysfunction. The concentration of amitriptyline used in this study was close to the intake level of patients with severe depression. Thus, it is possible that TCAs might be causally related with the pathogenesis of PD. A further detailed investigation is required to examine whether there is a causal relationship between chronic use of high dose TCAs and the pathogenesis of PD.
Acknowledgments
This work was support by grants from the National Research Foundation of Korea (2012R1A1A2007688 and 2012R1A2A2A 01046822).
REFERENCES
- Alonso A., Rodriguez L.A.G., Logroscino G., Hernan M.A. Use of antidepressants and the risk of Parkinson’s disease: a prospective study. J. Neurol. Neurosurg. Psychiatry. 2009;80:671–674. doi: 10.1136/jnnp.2008.152983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif I.A., Khan H.A. Environmental toxins and Parkinson’s disease: Putative roles of impaired electron transport chain and oxidative stress. Toxicol. Ind. Health. 2010;26:121–128. doi: 10.1177/0748233710362382. [DOI] [PubMed] [Google Scholar]
- Bautista-Ferrufino M.R., Cordero M.D., Sanchez-Alcazar J.A., Illanes M., Fernandez-Rodriguez A., Navas P., de Miguel M. Amitriptyline induces coenzyme Q deficiency and oxidative damage in mouse lung and liver. Toxicol. Lett. 2011;204:32–37. doi: 10.1016/j.toxlet.2011.03.033. [DOI] [PubMed] [Google Scholar]
- Benito-Leon J., Louis E.D., Bermejo-Pareja F., Cent N.D.I. Risk of incident Parkinson’s disease and parkinsonism in essential tremor: a population based study. J. Neurol. Neurosurg. Psychiatry. 2009;80:423–425. doi: 10.1136/jnnp.2008.147223. [DOI] [PubMed] [Google Scholar]
- Bishnoi M., Chopra K., Kulkarni S.K. Theophylline, adenosine receptor antagonist prevents behavioral, biochemical and neurochemical changes associated with an animal model of tardive dyskinesia. Pharmacol. Rep. 2007a;59:181–191. [PubMed] [Google Scholar]
- Bishnoi M., Chopra K., Kulkarni S.K. Neurochemical changes associated with chronic administration of typical anti-psychotics and its relationship with tardive dyskinesia. Methods Find. Exp. Clin. Pharmacol. 2007b;29:211–216. doi: 10.1358/mf.2007.29.3.1075355. [DOI] [PubMed] [Google Scholar]
- Chan H.N., Fam J., Ng B.Y. Use of antidepressants in the treatment of chronic pain. Ann. Acad. Med. Singapore. 2009;38:974–979. [PubMed] [Google Scholar]
- Crome P. Poisoning due to tricyclic antidepressant over-dosage. Clinical presentation and treatment. Med. Toxicol. 1986;1:261–285. doi: 10.1007/BF03259843. [DOI] [PubMed] [Google Scholar]
- Dick F.D., De Palma G., Ahmadi A., Scott N.W., Prescott G.J., Bennett J., Semple S., Dick S., Counsell C., Mozzoni P., et al. Environmental risk factors for Parkinson’s disease and parkinsonism: the Geoparkinson study. Occup. Environ. Med. 2007;64:666–672. doi: 10.1136/oem.2006.027003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estebe J.P., Myers R.R. Amitriptyline neurotoxicity - Dose-related pathology after topical application to rat sciatic nerve. Anesthesiol. 2004;100:1519–1525. doi: 10.1097/00000542-200406000-00026. [DOI] [PubMed] [Google Scholar]
- Foltynie T., Sawcer S., Brayne C., Barker R.A. The genetic basis of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry. 2002;73:363–370. doi: 10.1136/jnnp.73.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire C., Koifman S. Pesticide exposure and Parkinson’s disease: Epidemiological evidence of association. Neurotoxicol. 2012;33:947–971. doi: 10.1016/j.neuro.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Ghibelli L., Diederich M. Multistep and multitask Bax activation. Mitochondrion. 2010;10:604–613. doi: 10.1016/j.mito.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Haller I., Lirk P., Keller C., Wang G.K., Gerner P., Klimaschewski L. Differential neurotoxicity of tricyclic antidepressants and novel derivatives in vitro in a dorsal root ganglion cell culture model. Eur. J. Anaesthesiol. 2007;24:702–708. doi: 10.1017/S0265021507000154. [DOI] [PubMed] [Google Scholar]
- Han S.I., Kim Y.S., Kim T.H. Role of apoptotic and necrotic cell death under physiologic conditions. BMB Rep. 2008;41:1–10. doi: 10.5483/bmbrep.2008.41.1.001. [DOI] [PubMed] [Google Scholar]
- Hatcher J.M., Pennell K.D., Miller G.W. Parkinson’s disease and pesticides: a toxicological perspective. Trends Pharmacol. Sci. 2008;29:322–329. doi: 10.1016/j.tips.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins S.C., Pilkington G.J. The in vitro effects of tricyclic drugs and dexamethasone on cellular respiration of malignant glioma. Anticancer Res. 2010;30:391–397. [PubMed] [Google Scholar]
- Hirtz D., Thurman D.J., Gwinn-Hardy K., Mohamed M., Chaudhuri A.R., Zalutsky R. How common are the “common” neurologic disorders? Neurology. 2007;68:326–337. doi: 10.1212/01.wnl.0000252807.38124.a3. [DOI] [PubMed] [Google Scholar]
- Hroudova J., Fisar Z. Activities of respiratory chain complexes and citrate synthase influenced by pharmacologically different antidepressants and mood stabilizers. Neuroendocrinol. Lett. 2010;31:336–342. [PubMed] [Google Scholar]
- Hroudova J., Fisar Z. In vitro inhibition of mitochondrial respiratory rate by antidepressants. Toxicol. Lett. 2012;213:345–352. doi: 10.1016/j.toxlet.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Huang Y.Y., Peng C.H., Yang Y.P., Wu C.C., Hsu W.M., Wang H.J., Chan K.H., Chou Y.P., Chen S.J., Chang Y.L. Desipramine activated Bcl-2 expression and inhibited lipopolysaccharide-induced apoptosis in hippocampus-derived adult neural stem cells. J. Pharmacol. Sci. 2007;104:61–72. doi: 10.1254/jphs.fp0061255. [DOI] [PubMed] [Google Scholar]
- Karbowski M., Norris K.L., Cleland M.M., Jeong S.Y., Youle R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- Kim H.L., Ra H., Kim K.R., Lee J.M., Im H., Kim Y.H. Poly(ADP-ribosyl)ation of p53 contributes to TPEN-induced neuronal apoptosis. Mol. Cells. 2015;38:312–317. doi: 10.14348/molcells.2015.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa N., Oda M., Nobutaka I., Satoh H., Totoki T., Morimoto M. A proposed mechanism for amitriptyline neurotoxicity based on its detergent nature. Toxicol. Appl. Pharmacol. 2006;217:100–106. doi: 10.1016/j.taap.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Lemasters J.J. Mechanisms of hepatic toxicity - V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am. J. Physiol.-Gastrointestin. Liver Physiol. 1999;276:G1–G6. doi: 10.1152/ajpgi.1999.276.1.G1. [DOI] [PubMed] [Google Scholar]
- Lirk P., Haller I., Hausott B., Ingorokva S., Deibl M., Gerner P., Klimaschewski L. The neurotoxic effects of amitriptyline are mediated by apoptosis and are effectively blocked by inhibition of caspase activity. Anesth. Analg. 2006;102:1728–1733. doi: 10.1213/01.ane.0000216018.62549.bb. [DOI] [PubMed] [Google Scholar]
- Lohr J.B., Kuczenski R., Niculescu A.B. Oxidative mechanisms and tardive dyskinesia. CNS Drugs. 2003;17:47–62. doi: 10.2165/00023210-200317010-00004. [DOI] [PubMed] [Google Scholar]
- Mammucari C., Rizzuto R. Signaling pathways in mitochondrial dysfunction and aging. Mech. Ageing Dev. 2010;131:536–543. doi: 10.1016/j.mad.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K., Kabuto H., Makino H., Ogawa N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods. 1997;73:45–48. doi: 10.1016/s0165-0270(96)02211-x. [DOI] [PubMed] [Google Scholar]
- Mena M.A., de Yebenest J.G. Drug-induced parkin sonism. Exp. Opin. Drug Safety. 2006;5:759–771. doi: 10.1517/14740338.5.6.759. [DOI] [PubMed] [Google Scholar]
- Mena M.A., Deyebenes M.J.G., Tabernero C., Casarejos M.J., Pardo B., Deyebenes J.G. Effects of calcium-antagonists on the dopamine system. Clin. Neuropharmacol. 1995;18:410–426. doi: 10.1097/00002826-199510000-00004. [DOI] [PubMed] [Google Scholar]
- Meredith G.E., Kang U.J. Behavioral models of Parkinson’s disease in rodents: A new look at an old problem. Mov. Disord. 2006;21:1595–1606. doi: 10.1002/mds.21010. [DOI] [PubMed] [Google Scholar]
- Meredith G.E., Sonsalla P.K., Chesselet M.F. Animal models of Parkinson’s disease progression. Acta Neuropathologica. 2008;115:385–398. doi: 10.1007/s00401-008-0350-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Panda P.K., Sinha N., Das D.N., Bhutia S.K. Autophagy and apoptosis: where do they meet? Apoptosis. 2014;19:555–566. doi: 10.1007/s10495-014-0967-2. [DOI] [PubMed] [Google Scholar]
- Ogawa N., Hirose Y., Ohara S., Ono T., Watanabe Y. A simple quantitative bradykinesia test in Mptp-treated mice. Res. Commun. Chem. Pathol. Pharmacol. 1985;50:435–441. [PubMed] [Google Scholar]
- Peng T.I., Jou M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N Y Acad. Sci. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- Priyadarshi A., Khuder S.A., Schaub E.A., Priyadarshi S.S. Environmental risk factors and Parkinson’s disease: a metaanalysis. Environ. Res. 2001;86:122–127. doi: 10.1006/enrs.2001.4264. [DOI] [PubMed] [Google Scholar]
- Rauch F., Schwabe K., Krauss J.K. Effect of deep brain stimulation in the pedunculopontine nucleus on motor function in the rat 6-hydroxydopamine Parkinson model. Behav. Brain Res. 2010;210:46–53. doi: 10.1016/j.bbr.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Rollema H., Skolnik M., Dengelbronner J., Igarashi K., Usuki E., Castagnoli N. Mpp(+)-like neurotoxicity of a pyridinium metabolite derived from Haloperidol - in-vivo microdialysis and in-vitro mitochondrial studies. J. Pharmacol. Exp. Ther. 1994;268:380–387. [PubMed] [Google Scholar]
- Sheridan C., Delivani P., Cullen S.P., Martin S.J. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome c release. Mol. Cell. 2008;31:570–585. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Shin H.W., Chung S.J. Drug-induced Parkinsonism. J. Clin. Neurol. 2012;8:15–21. doi: 10.3988/jcn.2012.8.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M.J., Lee E.H.Y. Nitric oxide donors protect cultured rat astrocytes from 1-methyl-4-phenylpyridinium-induced toxicity. Free Rad. Biol. Med. 1998;24:705–713. doi: 10.1016/s0891-5849(97)00329-8. [DOI] [PubMed] [Google Scholar]
- Van Houten B., Woshner V., Santos J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) 2006;5:145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Warner C.H., Bobo W., Warner C., Reid S., Rachal J. Antidepressant discontinuation syndrome. Am. Fam. Phys. 2006;74:449–456. [PubMed] [Google Scholar]
- Wu Y.T., Tan H.L., Shui G., Bauvy C., Huang Q., Wenk M.R., Ong C.N., Codogno P., Shen H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010;285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschocke J., Zimmermann N., Berning B., Ganal V., Holsboer F., Rein T. Antidepressant drugs diversely affect autophagy pathways in astrocytes and neurons--dissociation from cholesterol homeostasis. Neuropsychopharmacol. 2011;36:1754–1768. doi: 10.1038/npp.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]