

Abstract
Given the rise of parasite resistance to all currently used antimalarial drugs, the identification of novel chemotypes with unique mechanisms of action is of paramount importance. Since Plasmodium expresses a number of aspartic proteases necessary for its survival, we have mined antimalarial datasets for drug-like aspartic protease inhibitors. This effort led to the identification of spiropiperidine hydantoins, bearing similarity to known inhibitors of the human aspartic protease β-secretase (BACE), as new leads for antimalarial drug discovery. Spiropiperidine hydantoins have a dynamic structure-activity relationship profile with positions identified as being tolerant of a variety of substitution patterns as well as a key piperidine N-benzyl phenol pharmacophore. Lead compounds 4e (CWHM-123) and 12k (CWHM-505) are potent antimalarials with IC50 values against Plasmodium falciparum 3D7 of 0.310 μM and 0.099 μM, respectively, and the former features equivalent potency on the chloroquine-resistant Dd2 strain. Remarkably, these compounds do not inhibit human aspartic proteases BACE, cathepsins D and E, or Plasmodium plasmepsins II and IV despite their similarity to known BACE inhibitors. Although the current leads suffer from poor metabolic stability, they do fit into a drug-like chemical property space and provide a new class of potent antimalarial agents for further study.
Keywords: aspartic protease inhibitors, spiropiperidine hydantoins, antiplasmodial, antimalarial
Graphical Abstract
1. Introduction
Malaria is caused by the parasite Plasmodium. In 2013, there were approximately 198 million cases of malaria leading to ~584,000 deaths, being particularly deadly to young children in sub-Saharan Africa.1 P. falciparum, the most lethal species, has developed varying degrees of resistance to all currently used antimalarial drugs.2–5 Approaches to combat parasite resistance include combination of antimalarial drugs as standard treatment regimens, as well as identification of new antimalarial drugs with unique mechanisms of action that can be combined with existing antimalarial drugs.
Plasmodium expresses a number of aspartic proteases necessary for its survival, including essential aspartic proteases Plasmepsin V (PMV or PM-5) and signal peptide peptidase (PfSPP).6–11 While a number of potent peptidomimetic inhibitors of Plasmodium aspartic proteases have been identified,7, 12–14 we have focused on repurposing classes of drug-like aspartic protease inhibitors developed by the pharmaceutical industry for human aspartic proteases such as β-secretase (BACE)15, 16 or renin.17
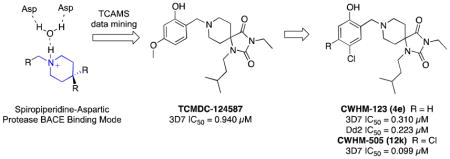
We have hypothesized that maintaining core structural motifs known to bind the aspartate residues in the active site may allow identification and optimization of novel classes of antimalarial compounds. Accordingly, we mined the Tres Cantos Anti-Malarial dataset (TCAMS) representing thousands of compounds18 for drug-like aspartic protease inhibitors. For example, we recently reported our identification and initial optimization of aminohydantoins as novel antimalarial compounds with selectivity for Plasmodium and in vivo antimalarial efficacy (e.g., CWHM-117) originating from BACE inhibitor 1 and database hit TCMDC-136879 (Figure 1a).19
Figure 1.

Strategy to identify drug-like aspartic protease inhibitors as novel antimalarials.
Spiropiperidine-containing compounds such as 2 and 3 have been reported as non-peptidomimetic BACE inhibitors16, 20–22 and represent a novel scaffold for development of new antimalarial aspartic protease inhibitors (Figure 1b). The reported x-ray crystal structure of 2 (3FKT)16 demonstrates the mechanism by which the protonated piperidine nitrogen forms a salt bridge with a water molecule in the active site. Similarly, other related piperidine and pyrrolidine BACE, renin and HIV protease inhibitor crystal structures demonstrate similar binding modes,17, 23 leading us to hypothesize that the spiropiperidine scaffold may be an appropriate core for mining antimalarial phenotypic screening databases. Substructure-based searching of the TCAMS revealed a single hit, TCMDC-124587 (4a), with a reported XC50 of 0.840 μM. Given its modest molecular weight, favorable CLogP, and submicromolar antimalarial potency, an effort to validate this hit and evaluate the potential of this class of spiropiperidines as antimalarials was initiated.
2. Results and discussion
2.1. Validation of hit and initial SAR
Searches of commercially available compound databases revealed that TCMDC-124587 and closely-related analogs could be purchased from ChemBridge. Most commericially-available compounds were derivatized at the R8 position. Two iterations of sets of six spiropiperidines each, including TCMDC-124587, were purchased and evaluated for inhibition of parasite growth in P. falciparum 3D7-infected red blood cells. Key structure-activity relationships are shown in Figure 2. Of foremost importance, 4a was found to have similar 3D7 potency (IC50 = 0.940 μM) as reported in the screening dataset. Substituent position was found to be important. For example, moving the methoxy group from the 4′- to the 3′- or 5′-positions resulted in 6-fold loss or 2-fold improvement in potency, respectively (4b,c). While deletion of the methoxy group (4d) did not have a significant impact on potency, replacement with chlorine (4e) gave about a five-fold improvement in potency. Most striking is the dependence of potency on the presence of the phenol moiety. Capping the phenol with a methyl group (4g) or deletion (4f,h) led to 8- to 60-fold losses in potency.
Figure 2.

Preliminary R8 Structure-Activity Relationships. Reported potencies are IC50 values in P. falciparum 3D7 infected erythrocytes.
The antimalarial activity of lead compound was determined to not be due to general cytoxicity (HepG2 72 h cytoxicity IC50 = 37 μM), having a selectivity index of >100-fold. We were further encouraged by identification of 4e in the Novartis-GNF antimalarial screening hit collection (GNF-Pf-5345, reported EC50 = 0.349 μM), although only a few related compounds were present in this collection.24 These data, along with the demonstration of a discrete SAR, encouraged us to investigate this class further by resynthesizing lead compound 4e and broadening the SAR.
2.2. Synthesis
The synthesis of 4e and related analogs is shown in Scheme 1. The spirohydantoin core 6 was prepared as previously described.25 The R3 alkyl group was then incorporated by simple alkylation with potassium carbonate. Subsequent alkylation with sodium hydride and R1X, followed by deprotection of the BOC group afforded intermediate 9. Finally, reductive amination provided R8 analogs such as 4e, 10a–b, 10d–e, 10g–i, 11a–d and 12a–u. Additionally, some R8 analogs were prepared from 8 standard amide coupling or alkylation conditions.
Scheme 1.
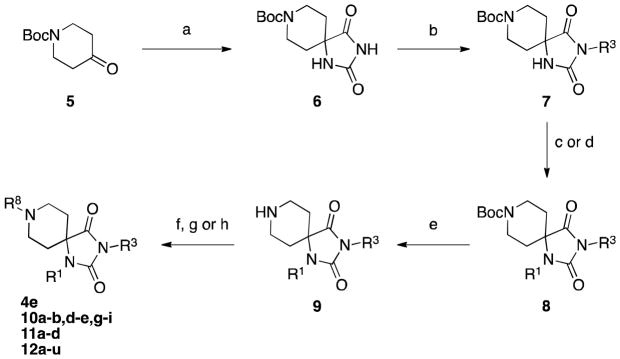
Reagents and Conditions: (a) KCN, (NH4)2CO3, aq. MeOH; (b) R3X, K2CO3, DMF; (c) NaH, R1X, DMF; (d) ArI, CuI, 2,2,6,6-tetramethyl-3,5-heptanedione, Cs2CO3; (e) HCl or TFA; (f) RCHO, Na(OAc)3BH, DMF; (g) RCO2H, EDC, HOBt, NEM, DCM; (h) R8Br, K2CO3, CH3CN.
Sterically hindered or otherwise incompatible R1 analogs were prepared instead by the approach reported by Carrera and Garvey26 shown in Scheme 2. In brief, the primary R1 amine is condensed with ketone 13 to furnish α-amino nitriles 14. Condensation with potassium cyanate and ring closure gives access to mono-substituted hydantoins 15. Subsequent alkylation, deprotection and reductive amination provided the final compounds 10c and 10f.
Scheme 2.
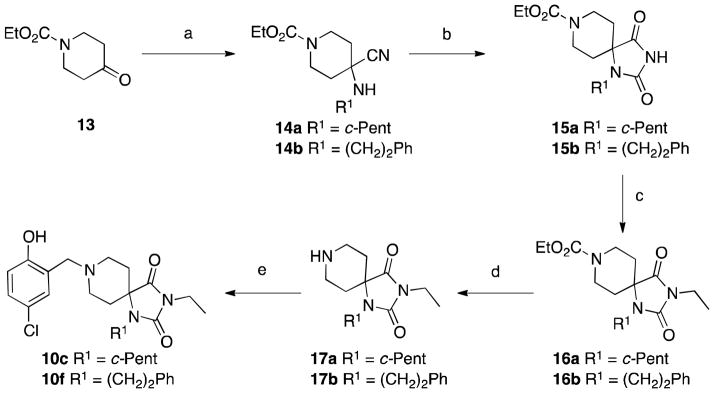
Reagents and Conditions: (a) R1NH2-HCl, KCN, aq. MeOH; (b) i. KOCN, AcOH, H2O; ii. aq. HCl; (c) NaH, EtI, DMF; (d) KOH, EtOH; (e) RCHO, Na(OAc)3BH, DMF
2.3. Structure-Activity Relationship Studies
Structure-activity relationships in the Pf3D7-infected red blood cell assay for R1 and R3 derivatives are shown in Figures 3 and 4. Resynthesized 4e was found to have a similar IC50 value as the commercial lot and was used a reference compound for subsequent SAR studies. Keeping R8 constant as the 5′-chloro-2′-hydoxybenzyl group, R1 and R3 were varied independently. R1 proved tolerant of modification provided that the substituents remained suitably large and flexible. For example, replacement of the isopentyl (4e) with hydrogen (10a) or ethyl (10b) led to a 20-fold or greater reduction in potency. Similarly, substitution with a phenyl ring (10d) proved detrimental to potency while cyclopentyl (10c) was well tolerated. This is presumably due to the rigidity of the phenyl ring since extension with one (10e) or two (10f) methylene linkers were roughly equipotent with the isopentyl group. Benzyl analogs with electron donating (10g) or withdrawing (10h,i) groups were tolerated with some modest effects on potency.
Figure 3. R1 Structure-Activity Relationships.
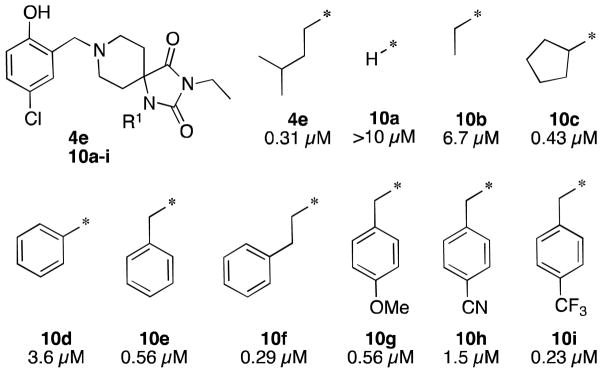
Reported potencies are IC50 values in P. falciparum 3D7 infected erythrocytes.
Figure 4. R3 Structure-Activity Relationships.
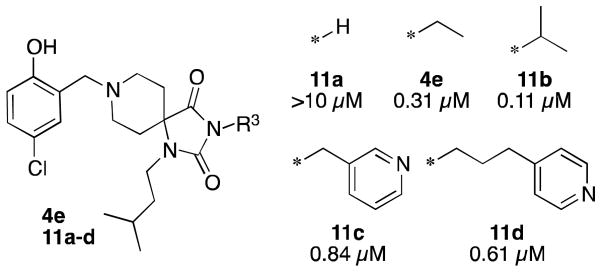
Reported potencies are IC50 values in P. falciparum 3D7 infected erythrocytes.
SAR at the R3 position also showed tolerance for a variety of lipophilic groups (Figure 4). Deletion of the ethyl group (11a) led to a dramatic loss in activity. Conversely, the ethyl group could be replaced with larger groups such as isopropyl (11b), which improved potency three-fold, or CH2-pyridyl (11c) and (CH2)3-pyridyl (11d) with only modest two-three-fold losses of potency.
Given the relative flexibility in SAR in the R1 and R3 positions, we elected to focus on expanding the more sensitive SAR in the R8 position (Figure 5). Moving the phenolic hydroxyl group to the 3′- or 4′-positions proved to be increasingly detrimental to 3D7 potency (12a,b) but not as dramatically as deletion of the hydroxyl group (4f). In contrast, the chlorine atom could be replaced by a variety of substituents. For example, while F is not an equivalent replacement (12c), bromo, methyl, and phenyl replacements are all well tolerated (12d,e,f).
Figure 5. Preliminary R8 Structure-Activity Relationships.
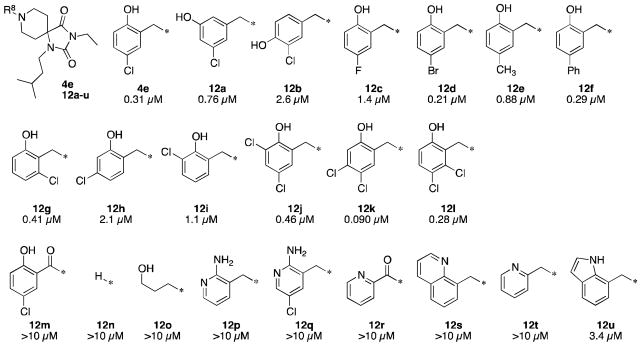
Reported potencies are IC50 values in P. falciparum 3D7 infected erythrocytes.
Positioning of the chlorine atom was found to be important for optimal 3D7 potency for this series. 6′-Chloro analog 12g was found to be essentially equipotent to the 5′-chloro 4e. In contrast, the 3′-chloro and 4′-chloro analogs (12h,i) were three- to seven-fold less potent. 3′,5′-Dichloro and 5′,6′-dichloro analogs (12j,l) were equipotent to the 5′-chloro compound 4e. Interestingly, with an IC50 of 0.090 μM, 4′,5′-dichloro analog 12k proved to be the most potent compound we identified in this series.
To further refine our understanding of the importance of the basic amine and benzylic phenol functionalities, we prepared amide analog 12m. The lack of 3D7 potency of analog 12m, a direct comparator to lead 4e, demonstrates the importance of basic amine to the anti-plasmodium pharmacophore. This result is consistent with the hypothesis that these compounds may be inhibiting an aspartic protease, similar to their BACE-inhibiting predecessors 2 and 3. However, the basic amine functionality alone is not sufficient for potency as demonstrated by deletion analog 12n and phenol replacements and isosteres (12o-u) which were not efficacious up to 10 μM. The only phenol replacement to give a hint of potency was indole 12u.
2.4. Inhibition of Aspartic Proteases, Cytotoxicity and Mechanism of Action
We evaluated twelve of these compounds for inhibition against a panel of aspartic proteases: human proteases BACE-1, cathepsin D (CatD) and cathepsin E (CatE), as well as plasmodium proteases plasmepsin II and IV (PM-II and PM-IV). Remarkably, all twelve compounds had no inhibitory activity on these aspartic proteases up to 10 μM. Our previously described series,19 the aminohydantoins, represented by CWHM-117, are also not potent against human aspartic proteases, but exhibit low nanomolar inhibition of plasmepsins II and IV. This selectivity result is encouraging from a toxicology and drug discovery standpoint but calls into question whether these compounds are acting through an aspartic protease mechanism.
In addition to compound 4e, compounds 12f, 12h, and 12k were evaluated for cytotoxicity in HepG2 cells. All three compounds exhibited IC50 values >50 μM, corresponding to a selectivity index of greater than 500 for 12k.
The mechanism of action of these compounds is currently unknown. Notably, spiropiperidine 4e is equipotent against both chloroquine-sensitive (3D7) and multidrug-resistant (Dd2) strains of P. falciparum (3D7 IC50 = 0.31 μM; Dd2 IC50 = 0.22 μM). The Dd2 strain is resistant to antimalarial drugs chloroquine, quinine, pyrimethamine, and sulfadoxine.27 Thus, the mechanism of action of these spiropiperidine hydantoins appears to be unique and is not due to pathways affected by these existing agents.
2.5. Pharmacokinetic Profile
Finally, we evaluated the pharmacokinetic (PK) properties of this novel series of antimalarials. A subset of analogs were assessed for metabolic stability in mouse, rat and human liver microsome assays (MLM, RLM and HLM, respectively). Figure 6 depicts relationships between antimalarial activity, metabolic stability and lipophilicity. All of the compounds evaluated herein fall within generally acceptable lipophilicity ranges for druglike compounds (Fig. 6a). Not surprisingly, there is a correlation between lipophilicity and 3D7 potency with the more lipophilic compounds being generally more potent.
Figure 6. Relationship between 3D7 potency, metabolic stability and lipophilicity.
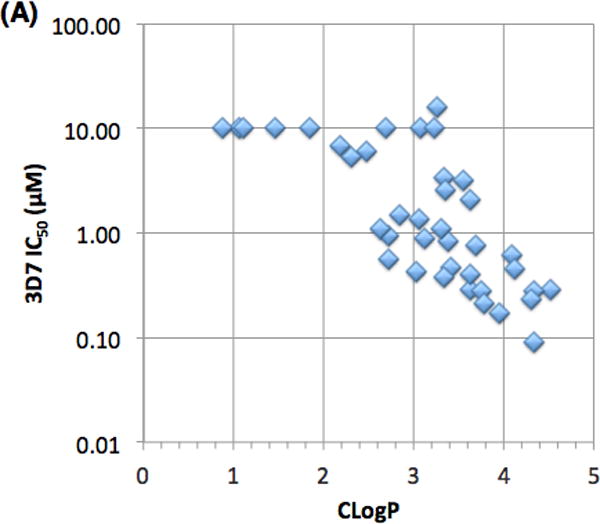
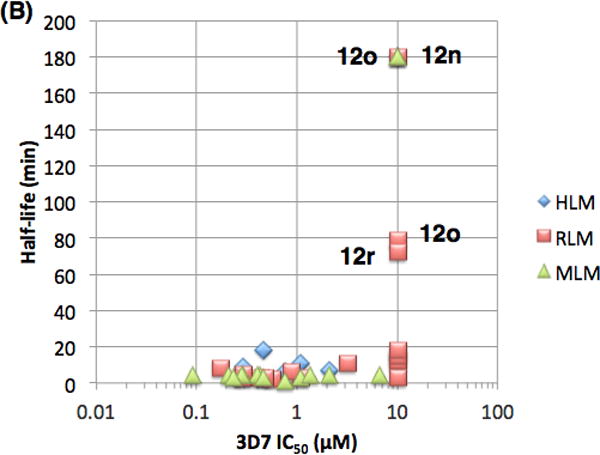
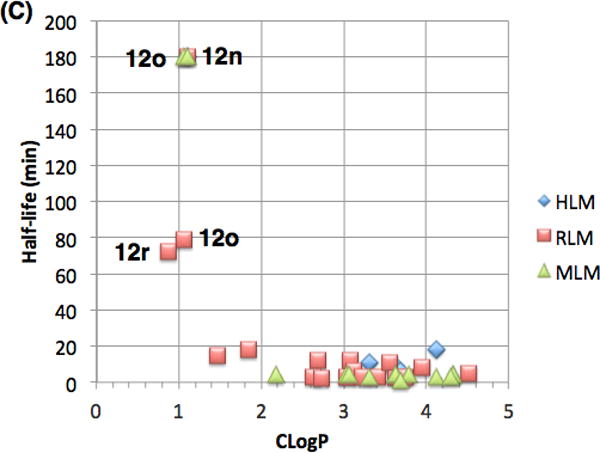
(A) Plot of Pf 3D7 potency as a function of CLogP. (B) Plot of Pf 3D7 potency versus metabolic stability in mouse, rat and human liver microsomes (MLM, RLM and HLM). (C) Plot of metabolic stability in liver microsomes as a function of CLogP.
Most of the compounds evaluated have poor metabolic stability in liver microsomes with half-lives typically less than 10 minutes, regardless of species (Fig 6b). A handful of analogs, however, are metabolically stable (12n,o,r) but are not potent in the 3D7 assay. These stable compounds all have CLogP values of approximately 1.0 and do not contain an N-benzyl group. This data suggests that the metabolic stability for this series of compounds requires either compounds with low CLogP (less than 2), non-benzylic amines or a combination of both features.
Lead compound 4e was also evaluated in vivo in a rat PK experiment to determine if in vitro liver microsome assays are predictive of in vivo PK properties. Compound 4e has short half-lives in vitro (MLM, RLM and HLM t1/2 = 4.0, 3.0 and 9.0 min, respectively) and in vivo (rat PK t1/2 = 0.28 h, CL = 152 mL/min/kg, Vd = 3.7 L/kg and F = 10%). However, the modest oral bioavailability is encouraging.
3. Conclusions
We have identified spiropiperidine hydantoins as a novel series of antimalarial compounds with oral bioavailability but short half-lives. We have explored structure-activity relationships for the three pendant groups and found that the R1 and R3 positions tolerate a variety of functionality, suggesting that modulation of these positions should allow modulation of physiochemical properties without detrimental effects on potency. However, the R8 benzylic phenol was found to be very sensitive to modification. We were able to demonstrate ~10-fold improvement in antimalarial activity through addition of chlorine atoms in the 4′ and 5′ positions, but replacement of the phenol or basic amine functionality resulted in dramatic losses in antimalarial potency. Unfortunately, this dependency on a benzylic amine for potency results in a series of compounds that face significant metabolic stability challenges that will need to be overcome in lead optimization. Progress towards understanding the mechanism of action and identification of more metabolically stable compounds will be reported in due course.
4. Experimental
4.1 General
Commercially available reagents and solvents were used without further purification unless stated otherwise. LC-MS analyses were performed on an Agilent 1100 or 1200HPLC/MSD electrospray mass spectrometer in positive ion mode with scan range was 100–1000d. Preparative normal phase chromatography was performed on a Biotage SP1 with prepacked Biotage or Varian silica gel cartridges. Preparative reverse phase HPLC was performed on a Shimadzu LC-20AP or Biotage SP1 equipped with a C18 column and a methanol/water or acetonitrile/water/0.05% TFA gradient. The purity of tested compounds was ≥95% as determined by HPLC analysis conducted on an Agilent 1100 or 1260 system using a reverse phase C18 column with diode array detector unless stated otherwise. NMR spectra were recorded on a Bruker 400 MHz spectrometer. The signal of the deuterated solvent was used as internal reference. Chemical shifts (δ) are given in ppm and are referenced to residual not fully deuterated solvent signal. Coupling constants (J) are given in Hz.
4.2. Purchased Compounds
Compounds 4a–g and 11c–d were purchased from ChemBridge (www.hit2lead.com). Compound 4e was resynthesized as described in section 4.3.
4.3. Synthesis of CWHM-123 (4e)
tert-Butyl 2,4-dioxo-1,3,8-triazaspiro[4.5]decane-8-carboxylate (6)
A solution of 11.25 g KCN in 25 mL water was added drop wise to a suspension of 16.24 g 1-Bocpiperidone (Oakwood; 80 mmol) and 16.9 g ammonium carbonate in 45 mL water and 55 mL MeOH. During the addition, everything dissolved. The reaction flask was closed up with a balloon. After several hours a ppt began to form. The mixture was stirred at room temp over the weekend. After 72 h, the thick reaction mixture was filtered, washed with small portions of water and dried to give the desired product as a white solid: 16.1 g, 59.8 mmol, 75% yield. HPLC purity >98%. ES-MS m/z 292 (M+Na).
tert-Butyl 3-ethyl-2,4-dioxo-1,3,8-triazaspiro[4.5]decane-8-carboxylate (7a)
A mixture of 6 (3.03 g, 11.3 mmol) and potassium carbonate in DMF (30 mL) was treated with ethyl iodide (1.0 mL, 12.4 mmol) dropwise at room temp. After 5 h, the reaction was diluted with EtOAc, washed with brine, dried over sodium sulfate, filtered and concentrated to give the crude product. The crude solid was recrystallized from EtOAc/heptane to give the title compound as a white solid (2.53 g, 8.51 mmol, 75% yield). HPLC purity >98%. ES-MS m/z 298 (M+H).
3-ethyl-1-isopentyl-1,3,8-triazaspiro[4.5]decane-2,4-dione hydrochloride (12n)
7a (3.16 g, 10.6 mmol) was dissolved in 30 mL DMF (anhydrous) and was treated with NaH (60% disp. in mineral oil, 483 mg, 12.1 mmol) at room temp. After 15 min of stirring, 1-iodo-3-methylbutane (1.7 mL, 12.9 mmol) was added dropwise. After 5 days, the reaction was quenched with water and partitioned between EtOAc and satd ammonium chloride. The organic layer was washed with satd ammonium chloride twice, and was dried with sodium sulfate, filtered and concentrated. The crude product was purified by silica gel chromatography (0–30% EtOAc/hexane) to give 3.1 g of tert-butyl 3-ethyl-1-isopentyl-2,4-dioxo-1,3,8-triazaspiro[4.5]decane-8-carboxylate as a clear oil. The oil was then dissolved in 10 mL of diethyl ether, and 35 mL of 1 M HCl was added. The reaction was stirred at room temp for 48 h. The precipitate was then filtered and washed with diethyl ether, yielding the title compound as a white, powdery HCl salt. (2.13 g, 7.02 mmol, 66% yield). HPLC purity > 90%. ES-MS m/z 268 (M+H). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.26 (br. s., 1 H), 3.39 (q, J=7.2 Hz, 2 H), 3.32 (d, J=6.9 Hz, 4 H), 3.18 (t, J=1.0 Hz, 2 H), 2.36 (dt, J=14.1, 9.2 Hz, 2 H), 1.86 (d, J=14.4 Hz, 2 H), 1.55 (spt, J=1.0 Hz, 1 H), 1.45 (q, J=1.0 Hz, 2 H), 1.08 (t, J=1.0 Hz, 3 H), 0.91 (d, J=1.0 Hz, 6 H).
8-(5-chloro-2-hydroxybenzyl)-3-ethyl-1-isopentyl-1,3,8-triazaspiro[4.5]decane-2,4-dione hydrochloride (CWHM-123; 4e)
A mixture of 299 hydrochloride (200 mg, 0.658 mmol) and 5-chloro-2-hydroxybenzaldehyde (113 mg, 0.724 mmol) in DCE (2 mL) was treated with sodium triacetoxyborohydride (279 mg, 1.32 mmol) followed by DMF (1 mL). The suspension was stirred at room temp overnight. The reaction was quenched with water, acidified with TFA, concentrated and purified by reverse phase HPLC (10 to 70% acetonitrile/water/0.05% TFA). Excess acetonitrile was removed under vacuum and the resultant aqueous solution was neutralized with saturated sodium bicarbonate, extracted with EtOAc, washed with brine, dried over sodium sulfate and concentrated to give a white foam (191 mg). The white foam was dissolved in diethyl ether and treated with 1 N HCl in diethyl ether (0.6 mL). The white ppt was filtered, washed with ether and dried to give the title compound a white HCl salt (194 mg, 0.436 mmol, 66% yield). HPLC purity > 95%. ES-MS m/z 408 (M+H). 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 7.63 (d, J = 2.45 Hz, 1H), 7.34 (dd, J = 2.73, 8.69 Hz, 1H), 7.00 (d, J = 8.85 Hz, 1H), 4.20 – 4.32 (m, 2H), 3.34 – 3.47 (m, 6H), 3.13 – 3.20 (m, 2H), 2.42 – 2.48 (m, 2H), 1.90 – 1.98 (m, 2H), 1.49 – 1.62 (m, 1H), 1.39 – 1.48 (m, 2H), 1.08 (t, J = 7.18 Hz, 3H), 0.90 (d, J = 6.59 Hz, 6H).
4.4. Supporting Information
Synthesis and characterization of all compounds and biological assay methods are included in the supporting information.
Supplementary Material
Acknowledgments
The authors would like to thank Eva Istvan for supplying the PM-II and PM-IV DNA constructs used to express PM-II and PM-IV and Anna Oksman for supplying red blood cells for the 3D7 assay.
Funding Sources
Research reported in this publication at Saint Louis University, was supported by Saint Louis University and the National Institute Of Allergy And Infectious Diseases of the National Institutes of Health under Award Number R01AI106498. Research reported in this publication at the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, was supported by Bureau of Science and Information Technology of Guangzhou Municipality under Grant Number 2009Z1-E841 and Natural Science Foundation of China under Grant Number 81361120380. DEG is supported by the National Institute Of Allergy And Infectious Diseases of the National Institutes of Health under Award Number AI047798. The content is solely the responsibility of the authors and does not necessarily represent the official views of Saint Louis University, the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, the Natural Science Foundation of China, Washington University or the National Institutes of Health.
Abbreviations
- PM
plasmepsin
- SPP
signal peptide peptidase
- HIV
human immunodeficiency virus
- SAR
structure-activity relationships
- BACE
beta-site APP cleaving enzyme 1 or beta-secretase
- TCAMS
Tres Cantos Antimalarial Set
- RBC
red blood cells
- CatD
cathepsin D
- CatE
cathepsin E
- MLM
mouse liver microsomes
- RLM
rat liver microsomes
- HLM
human liver microsomes
- PK
pharmacokinetics
Footnotes
Author Contributions
All authors have given approval to the final version of the manuscript. The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Malaria Report. World Health Organization; 2014. [Google Scholar]
- 2.Dondorp AM, Yeung S, White L, Nguon C, Day NP, Socheat D, von Seidlein L. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol. 2010;8:272–280. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- 3.Hyde JE. Drug-resistant malaria. Trends Parasitol. 2005;21:494–498. doi: 10.1016/j.pt.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, White NJ. C. Tracking Resistance to Artemisinin, Spread of artemisinin resistance in Plasmodium falciparum malaria. The New England journal of medicine. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lubell Y, Dondorp A, Guerin PJ, Drake T, Meek S, Ashley E, Day NP, White NJ, White LJ. Artemisinin resistance - modelling the potential human and economic costs. Malaria journal. 2014;13:452. doi: 10.1186/1475-2875-13-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coombs GH, Goldberg DE, Klemba M, Berry C, Kay J, Mottram JC. Aspartic proteases of Plasmodium falciparum and other parasitic protozoa as drug targets. Trends Parasitol. 2001;17:532–537. doi: 10.1016/s1471-4922(01)02037-2. [DOI] [PubMed] [Google Scholar]
- 7.Meyers MJ, Goldberg DE. Recent advances in plasmepsin medicinal chemistry and implications for future antimalarial drug discovery efforts. Curr Top Med Chem. 2012;12:445–455. doi: 10.2174/156802612799362959. [DOI] [PubMed] [Google Scholar]
- 8.Boddey JA, Hodder AN, Gunther S, Gilson PR, Patsiouras H, Kapp EA, Pearce JA, de Koning-Ward TF, Simpson RJ, Crabb BS, Cowman AF. An aspartyl protease directs malaria effector proteins to the host cell. Nature. 2010;463:627–631. doi: 10.1038/nature08728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo I, Babbitt S, Muralidharan V, Butler T, Oksman A, Goldberg DE. Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature. 2010;463:632–636. doi: 10.1038/nature08726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harbut MB, Patel BA, Yeung BK, McNamara CW, Bright AT, Ballard J, Supek F, Golde TE, Winzeler EA, Diagana TT, Greenbaum DC. Targeting the ERAD pathway via inhibition of signal peptide peptidase for antiparasitic therapeutic design. Proc Natl Acad Sci U S A. 2012;109:21486–21491. doi: 10.1073/pnas.1216016110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Chen H, Bahamontes-Rosa N, Kun JF, Traore B, Crompton PD, Chishti AH. Plasmodium falciparum signal peptide peptidase is a promising drug target against blood stage malaria. Biochem Biophys Res Commun. 2009;380:454–459. doi: 10.1016/j.bbrc.2009.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciana CL, Siegrist R, Aissaoui H, Marx L, Racine S, Meyer S, Binkert C, de Kanter R, Fischli C, Wittlin S, Boss C. Novel in vivo active anti-malarials based on a hydroxy-ethylamine scaffold. Bioorganic & Medicinal Chemistry Letters. 2013;23:658–662. doi: 10.1016/j.bmcl.2012.11.118. [DOI] [PubMed] [Google Scholar]
- 13.Sleebs BE, Gazdik M, O’Neill MT, Rajasekaran P, Lopaticki S, Lackovic K, Lowes K, Smith BJ, Cowman AF, Boddey JA. Transition state mimetics of the Plasmodium export element are potent inhibitors of Plasmepsin V from P. falciparum and P. vivax. J Med Chem. 2014;57:7644–7662. doi: 10.1021/jm500797g. [DOI] [PubMed] [Google Scholar]
- 14.Sleebs BE, Lopaticki S, Marapana DS, O’Neill MT, Rajasekaran P, Gazdik M, Gunther S, Whitehead LW, Lowes KN, Barfod L, Hviid L, Shaw PJ, Hodder AN, Smith BJ, Cowman AF, Boddey JA. Inhibition of Plasmepsin V activity demonstrates its essential role in protein export, PfEMP1 display, and survival of malaria parasites. PLoS biology. 2014;12:e1001897. doi: 10.1371/journal.pbio.1001897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malamas MS, Erdei J, Gunawan I, Turner J, Hu Y, Wagner E, Fan K, Chopra R, Olland A, Bard J, Jacobsen S, Magolda RL, Pangalos M, Robichaud AJ. Design and Synthesis of 5,5′-Disubstituted Aminohydantoins as Potent and Selective Human β-Secretase (BACE1) Inhibitors. Journal of Medicinal Chemistry. 2010;53:1146–1158. doi: 10.1021/jm901414e. [DOI] [PubMed] [Google Scholar]
- 16.Barrow JC, Stauffer SR, Rittle KE, Ngo PL, Yang Z, Selnick HG, Graham SL, Munshi S, McGaughey GB, Holloway MK, Simon AJ, Price EA, Sankaranarayanan S, Colussi D, Tugusheva K, Lai MT, Espeseth AS, Xu M, Huang Q, Wolfe A, Pietrak B, Zuck P, Levorse DA, Hazuda D, Vacca JP. Discovery and X-ray crystallographic analysis of a spiropiperidine iminohydantoin inhibitor of beta-secretase. J Med Chem. 2008;51:6259–6262. doi: 10.1021/jm800914n. [DOI] [PubMed] [Google Scholar]
- 17.Lorthiois E, Breitenstein W, Cumin F, Ehrhardt C, Francotte E, Jacoby E, Ostermann N, Sellner H, Kosaka T, Webb RL, Rigel DF, Hassiepen U, Richert P, Wagner T, Maibaum J. The discovery of novel potent trans-3,4-disubstituted pyrrolidine inhibitors of the human aspartic protease renin from in silico three-dimensional (3D) pharmacophore searches. J Med Chem. 2013;56:2207–2217. doi: 10.1021/jm3017078. [DOI] [PubMed] [Google Scholar]
- 18.Gamo F-J, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera J-L, Vanderwall DE, Green DVS, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. doi: 10.1038/nature09107. ChEMBL-NTD ( http://www.ebi.ac.uk/chemblntd) [DOI] [PubMed] [Google Scholar]
- 19.Meyers MJ, Tortorella MD, Xu J, Qin L, He Z, Lang X, Zeng W, Xu W, Qin L, Prinsen MJ, Sverdrup FM, Eickhoff CS, Griggs DW, Oliva J, Ruminski PG, Jacobsen EJ, Campbell MA, Wood DC, Goldberg DE, Liu X, Lu Y, Lu X, Tu Z, Lu X, Ding K, Chen X. Evaluation of Aminohydantoins as a Novel Class of Antimalarial Agents. ACS Medicinal Chemistry Letters. 2014;5:89–93. doi: 10.1021/ml400412x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brodney MA. Preparation of lactams as beta secretase inhibitors. Pfizer Inc; USA: 2012. p. 59. [Google Scholar]
- 21.Brodney MA, Barreiro G, Ogilvie K, Hajos-Korcsok E, Murray J, Vajdos F, Ambroise C, Christoffersen C, Fisher K, Lanyon L, Liu J, Nolan CE, Withka JM, Borzilleri KA, Efremov I, Oborski CE, Varghese A, O’Neill BT. Spirocyclic Sulfamides as β-Secretase 1 (BACE-1) Inhibitors for the Treatment of Alzheimer’s Disease: Utilization of Structure Based Drug Design, WaterMap, and CNS Penetration Studies To Identify Centrally Efficacious Inhibitors. J Med Chem. 2012;55:9224–9239. doi: 10.1021/jm3009426. [DOI] [PubMed] [Google Scholar]
- 22.Brodney MA, Efremov IV, Helal CJ, O’Neill BT. Lactams as β-secretase inhibitors and their preparation, pharmaceutical compositions and use in the treatment of neurodegenerative diseases. Pfizer Inc; USA: 2010. p. 106. [Google Scholar]
- 23.Stachel SJ, Steele TG, Petrocchi A, Haugabook SJ, McGaughey G, Katharine Holloway M, Allison T, Munshi S, Zuck P, Colussi D, Tugasheva K, Wolfe A, Graham SL, Vacca JP. Discovery of pyrrolidine-based beta-secretase inhibitors: lead advancement through conformational design for maintenance of ligand binding efficiency. Bioorg Med Chem Lett. 2012;22:240–244. doi: 10.1016/j.bmcl.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 24.K.G. ChEMBL-NTD (http://www.ebi.ac.uk/chemblntd). Novartis-GNF Malaria Box, R Borboa, C Francek, Z Chen, J Buenviaje, D Plouffe, E Winzeler, A Brinker, T Diagana, J Taylor, R Glynne, A Chatterjee, K Kuhen. Genomics Institute of the Novartis Research Foundation (GNF), 10675 John Jay Hopkins Drive, San Diego CA 92121, USA and Novartis Institute for Tropical Disease, 10 Biopolis Road, Chromos # 05–01, 138 670 Singapore.
- 25.Spyvee M, Chen Q, Carlson E, Kusano K. Imidazoazepinone compounds that suppress the PGE2/EP4 signaling and their preparation and use in the treatment of diseases. Eisai R&D Management Co., Ltd; Japan: 2011. p. 178. [Google Scholar]
- 26.Carrera GM, Garvey DS. Synthesis of novel substituted spirohydantoins. Journal of Heterocyclic Chemistry. 1992;29:847–850. [Google Scholar]
- 27.Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nature reviews Drug discovery. 2004;3:509–520. doi: 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.