

Abstract
Chronic alcohol exposure inhibits insulin and insulin-like growth factor signaling in the liver and brain by impairing the signaling cascade at multiple levels. These alterations produced by alcohol cause severe hepatic and central nervous system insulin resistance as the cells fail to adequately transmit signals downstream through Erk/mitogen-activated protein kinase (MAPK), which is needed for DNA synthesis and liver regeneration, and phosphatidylinositol 3-kinase (PI3K), which promotes growth, survival, cell motility, glucose utilization, plasticity, and energy metabolism. The robust inhibition of insulin signaling in liver and brain is augmented by additional factors involving the activation of phosphatases such as phosphatase and tensin homologue (PTEN), which further impairs insulin signaling through PI3K/Akt. Thus, intact insulin signaling is important for neuronal survival. Chronic alcohol consumption produces steatohepatitis, which also promotes hepatic insulin resistance, oxidative stress and injury, with the attendant increased generation of “toxic lipids” such as ceramides that increase insulin resistance. The PI3K/Akt signaling cascade is altered by direct interaction with ceramides as well as through PTEN upregulation as a downstream target gene of enhanced p53 transcriptional activity. Cytotoxic ceramides transferred from the liver to the blood can enter the brain due to their lipid-soluble nature, and thereby exert neurodegenerative effects via a liver–brain axis. We postulate that the neurotoxic and neurodegenerative effects of liver-derived ceramides activate pro-inflammatory cytokines and increase lipid adducts and insulin resistance in the brain to impair cognitive and motor function. These observations are discussed in the context of insulin sensitizers as potential cytoprotective agents against liver and brain injury induced by alcohol.
Introduction
Insulin produces its effect on hepatocyte growth and survival by activation of a signal transduction cascade, which involves ligand interaction with cell surface receptors and the sequential activation of tyrosine kinases. 1 Important components of this cascade include the insulin receptor (IR), insulin receptor substrate-1 (IRS-1), and substrate-2 (IRS-2). Indeed, IRS-1 transmits the insulin signal following tyrosylphosphorylation (PY) by the IR tyrosine kinase. Following this event, PY-IRS-1 interacts with growth factor receptor bound protein 2, Grb2, 2 son of sevenless which activates the Ras/Raf/MAPKK/MAPK/Erk cascade. 3 The functional consequences are hepatocyte proliferation and enhanced gene expression. Recent investigations demonstrate that the activation of signaling through IRS-1 is essential for DNA synthesis and cell cycle progression. 4,5 Thus, the effects of IRS-1 overexpression on cell growth seemed to depend on the constitutive activation of mitogenic signal transduction cascades. 4–7
Another arm of the signaling cascade involves the binding of PY-IRS-1 to phosphatidylinositol-3 kinase (PI3K). The lipid kinase, PI3K, regulates cellular processes including survival, growth, and proliferation. In this regard, PI3K phosphorylates the 3′hydroxyl-group on the inositol ring of the downstream lipid component phosphatidylinositol-4-5-biphosphate (PIP2) to produce a second messenger, namely PIP3. This molecule activates Akt, which is a serine threonine kinase, and Akt enhances cell survival and proliferation by inhibiting the activities of the fork-head family of transcription factors. Therefore, insulin signaling through PI3K inhibits apoptosis by activating Akt/protein kinase B, 8,9 which phosphorylates GSK-3β 10–12 and BAD, 13 rendering them inactive; low levels of Akt kinase and high levels of activated GSK-3β and/or BAD are associated with increased programmed cell death. There are several PIP3 phosphatases that act to inhibit PI3K signaling, such as phosphatase and tensin homolog (PTEN). This protein phosphatase takes PIP3 back to the 4, 5-biphosphate (PIP2) form. Insulin signaling may be inhibited through alterations in PTEN expression at the transcriptional and protein levels in the liver. 14 In this regard, high levels of biologically active PTEN are associated with reduced levels of phospho-Akt, which subsequently increases GSK3β and BAD activity, thereby inhibiting survival signals to promote apoptosis.
We propose a novel hypothesis that p53 activation may be important in the pathogenesis of alcoholic liver disease (ALD) 15 and facilitate disease progression by impacting on oxidative stress, apoptosis and cellular metabolism, as outlined in Figure 1. Indeed, p53 appears to modulate alcohol-induced hepatocyte apoptosis, since it was completely abrogated in mice with a p53 null background. 16 It is also noteworthy that p53 is essential to the regulation of oxidative stress. Under normal conditions or after mild stress events, low p53 levels drive the transcription of antioxidant genes that decrease reactive oxygen species (ROS) and protect the cells from DNA damage. 17 However, p53 activation under severe or extended stress (e.g. chronic alcohol consumption) leads to the upregulation of pro-oxidant (such as p66shc) and pro-apoptotic genes (p53 upregulated modulator of apoptosis (PUMA)-α, a direct target of p53), and results in elevated ROS levels as well as apoptosis. 18 Furthermore, ROS provide a major stimulus for p53 stabilization and the subsequent induction of apoptosis by a feed-forward regulatory loop. 19 Recent observations reveal that p53 not only orchestrates various forms of cell death but also regulates cellular energy metabolism and suppresses the PI3K/AKT survival pathway. Activation of metabolically active p53-downstream target genes and interference with the PI3K/AKT pathway reduce glycolysis and favor oxidative phosphorylation by the mitochondria, which increases the likelihood of further ROS generation. It has been shown that p53 downregulates phosphoglycerate mutase (PGM), an important enzyme in glycolysis, 20 upregulates TP53 induced glycolysis and apoptosis regulator (TIGAR), 21 which functions as a fructose-bisphosphatase, decreasing the intracellular concentration of fructose-2,6-bisphosphate, which is the most potent allosteric activator of the glycolytic enzyme 6-phosphofructose-1-kinase. Furthermore, p53 promotes oxidative phosphorylation by inducing cytochrome c oxidase 2 expression, 22 a key regulator of the cytochrome c oxidase. This complex is embedded in the inner mitochondrial membrane and represents a major site of oxygen use in mammalian cells. The suppression of the PI3K/Akt pathway is mediated primarily by PTEN, which is also a p53-activated downstream gene. 23 By stimulating PTEN gene expression, p53 inhibits Akt, leading to reduced hepatocyte and neuronal survival. 24
Figure 1.

Hepatocellular consequences of insulin resistance induced by chronic alcohol consumption, emphasizing the major concepts developed from our research. Central to the hypothesis of how alcohol contributes to liver injury and attenuates the repair process (regeneration) is the induction of insulin resistance. This cascade leads to a series of downstream adverse events involving cell survival, proliferation and apoptosis. Note the central role of p53 in this process. ROS, PUMA, P13K, PTEN.
Results and discussion
Alcohol, insulin resistance and the liver
The potential role of PTEN phosphatase in alcohol impaired survival signaling in the liver of chronic alcohol-fed rats
Previous investigations from our laboratory demonstrated that alcohol inhibition of insulin/insulin-like growth factor (IGF)1 signaling through IRS-1 impaired liver regeneration by decreased DNA synthesis due to lack of Erk/mitogen-activated protein kinase (MAPK) activation. Further studies revealed that with IRS-1 overexpression either in transgenic mice or hepatocyte-derived cell lines, alcohol prominently inhibited growth pathways. 25 Here, we observed that chronic alcohol exposure impaired survival mechanisms in the previously normal liver by the constitutive inhibition of PI3K activity: an effect that is mediated by increased levels of PTEN phosphatase expression and function. In these experiments, Long–Evans (LE) rats were fed the Lieber–DiCarli diet (37% calories derived from alcohol 9.2% v/v) and pair-fed controls for 8 weeks followed by a histological analysis of liver injury and insulin survival signaling through PI3K/Akt and downstream effector molecules.
Alcohol-induced PTEN expression in vivo
Since PTEN is a negative regulator of PI3K-mediated cell growth and survival signaling, we evaluated its expression levels in the liver. PTEN functions by dephosphorylating the third position of the phosphatidylinositol and, therefore, may serve as a potential inhibitor of PI3K-mediated cell survival. Western blot analysis revealed the expected 54 kd PTEN protein in liver samples from alcohol-fed and control LE rats, as shown in Figure 2.
Figure 2.
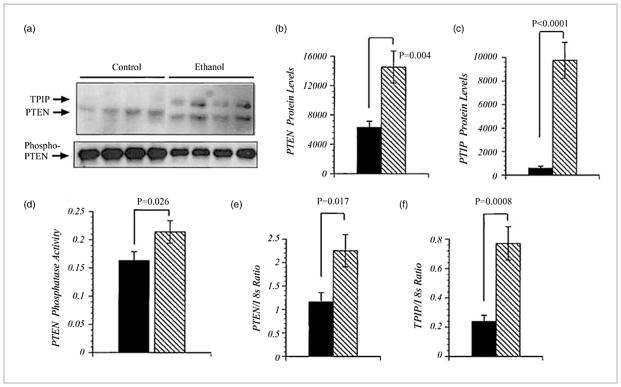
Increased phosphatase and tensin homologue (PTEN) expression in liver tissue from alcohol-exposed rats. (a) Western blot autoradiograph demonstrating the expression of the PTEN and TPIP proteins in liver homogenates. (b and c) Mean levels (± SEM) of PTEN or TPIP measured by digital image analysis of Western blot signals. (d) Levels of PTEN phosphatase activity measured in immunoprecipitates using the Biomol Green assay. (e and f) A real-time quantitative polymerase chain reaction was used to examine the mRNA levels of PTEN, TPIP, and 18 s. The actual levels of expression were determined from standard curves generated with recombinant plasmid DNA. PTEN and TPIP mRNA levels were normalized to 18 s to control for slight differences in template loading or cellular RNA abundance. Means ± SEM of the calculated PTEN/18 s and TPIP/18 s copy number ratios for each group.
In addition, a second slower migrating band was identified as the PTEN isoform TPIP. 26 There are significantly higher mean levels of PTEN and TPIP in alcohol-fed rats relative to controls (Fig. 2b,c). Because previous reports demonstrated that PTEN phosphorylation inhibited phosphatase activity 27 further studies examined phospho-PTEN levels by antiphosphoserine/phosphothreonine Western blot analysis of PTEN immunoprecipitates (Fig. 2a). Such studies revealed reduced levels of phosphos-PTEN in livers from alcohol-fed LE rats relative to controls (P = 0.003). In addition, the samples from alcohol-fed rats had significantly increased levels of phosphatase activity in PTEN immunoprecipitates (Fig. 2d). The biological consequence of enhanced PTEN expression in insulin resistance.
What are the possible mechanisms of the adverse effect of alcohol on survival signaling? In contrast with chronic alcohol exposure as shown above, there is no increase in PTEN protein levels and Huh-7 cells. However, there is a shift to an increased interaction of PI3K p85α with PTEN. Furthermore, acute alcohol exposure alone rapidly increases this association in as short a time as 5 min in the absence of insulin stimulation. In this context, alcohol substantially augments the interaction of PI3K p85α with PTEN under conditions of insulin stimulation. Taken together, acute alcohol exposure both in vitro and in vivo has surprising and substantial effects on insulin-mediated survival signaling in the liver and rapidly increases the disassociation of PTEN to the p85α regulatory subunit of PI3K, even though the cellular levels of PTEN remain unchanged. This shifts the balance from the IRS-1/PI3K p85α towards the PTEN/PI3K p85α complex through a direct interaction that subsequently leads to the reduced phosphorylation of Akt, thus activating GSK3β and BAD to promote programmed cell pathways, as demonstrated by the scheme in Figure 3. These studies contribute to a better understanding of the toxic effects of acute and chronic alcohol exposure on the liver.
Figure 3.

Hypothetical model of acute alcohol effects on insulin signaling through phosphatidylinositol 3-kinase (PI3K)/Akt and the role of phosphatase and tensin homologue (PTEN). In the absence of alcohol exposure, as shown on the left, the insulin signal is transmitted through insulin receptor substrate-1 (IRS-1) followed by binding of the p85α subunit of PI3K. This event activates the kinase and leads to Akt phosphorylation. The phosphorylation of Akt results in the phosphorylation of GSK3β and BAD, rendering them inactive. The functional effects are increased pro-survival signals. In the presence of acute alcohol exposure there is preferential binding of the p85α subunit of PI3K to PTEN, which competes for its binding to IRS-1. The net effect is the reduced phosphorylation of Akt as well as GSK3β and BAD, rendering the latter two signaling molecules active. Under these conditions there is a generation of pro-apoptotic signals and hepatocytes are more susceptible to injury due to the acute effects of alcohol.
PPARδ (peroxisome proliferator-activated receptor δ) agonist attenuates alcohol-induced hepatic insulin resistance and improves liver injury and repair in the LE rat model
Previous studies demonstrated that chronic alcohol-induced liver injury was partly mediated by hepatic insulin resistance and oxidative stress with increased DNA damage and lipid peroxidation. 28 In light of the known therapeutic actions of PPAR agonists, we evaluated the effectiveness of PPAR agonists in restoring insulin responsiveness, insulin signaling, histology, and regenerative responses in livers of chronic alcohol-fed rats. We focused on the δ class of PPAR agonists because it was already shown to have positive effects on hepatic insulin sensitivity. Our hypothesis was that by improving insulin signaling and insulin responsive gene expression, alcohol’s adverse effects on liver injury and repair 24,29 would be partially attenuated. The results presented below demonstrate that treatment with a PPARδ agonist can effectively reduce injury, oxidative stress, and DNA damage, and substantially improved the regenerative response in livers of chronic alcohol-fed LE rats.
PPARδ agonist reverses alcohol-induced liver pathology
Livers from control rats fed liquid diet or chow had well-organized lobular architectures with minimal steatosis, nuclear size variation, or hepatocyte drop-out (data not shown). In contrast, alcohol exposed livers exhibited microvesicular and macrovesicular steatosis with multifocal intralobular lymphomononuclear cell inflammation and scattered areas of apoptosis or hepatocyte drop-out. In addition, alcohol feeding caused liver architectural disarray with a loss of regular chords and increased variability in hepatocyte nuclear size. In alcohol-fed rats, treatment with the PPARδ agonist L-165,041 reduced the liver architectural disarray, micro- and macrovesicular steatosis, and apoptotic cell death. 30
Effects of alcohol on insulin and IGF-1 receptor binding
We measured insulin and IGF-1 receptor binding and affinity in liver tissue excised at the time of a two-thirds hepatectomy to assess hepatic insulin resistance after 8 weeks on the liquid diets, and 3 weeks of L-165,041 or vehicle treatment. The top-level insulin binding to its own receptor (BMAX ± SD) was 7.23 ± 2.66 in the controls, compared with 2.59 ± 0.61 in the alcohol-exposed rats ( P < 0.001 relative to control). IR binding affinity (Kd ± SEM) was significantly higher in controls (51.38 ± 18.63) than in the alcohol-exposed livers (165.5 ± 73; P < 0.001). (Note that higher affinity binding is associated with a lower Kd). L-165,041 treatment increased the BMAX (4.21 ± 0.45) and decreased the Kd (33.85 ± 9.91) in the alcohol-exposed livers (both P < 0.001 relative to corresponding vehicle-treated rats), consistent with the hypothesis that PPARδ improves hepatic insulin sensitivity.
PPARδ agonist augments hepatic regenerative response during chronic alcohol consumption
Since the PPARδ agonist treatments reduced hepatic insulin resistance in the setting of chronic alcohol exposure, we extended our investigations to determine whether it would also enhance liver regeneration. DNA synthesis was assessed by bromodeoxyuridine (BrdU) incorporation into nuclear DNA. After 24 h subsequent to the two-thirds hepatectomy, approximately 35% of hepatocytes were BrdU labeled in the control rats, whereas only 4% of hepatocytes were labeled in the alcohol-fed group (Fig. 4b). Correspondingly, Western blot analysis demonstrated lower levels of proliferating cell nuclear antigen (PCNA) in alcohol-fed relative to control livers, 24 h after the two-thirds hepatectomy (Fig. 4a,c). Interestingly, PPARδ agonist treatments partially reversed the inhibitory effects of alcohol on DNA synthesis, as demonstrated by the fourfold to fivefold increases in BrdU immunohistochemical staining relative to vehicle-treatment (Fig. 4b).
Figure 4.

Peroxisome proliferator-activated receptor (PPAR)δ agonist improves ethanol-impaired liver regeneration. Adult male Long–Evans rats were fed with liquid diets containing 0% or 37% ethanol, treated with a PPARδ agonist, and then subjected to a two-thirds hepatectomy. Subsequently, 24 h later, the remnant regenerating livers were used to measure (a,c) proliferating cell nuclear antigen (PCNA) (a, d) aspartyl-(asparaginyl)-β-hydroxylase (AAH) (a,e) glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and the (a) p85 subunit of PI3 kinase (negative control) expression by (a) Western blot analysis and (c–e) digital image quantification (arbitrary densitometry units). Two h before they were killed, the rats were injected with bromodeoxyuridine (BrdU). The histological sections were immunostained to detect nuclear BrdU. Under code, BrdU positive and negative hepatocytes were enumerated in 10 adjacent 100 × magnification fields per slide. (b) The percentages of BrdU-labeled nuclei in regenerating livers from control, ethanol exposed and ethanol exposed + PPARδ agonist treated rats. For all studies, inter-group comparisons were made using one-way repeated measures ANOVA and the Tukey–Kramer post-hoc test of significance. P-values are indicated in each panel. Note that PPARδ agonist treatments significantly increased DNA synthesis, and PCNA, AAH, and GAPDH expression during liver regeneration. (
 ) BrdU(+)/total hepatocytes; (▨) PCNA; (▩) AAH; (▥) GAPDH.
) BrdU(+)/total hepatocytes; (▨) PCNA; (▩) AAH; (▥) GAPDH.
DNA damage in alcohol-fed rat livers
In addition to impairing insulin responsiveness, alcohol mediates its adverse effects in liver by causing oxidative stress, lipid peroxidation, and DNA damage. 28,29 PPAR agonists enhance insulin sensitivity and also reduce inflammation and oxidative stress. 31 We determined the effects of PPARδ agonist treatment on alcohol-induced DNA damage by examining 8-Hydroxydeoxyguanosine (8-OHdG) immunoreactivity. The studies demonstrated higher levels of 8-OHdG in alcohol-exposed rats relative to the livers of control rats by immunohistochemical staining (Fig. 5). PPARδ agonist treatments strikingly reduced 8-OHdG immunoreactivities in the alcohol-exposed livers (Fig. 5). During peak regeneration (24 h point), the control livers exhibited only rare nuclear 8-OHdG labeling, while the alcohol-exposed livers had abundant nuclear 8-OHdG immunoreactivity. PPARδ agonist treatments substantially reduced, but failed to abolish, hepatic nuclear DNA damage in chronic alcohol-fed rats (Fig. 5). The beneficial effect of the PPARδ agonist most likely aided in restoring the hepatic regenerative response, despite continued chronic exposure to alcohol.
Figure 5.

Peroxisome proliferator-activated receptor (PPAR)δ agonist treatment partially reversed ethanol-induced oxidative stress. Regenerating Long–Evans rat livers were used to measure oxidative stress (original magnifications 100 ×). The ratios of 8-OHdG-positive nuclei to total nuclei in 10–100 × microscopic fields were determined The graph (lower right panel) shows the mean relative densities of 8-Oxo-2′-deoxyguanosine (8-OHdG) immunoreactive nuclei in regenerating livers from control, ethanol exposed, and ethanol exposed + PPARδ agonist treated rats. Inter-group comparisons were made using one-way repeated measures ANOVA and the Tukey–Kramer post hoc significance test. P -values are indicated within each panel. (▧) (8-OHdG)/total hepatocytes.
It is noteworthy that, while some hepatotoxic effects of alcohol are due to impaired insulin signaling, others are mediated by alcohol’s cytotoxic metabolites, principally acetaldehyde, together with oxidative stress, mitochondrial dysfunction, lipid peroxidation, and protein and DNA adduct formation. 32 In the LE rat model, the livers had alcohol-induced histopathological lesions as previously described. 24,28 Since PPAR agonists have dual actions in their capacity to enhance insulin responsiveness and reduced cellular inflammation, 31 we hypothesized that PPARδ agonist treatments would, in addition to improving insulin sensitivity, abrogate inflammation and DNA damage in the liver. These results potentially offer a new strategy for mitigating chronic alcohol-induced liver injury and hepatic dysfunction.
Alcohol, insulin resistance and the brain
Previous studies have shown that chronic alcohol feeding of adult LE rats causes major central nervous system abnormalities that link neuronal loss and impaired acetylcholine homeostasis to the ethanol inhibition of insulin and IGF signaling and increased oxidative stress. We have now characterized the integrity of insulin and IGF signaling mechanisms and assessed the molecular indices of neurodegeneration in the cerebellar vermis and anterior cingulate gyrus of human alcoholics. We found that alcoholic cerebella had increased neuronal loss, gliosis, lipid peroxidation, and DNA damage relative to control. Quantitative RT-PCR studies demonstrated reduced expression of insulin, IR and IGF-II receptor in the anterior cingulate, and reduced expression of insulin, IGF-I, and their corresponding receptors in the vermis. Competitive equilibrium binding assays revealed significantly reduced specific binding to the insulin, IGF-I, and IGF-II receptors in both the anterior cingulate and vermis of alcoholic brains (Fig. 6). These effects of chronic alcohol abuse were associated with significantly reduced expression of choline acetyltransferase, which is needed for acetylcholine biosynthesis. The results suggest that alcoholic neurodegeneration in humans is associated with insulin and IGF resistance with the attendant impairment of neuronal survival mechanisms and acetylcholine homeostasis. 33
Figure 6.

Chronic alcohol abuse impairs insulin receptor binding in the human brain. Equilibrium binding assays were performed by incubating membrane protein extracts of (a, c, e) anterior cingulate gyrus or (b, d, f) cerebellar vermis overnight at 4°C with 50 pmol [125I]-labeled insulin, insulin-like growth factor (IGF-I), or IGF-II as tracer, in the presence or absence of 100 mmol cold ligand. The membrane bound tracer was precipitated by adding bovine gamma globulin and polyethylene glycol-8000 to the reactions and centrifuging the samples (14 000 g ). The radioactivity present in the supernatant fractions (containing unbound/free ligand) and the pellets (containing bound ligand) was measured in a gamma counter. Specific binding (fmol/mg) was calculated using the GraphPad Prism version 4.00 for Windows, GraphPad Software, San Diego, CA, USA, http://www.graphpad.com. Graphs depict mean ± SEM of results obtained for (a, b) insulin (c, d) IGF-1, and (e, f) IGF-2 specific binding with six cases per group. Data were analyzed statistically using Student’s t-tests. Significant P-values are indicated over the bar graphs. (■) control; (▨) alcoholic rats.
Alcohol, insulin resistance and toxic ceramides: liver–brain axis of neurodegeneration
Role of steatohepatitis
Clinically, alcoholic, non-alcoholic, and viral steatohepatitis can be associated with cognitive and neuropsychiatric dysfunction. 34–40 Insulin signaling mechanisms, pro-ceramide gene expression, and ceramide levels have been examined in various experimental models of steatohepatitis and deficits in learning and memory by Morris water maze testing have been demonstrated. 41–43 Steatohepatitis is associated with liver and brain insulin resistance manifested by reduced IR binding, IR gene expression, IR tyrosine kinase activation, and insulin responsive gene expression, including those required for metabolism or neurotransmitter synthesis; 28,41–43 in addition, there was increased oxidative stress. Using qualitative real-time polymerase chain reaction analysis, it was found that steatohepatitis was associated with the upregulated expression of multiple pro-ceramide genes in liver involving both de novo synthesis and generation from sphinogomyelin via the degradation pathway. ELISA and dot blot analyses demonstrated higher mean levels of ceramide immunoreactivity in the liver and blood as well. 44
Experimentally, ethanol-fed rats with steatohepatitis developed CNS insulin resistance with neurodegeneration and cognitive impairment, 28,30,45 and in that model, pro-ceramide gene expression was increased in both the liver and brain. More important, in human alcoholics, white matter atrophy and degeneration with reduced expression of myelin-associated genes, and increased levels of oxidative stress were associated with increased expression of pro-ceramide genes. 33
Toxic lipids, including ceramides, readily cross the blood–brain barrier and cause insulin resistance by interfering with critical phosphorylation events 46 and activate pro-inflammatory cytokines. 47–49 In this regard, we conducted several experiments designed to address our hypothesis on the potential role of extra-CNS (liver)-derived ceramides as mediators of neurodegeneration. In vitro experiments revealed that C2 or C6 ceramide exposures caused neuronal insulin resistance with reduced viability and neurotransmitter function, and increased mitochondrial dysfunction, oxidative stress, DNA damage, and lipid peroxidation. 50 Moreover, preliminary studies demonstrated that in vivo intraperitoneal (i.p.) ceramide treatments caused brain insulin resistance, neurodegeneration, and cognitive-motor deficits mimicking features of chronic alcohol feeding; increased pro-ceramide gene expression in the brain was found. Therefore, ceramides generated or delivered from extra-CNS sources will cause brain insulin resistance, thereby mediating neurodegeneration. In addition, exposure to exogenous ceramides may increase CNS expression of pro-ceramide genes.
Thus, chronic alcohol exposure resulting in steatohepatitis, ER stress, and persistent hepatic injury promotes lipolysis and the increased generation of toxic lipids, including ceramides. Ceramides promote liver injury by causing insulin resistance, oxidative stress, and pro-inflammatory cytokine activation. Lipid soluble ceramides released into peripheral blood can readily cross the blood–brain barrier. In the CNS ceramides initiate a complex cascade of neurodegeneration mediated by insulin resistance, inflammation, and oxidative stress. This important CNS insulin resistance leads to neuronal loss and impaired oligodendrocyte function. Furthermore, myelin degradation leads to increased CNS toxic lipid production and the worsening of neurodegeneration. Finally, the direct neurotoxic effects of alcohol and its metabolites that impair mitochondrial function, reduce membrane integrity, and increase oxidative stress also contribute to neurodegeneration.
The net effect from brain insulin resistance is neuronal loss and impaired neurotransmitter function required for plasticity, learning, and memory. Brain insulin resistance impairs oligodendrocyte survival and function, resulting in reduced myelin integrity, and the increased generation of ceramides that further increase brain insulin resistance, neuro-inflammation, oxidative stress, and neurodegeneration. These concepts open exciting new possibilities on disease mechanisms and strategies for developing non-invasive tools to monitor proneness to and progression of alcoholic neurodegeneration. The overarching hypothesis about how alcohol abuse promotes neurodegeneration both directly and via the liver–brain axis is diagrammed in Figure 7 and links these two organ systems to the pathogenesis of alcohol-induced tissue damage.
Figure 7.
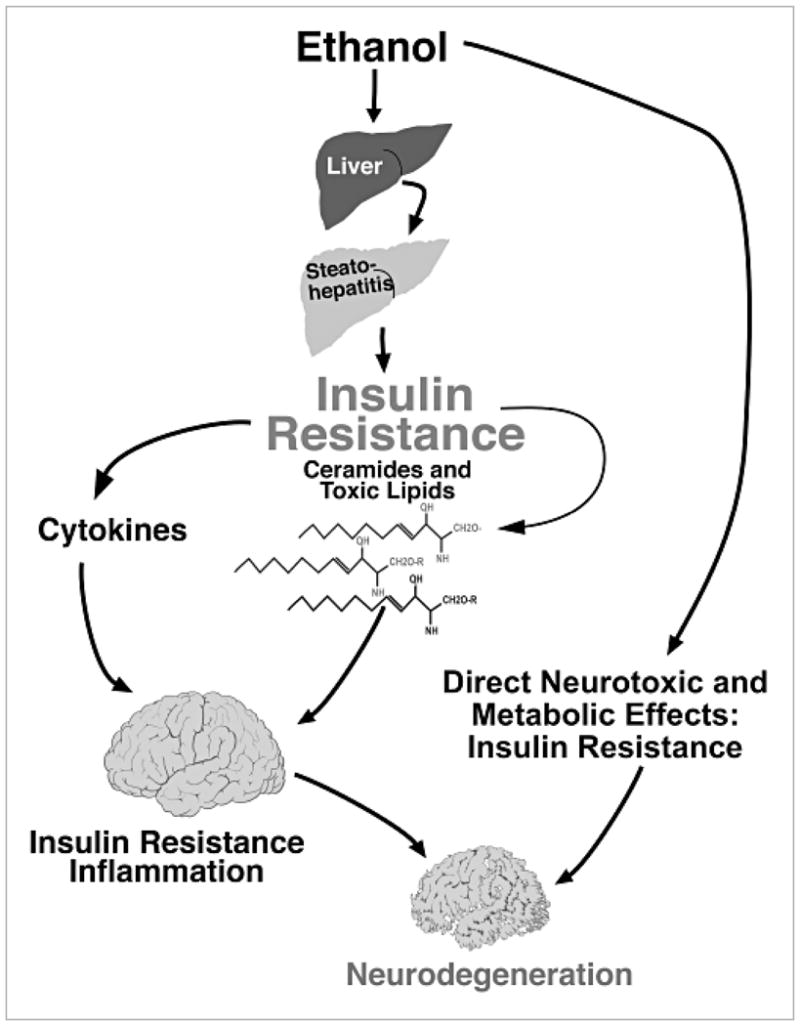
Liver–brain axis of neurodegeneration hypothesis. Progressive hepatic steatosis incites inflammation and pro-inflammatory cytokine activation. Attendant insulin resistance initiates a lipolysis and lipid disequilibrium cascade, leading to the increased production and accumulation of ceramides. The ceramides are cytotoxic and promote insulin resistance, and their lipid solubility enables ready transfer across the blood–brain barrier to cause central nervous system insulin resistance and neurodegeneration with loss of neurons and oligodendrocytes. Ethanol’s lipid solubility enables it to exert direct neurotoxic effects and cause brain insulin resistance.
Acknowledgments
This study was supported in part by grants AA-02666 (JRW), AA08169 (JRW), AA11431 (SMD), and AA12908 (SMD) from the National Institutes of Health.
Footnotes
Conflicts of interest: the authors have no conflict of interest.
References
- 1.Ullrich A, Bell JR, Chen EY, et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature. 1985;313:756–61. doi: 10.1038/313756a0. [DOI] [PubMed] [Google Scholar]
- 2.Bonini JA, Colca JR, Dailey C, White M, Hofmann C. Compensatory alterations for insulin signal transduction and glucose transport in insulin-resistant diabetes. Am J Physiol. 1995;269 (4 Pt 1):E759–65. doi: 10.1152/ajpendo.1995.269.4.E759. [DOI] [PubMed] [Google Scholar]
- 3.Skolnik EY, Batzer A, Li N, et al. The function of GRB2 in linking the insulin receptor to Ras signaling pathways. Science. 1993;260:1953–5. doi: 10.1126/science.8316835. [DOI] [PubMed] [Google Scholar]
- 4.Ito T, Sasaki Y, Wands JR. Overexpression of human insulin receptor substrate 1 induces cellular transformation with activation of mitogen-activated protein kinases. Mol Cell Biol. 1996;16:943–51. doi: 10.1128/mcb.16.3.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanaka S, Ito T, Wands JR. Neoplastic transformation induced by insulin receptor substrate-1 overexpression requires an interaction with both Grb2 and Syp signaling molecules. J Biol Chem. 1996;271:14610–16. doi: 10.1074/jbc.271.24.14610. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki Y, Zhang XF, Nishiyama M, Avruch J, Wands JR. Expression and phosphorylation of insulin receptor substrate 1 during rat liver regeneration. J Biol Chem. 1993;268:3805–8. [PubMed] [Google Scholar]
- 7.Tanaka S, Mohr L, Schmidt EV, Sugimachi K, Wands JR. Biological effects of human insulin receptor substrate-1 overexpression in hepatocytes. Hepatology. 1997;26:598–604. doi: 10.1002/hep.510260310. [DOI] [PubMed] [Google Scholar]
- 8.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 9.Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harwood AJ. Regulation of GSK-3: a cellular multiprocessor. Cell. 2001;105:821–4. doi: 10.1016/s0092-8674(01)00412-3. [DOI] [PubMed] [Google Scholar]
- 11.Orena SJ, Torchia AJ, Garofalo RS. Inhibition of glycogen-synthase kinase 3 stimulates glycogen synthase and glucose transport by distinct mechanisms in 3T3-L1 adipocytes. J Biol Chem. 2000;275:15765–72. doi: 10.1074/jbc.M910002199. [DOI] [PubMed] [Google Scholar]
- 12.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–32. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 13.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 14.Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100:387–90. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 15.Derdak Z, Lang CH, Villegas KA, et al. Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. J Hepatol. 2011;54:164–72. doi: 10.1016/j.jhep.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pani G, Fusco S, Colavitti R, et al. Abrogation of hepatocyte apoptosis and early appearance of liver dysplasia in ethanol-fed p53-deficient mice. Biochem Biophys Res Commun. 2004;325:97–100. doi: 10.1016/j.bbrc.2004.09.213. [DOI] [PubMed] [Google Scholar]
- 17.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–13. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–5. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 19.Hwang PM, Bunz F, Yu J, et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med. 2001;7:1111–17. doi: 10.1038/nm1001-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondoh H, Lleonart ME, Gil J, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–85. [PubMed] [Google Scholar]
- 21.Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 22.Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–3. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 23.Stambolic V, MacPherson D, Sas D, et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–25. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 24.Yeon JE, Califano S, Xu J, Wands JR, de la Monte SM. Potential role of PTEN phosphatase in ethanol-impaired survival signaling in the liver. Hepatology. 2003;38:703–14. doi: 10.1053/jhep.2003.50368. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee K, Mohr L, Wands JR, de la Monte SM. Ethanol inhibition of insulin signaling in hepatocellular carcinoma cells. Alcohol Clin Exp Res. 1998;22:2093–101. [PubMed] [Google Scholar]
- 26.Walker SM, Downes CP, Leslie NR. TPIP: a novel phosphoinositide 3-phosphatase. Biochem J. 2001;360 (Pt 2):277–83. doi: 10.1042/0264-6021:3600277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–8. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- 28.de la Monte SM, Yeon JE, Tong M, et al. Insulin resistance in experimental alcohol-induced liver disease. J Gastroenterol Hepatol. 2008;8(Pt 2):e477–86. doi: 10.1111/j.1440-1746.2008.05339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ronis MJ, Wands JR, Badger TM, de la Monte SM, Lang CH, Calissendorff J. Alcohol-induced disruption of endocrine signaling. Alcohol Clin Exp Res. 2007;31:1269–85. doi: 10.1111/j.1530-0277.2007.00436.x. [DOI] [PubMed] [Google Scholar]
- 30.Pang M, de la Monte S, Longato L, et al. PPARdelta agonist attenuates alcohol-induced hepatic insulin resistance and improves liver injury and repair. J Hepatol. 2009;50:1192–201. doi: 10.1016/j.jhep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moller DE, Berger JP. Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. Int J Obes Relat Metab Disord. 2003;27 (Suppl 3):S17–21. doi: 10.1038/sj.ijo.0802494. [DOI] [PubMed] [Google Scholar]
- 32.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43 (2 Suppl 1):S63–74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 33.de la Monte SM, Tong M, Cohen AC, Sheedy D, Harper C, Wands JR. Insulin and insulin-like growth factor resistance in alcoholic neurodegeneration. Alcohol Clin Exp Res. 2008;32:1630–44. doi: 10.1111/j.1530-0277.2008.00731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elwing JE, Lustman PJ, Wang HL, Clouse RE. Depression, anxiety, and nonalcoholic steatohepatitis. Psychosom Med. 2006;68:563–9. doi: 10.1097/01.psy.0000221276.17823.df. [DOI] [PubMed] [Google Scholar]
- 35.Karaivazoglou K, Assimakopoulos K, Thomopoulos K, et al. Neuropsychological function in Greek patients with chronic hepatitis C. Liver Int. 2007;27:798–805. doi: 10.1111/j.1478-3231.2007.01486.x. [DOI] [PubMed] [Google Scholar]
- 36.Kopelman MD, Thomson AD, Guerrini I, Marshall EJ. The Korsakoff syndrome: clinical aspects, psychology and treatment. Alcohol Alcohol. 2009;44:148–54. doi: 10.1093/alcalc/agn118. [DOI] [PubMed] [Google Scholar]
- 37.Loftis JM, Huckans M, Ruimy S, Hinrichs DJ, Hauser P. Depressive symptoms in patients with chronic hepatitis C are correlated with elevated plasma levels of interleukin-1beta and tumor necrosis factor-alpha. Neurosci Lett. 2008;430:264–8. doi: 10.1016/j.neulet.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perry W, Hilsabeck RC, Hassanein TI. Cognitive dysfunction in chronic hepatitis C: a review. Dig Dis Sci. 2008;53:307–21. doi: 10.1007/s10620-007-9896-z. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt KS, Gallo JL, Ferri C, et al. The neuropsychological profile of alcohol-related dementia suggests cortical and subcortical pathology. Dement Geriatr Cogn Disord. 2005;20:286–91. doi: 10.1159/000088306. [DOI] [PubMed] [Google Scholar]
- 40.Weiss JJ, Gorman JM. Psychiatric behavioral aspects of comanagement of hepatitis C virus and HIV. Curr HIV/AIDS Rep. 2006;3:176–81. doi: 10.1007/s11904-006-0013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de la Monte SM, Tong M, Lester-Coll N, Plater M, Jr, Wands JR. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis. 2006;10:89–109. doi: 10.3233/jad-2006-10113. [DOI] [PubMed] [Google Scholar]
- 42.Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J Alzheimers Dis. 2006;9:13–33. doi: 10.3233/jad-2006-9102. [DOI] [PubMed] [Google Scholar]
- 43.Lyn-Cook LE, Jr, Lawton M, Tong M, et al. Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. J Alzheimers Dis. 2009;16:715–29. doi: 10.3233/JAD-2009-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de la Monte SM, Longato L, Tong M, DeNucci S, Wands JR. The liver–brain axis of alcohol-mediated neurodegeneration: role of toxic lipids. Int J Environ Res Public Health. 2009;6:2055–75. doi: 10.3390/ijerph6072055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen AC, Tong M, Wands JR, de la Monte SM. Insulin and insulin-like growth factor resistance with neurodegeneration in an adult chronic ethanol exposure model. Alcohol Clin Exp Res. 2007;31:1558–73. doi: 10.1111/j.1530-0277.2007.00450.x. [DOI] [PubMed] [Google Scholar]
- 46.Arboleda G, Huang TJ, Waters C, Verkhratsky A, Fernyhough P, Gibson RM. Insulin-like growth factor-1-dependent maintenance of neuronal metabolism through the phosphatidylinositol 3-kinase-Akt pathway is inhibited by C2-ceramide in CAD cells. Eur J Neurosci. 2007;25:3030–8. doi: 10.1111/j.1460-9568.2007.05557.x. [DOI] [PubMed] [Google Scholar]
- 47.Bryan L, Kordula T, Spiegel S, Milstien S. Regulation and functions of sphingosine kinases in the brain. Biochim Biophys Acta. 2008;1781:459–66. doi: 10.1016/j.bbalip.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 49.Van Brocklyn JR. Sphingolipid signaling pathways as potential therapeutic targets in gliomas. Mini Rev Med Chem. 2007;7:984–90. doi: 10.2174/138955707782110123. [DOI] [PubMed] [Google Scholar]
- 50.Tong M, de la Monte SM. Mechanisms of ceramide-mediated neurodegeneration. J Alzheimers Dis. 2009;16:705–14. doi: 10.3233/JAD-2009-0983. [DOI] [PMC free article] [PubMed] [Google Scholar]