

Abstract
Neurofilament light (NFL) proteins in cerebrospinal fluid (CSF) are a marker of neuronal damage, especially subcortical axonal injury and white matter disease. Subjects with Alzheimer’s disease (AD) have shown elevated levels of CSF NFL as compared to controls. However, the presence of the APOE ε4 allele, an established risk factor for AD, was not found to associate with higher CSF NFL concentrations. We examined whether TOMM40 variants, which have been reported to influence age of onset of AD and are in linkage disequilibrium with APOE, have an effect on CSF NFL levels, in 47 healthy, cognitively intact individuals with or without APOE ε4. Our results show that the presence of APOE ε4 alone does not affect CSF NFL levels significantly; however APOE and TOMM40 appear to interact. Subjects with APOE ε4 have higher CSF NFL levels than non- ε4 carriers, only when they do not carry a short poly-T variant of TOMM40, which is associated with later age of onset of AD, and may act as protective against the dose effect of ε4.
Keywords: APOE, TOMM40, neurofilament light, cerebrospinal fluid, Alzheimer’s disease, memory performance, cortisol
1. Introduction
Neurofilaments are proteins that are found within the axons in neuronal cells, and play a critical role in maintaining morphological integrity of neurons and speed of impulses [1]; they are typically distinguished on the basis of their weight into heavy, medium and light. Axonal damage releases neurofilament proteins into interstitial fluid; these can be detected in the cerebrospinal fluid (CSF), which makes their determination a convenient marker of neuronal damage, as demonstrated by studies on boxing [2], amyotrophic lateral sclerosis [3], Alzheimer’s disease (AD) [4], and other neurological disorders [e.g., 5].
Neurofilaments are expressed predominantly in large caliber, myelinated axons [6], suggesting that elevated CSF levels of NFL reflect subcortical axonal injury and white matter disease, mostly consistent with subcortical dementia. However, elevated CSF concentrations of NFL have also been observed in other neurological disorders, compared to controls, including late- and early-onset AD, vascular dementia and frontotemporal dementia [e.g., 7–12]. In a recent meta-analysis, Petzold et al. [13] reviewed seven studies that compared 172 subjects with AD and 166 healthy age-matched controls, and found, overall, that CSF neurofilament light (NFL) values were higher for AD subjects than controls.
Since being originally proposed as a genetic marker for AD [14], the ε4 allele of the APOE gene has been established as the most robust susceptibility factor for this disease [15]. Despite this association, however, Sjögren et al. [16, 17] failed to observe any effect of APOE ε4 on CSF levels of NFL; these results suggest that the ε4 allele may not be implicated in subcortical axonal degeneration, despite its association with increased AD risk and, to a lower degree, with cerebrovascular disease [18].
Recently, a variable length deoxythymidine homopolymer (poly-T), rs10542523, in the TOMM40 gene, which is in linkage disequilibrium with APOE, was reported, in association with APOE, to influence risk of developing AD [19, 20]. Roses and colleagues [see also 21] demonstrated that APOE ε4 alleles are nearly exclusively linked to TOMM40 poly-T variants between 21 and 30 T residues in length (long variants; i.e., L), whereas APOE ε3 alleles, typically considered neutral for AD risk, can be linked to either short variants (20 or lower T residues in length; i.e., S) or very long variants (31 or over T residues in length; i.e., VL). In individuals carrying the ε3 allele, VL poly-T variants were found to associate with earlier onset age of AD than S poly-T variants. This introduces a potential modulating factor for AD risk and age of onset in the more than 65% of subjects in general population who do not carry the ε4 allele; VL poly-T variants may be a risk factor for AD in subjects who carry the “neutral” ε3 allele, whereas S poly-T variants may be risk protective.
Collectively, these findings, prompted us to examine whether TOMM40 poly-T variants might have an effect on CSF NFL concentrations, either in association with or independent of APOE alleles. We hypothesized that shorter TOMM40 poly-T variants, as they are linked to reduced risk for AD, should also be associated with lower CSF NFL concentrations than longer variants. As a secondary, exploratory aim, we examined whether CSF NFL levels correlated with other commonly used AD biomarkers (i.e., Aβ40, Aβ42, total tau and phosphorylated tau), with CSF cortisol levels, and with measures of memory performance. The present report complements previously reported studies in which we have examined CSF Aβ40, Aβ42, total (T) tau, phosphorylated (P) tau, and cortisol values in conjunction with APOE and TOMM40 poly-T variants [22, 23], therefore, these specific results will not be reported here.
Finally, as noted, CSF NFL is primarily a marker of subcortical axonal degeneration; AD is more commonly associated with cortical degeneration, but subcortical pathology is also present in AD. In fact, it has been reported that subcortical pathology – affecting areas such as the basal forebrain, which includes the nucleus basalis, and the brainstem nuclei, including the dorsal raphe nucleus and the locus coeruleus – is present at a very early stage in the continuum from old age to AD [24–26]. If TOMM40 poly-T variants, unlike APOE ε4, are found to influence CSF NFL levels, it is possible that the increased AD risk related to longer TOMM40 variants may reflect early subcortical, rather than cortical, degeneration.
2. Material and Methods
2.1 Subjects
This study was approved by the Institutional Review Boards of the Nathan S. Kline Institute for Psychiatric Research and the New York University School of Medicine. Participants in the study were volunteers recruited from the local communities who responded to advertisements in flyers and local newspapers. All subjects signed a consent form prior to being examined. Compensation of up to $450 was provided to the study participants. The total number of 133 participants completed the baseline visit; 51 of these subjects took part in the optional lumbar puncture procedure where CSF samples for indeces determination were collected. Out of these 51 subjects, a total of three subjects were excluded because they showed gross structural abnormalities (e.g., infarction and severe ventriculomegaly) in the MRI, and one had a Mini-Mental State Exam (MMSE) score below 28. These subjects were originally recruited for a study on the relationship between major depressive disorder (MDD) and Aβ levels, which will be reported separately. For this reason, 28 of the 47 remaining subjects were diagnosed with MDD, and 19 were controls. Group demographics are reported in Table 1.
Table 1.
Demographics reported by TOMM40 and APOE genotypes combination; means (SD) are reported for Age, Years of Education, and MMSE; gender is expressed in proportion of females; MDD is expressed in proportion of subjects with MDD. None of these differences are significant at the α = 0.05 level.
| With S poly-T | Without S poly-T | |||
|---|---|---|---|---|
|
|
||||
| ε4 positive (6) | ε4 negative (24) | ε4 positive (10) | ε4 negative (7) | |
|
| ||||
| Age | 66.2 (6.6) | 67.3 (6.5) | 68.3 (6.8) | 65.8 (5.0) |
| Education | 18.3 (3.2) | 16.2 (2.7) | 16.8 (2.2) | 16.0 (2.4) |
| MMSE | 29.7 (0.5) | 29.5 (0.7) | 29.7 (0.5) | 30.0 (0.0) |
| Gender | 33% | 42% | 50% | 71% |
| MDD | 67% | 50% | 70% | 71.4% |
Note: S poly-T = short TOMM40 poly-T. Education = Years of education.
2.2 NFL Determination
All CSF analyses were performed by certified laboratory technicians. All analyses were performed on fresh aliquots of CSF, avoiding extra freeze/thaw steps, and all CSF samples were analyzed in one batch on the same day. Two internal control samples (aliquots of pooled CSF) were analyzed on each plate, to assure reproducibility of the assays. CSF neurofilament light (NFL) protein was measured using a sandwich ELISA method (NF-light® ELISA kit, Uman Diagnostics AB, Umeå, Sweden). In short, the method is based on two monoclonal antibodies against NFL, and purified bovine NFL as standard. The level of quantification is between 50 and 10.000 ng/L.
2.3 Aβ and tau Determination
Quantities of CSF Aβ 40 and Aβ 42 were analyzed by electrochemiluminescence technology (Meso Scale Discovery, Gaithersburg, Maryland, USA), using the MS6000 Human Abeta Ultra-Sensitive Kit. Levels of CSF T tau were determined using a sandwich ELISA (Innotest hTAU-Ag, Innogenetics, Gent, Belgium) specifically constructed to measure all tau isoforms [27]. P tau was measured using a sandwich ELISA method (INNOTEST® PHOSPHO-TAU(181P), Innogenetics, Ghent, Belgium), as previously detailed [28].
2.4 Cortisol Determination
Levels of CSF Cortisol were determined by the Clinical Translational Research Laboratory at Emory University Hospital, Atlanta, GA. Cortisol determination was obtained by using a chemiluminescent immunoassay available on the Beckman Access analyzer and calibrated against the European GC/MS reference method.
2.5 Description of the APOE Genotyping
Five cc of whole blood for APOE genotyping and were collected and transferred into a cryogenic tube and frozen at −80° C until sent to the laboratory. APOE genotyping was performed at Dr. Frank Martiniuk’s laboratory (Molecular Core Lab, NYU-HHC CTSI). Genomic DNA samples were prepared from peripheral white blood cells. APOE genotype was determined by the standard PCR-RFLP methods using Hhal (Cfol) digestion of an APOE genomic PCR product spanning the polymorphic (cys/arg) sites at codon 112 and 158.
2.6 Description of the DNA extraction for determination of TOMM40
DNA was extracted from 2 ml of frozen whole blood and determination was also performed by Dr. Frank Martiniuk’s laboratory. After thawing, nucleated cells were separated from red cells by addition of 1 volume isotonic sucrose buffer and 3 volume water then placed on ice for 10 minutes, centrifuged for 10 minutes, and decanting supernatant. Cell pellet was washed with sucrose-water solution and re-centrifuged. Cell pellet was re-suspended in 0.5 ml low-TE with 25 ul of 20%SDS and proteinase K (10 mg/ml) and incubated at 50°C for 24 hrs. DNA was isolated with phenol; phenol-chloroform; chloroform and precipitated with sodium chloride-ethanol. DNA pellets were washed with 80% ethanol; dried under vacuum and re-suspended on 0.5 ml.
2.7 Description of the TOMM40 Polymorphic Assays
Rs10524523 poly-T length determination was performed by Polymorphic DNA Technologies, Inc. (Alameda, CA). The polymorphism rs10524523 is a homopolymer length polymorphism (polyT) located in an intronic region of TOMM40. For each genomic sample, PCR was used to amplify each polymorphism using 40 to 120 nanograms of genomic DNA per sample followed by bidirectional direct Sanger sequencing of the DNA templates on an ABI 3730xl sequencing platform and sequence data analysis. Details of the rs10524523 assay are found in Roses et al. [20].
2.8 Design and Analysis
The main purpose of this study was to test whether shorter TOMM40 poly-T variants influenced CSF NFL levels. The APOE and TOMM40 genotypes combinations per group are presented in Table 2. APOE had two levels: subjects in the ε4 carriers group had at least one ε4 allele (n= 16), whereas non-ε4 carriers had no ε4 alleles (n= 31). TOMM40 also had two levels: short carriers had at least one S allele (n= 30), whereas non-short carriers had no S allele (n= 17). To examine the effect of APOE and TOMM40 on CSF NFL, we conducted a 2 X 2, fully independent, analysis of variance (ANOVA). In addition, we also conducted an analogous ANOVA on age, years of education, MMSE, gender and MDD status to test for covariates. In case of significant covariates, we planned to employ an analysis of covariance (ANCOVA) to replace the ANOVA, including relevant covariates. To explore our secondary question, we conducted bivariate correlations between CSF NFL and CSF Aβ 40, Aβ 42, T tau, P tau, cortisol, and the Buschke Selective Reminding Task (BSRT) total and delayed recall scores, as measures of memory performance [29].
Table 2.
List of individual combinations of TOMM40 and APOE genotypes. Numbers in parentheses indicate number of subjects.
| With S poly-T | Without S poly-T | ||
|---|---|---|---|
|
| |||
| ε4 positive (6) | ε4 negative (24) | ε4 positive (10) | ε4 negative (7) |
|
| |||
| ε2/ε4, S-L (1) | ε2/ε3, S-L (1) | ε2/ε4, L-VL (1) | ε2/ε3, VL-VL (1) |
| ε2/ε4, S-VL (1) | ε2/ε3, S-VL (3) | ε3/ε4, L-L (1) | ε3/ε3, VL-VL (6) |
| ε3/ε4, S-L (1) | ε2/ε3, S-S (5) | ε3/ε4, L-VL (8) | |
| ε3/ε4, S-S (2) | ε3/ε3, S-VL (11) | ||
| ε3/ε4, S-VL (1) | ε3/ε3, S-S (4) | ||
Note: S poly-T = short TOMM40 poly-T.
2.9 Procedure
The study was conducted over four visits, generally each separated by one week. The first three visits were conducted at the Nathan Kline Institute, Orangeburg, NY and at the Clinical & Translational Science Institute, NYU Langone Medical Center. On visit 1, participants were explained the study procedures and informed of their rights; here they signed an informed consent form. Medical and psychiatric history and vital signs were also obtained at this stage. Participants also underwent a psychiatric evaluation, and global cognitive status was assessed using the MMSE and the Clinical Dementia Rating. Blood was also drawn for routine laboratory tests and for APOE and TOMM40 genotyping. During visit 2, subjects received an MRI scan of the head to quantify the magnitude of vascular brain pathology. At visit 3, participants underwent a comprehensive neuropsychological assessment, where the BSRT tests were administered. The BSRT constitutes a list of 16 nouns presented orally to the subject, at a rate of 2 seconds each. The subject is asked to recall as many words as possible and to indicate when no more can be recalled. Seven presentation and recall trials of the same list are given in immediate succession. After the initial presentation, the subject is presented only with those words that were not recalled on the immediately preceding recall trial, although they are asked to recall the entire list on each recall trial. Finally, after twenty minutes, a delayed recall test is also given in which the subject is asked to free recall as many words from the list as possible. One of four alternative versions of this test, using different word lists was assigned randomly to each participant. On the final visit (4), a lumbar puncture was performed by a neuroradiologist under fluoroscopy guidance. Subjects were asked to fast overnight prior to the lumbar puncture (LP) visit. After fasting, at 10 AM, 15 ml of clear CSF was collected into three polypropylene tubes using a fine 25G LP needle guided by fluoroscopy. Tubes were then immediately placed directly on ice for a maximum of 1 hour until samples were centrifuged at 4 degrees C at 1500 rpm for 10 minutes, then aliquots of 0.25 cc placed into 1.00 cc polypropylene cryogenic vials and labeled “A”, “B”, or “C”, and placed in Nunc 81-Cell Storage Boxes at −80 degrees C.
3 Results
The 2 X 2 ANOVAs did not show any significant effects of APOE, TOMM40 or an interaction on years of education, MMSE, gender and MDD status, therefore, we opted not to employ these variables as covariates in an ANCOVA to test the effects of APOE and TOMM40 variants on CSF NFL. In addition, MDD status had no effect on CSF NFL levels [t(45)=1.003, p=.322].
Although age did not show any significant effects of APOE, TOMM40 or an interaction, we found age to correlate significantly with CSF NFL [r=.444, p=.002], consistently with previous reports [e.g., 30]. As a consequence, we opted to include age as a covariate. We conducted a 2 X 2 ANCOVA on CSF NFL to test our primary hypothesis. The homogeneity-of-regression assumption was met, and results showed a significant interaction between APOE and TOMM40 [F(1,42)=4.735, p=.035], but no significant main effects (p>.05 for both APOE and TOMM40 main effects). However, the assumptions of normality (p=.008 for ε4-carriers) and homogeneity of variance were not met (p=.006). Therefore, we transformed the CSF NFL data by natural log and re-run the same ANCOVA on the transformed values; both assumptions were now met and the pattern of results was unchanged: the main effects did not reach significance, but the interaction did [F(1,42)=5.306, p=.026].
Post-hoc comparisons on untransformed data, conducted with Fisher’s LSD to correct for multiple testing, showed no significant simple effect of carrying the ε4 allele on CSF NFL concentrations when subjects also carried a S TOMM40 poly-T variant (p=.428) (Figure 1, left), whereas, we detected a significant simple effect of ε4 in subjects who did not carry a S poly-T variant (p=.018) (Figure 1, right). By comparing TOMM40 effects on CSF NFL in subjects with ε4, we also found significantly greater CSF NFL when the S-poly T was not present (i.e., subjects with L-L or L-VL) than for subjects with the S-poly T (S-S, S-L and S-VL) (p=.033; grey bars in Figure 1). No other post-hoc comparison reached significance. The same pattern of results was also found using transformed data.
Figure 1.

Left: mean CSF NFL concentrations in ng/L (and standard errors of the mean) in subjects with the S poly-T, as a function of APOE ε4. Right: mean CSF NFL concentrations in ng/L (and standard errors of the mean) in subjects without the S poly-T, as a function of APOE ε4. e4 positive refers to subjects with APOE ε4; e4 negative refers to subjects without APOE ε4. CSF NFL ranges are reported above each bar. ✩ = indicates a significant difference (α < .05).
Secondary analyses were bivariate correlations between CSF NFL and other CSF and cognitive indeces, conducted over the whole sample. With the exception of CSF Aβ 42 [p=.318], all other CSF indeces tested correlated with CSF NFL: CSF Aβ 40, r=.468, p=.001; CSF T tau, r=.477, p=.001; CSF P tau, r=.468, p=.001; cortisol, r=.500, p<.001. In addition, BSRT recall scores were negatively correlated with CSF NFL: Total Recall, r= −.423, p=.003 (Figure 2); Delayed Recall, r= −.314, p=.031. Finally, total length of the TOMM40 poly-T variants was not found to correlate significantly with CSF NFL, either in the whole sample, or in ε4-negatives, or in ε4-positives (lowest p value = .347).
Figure 2.
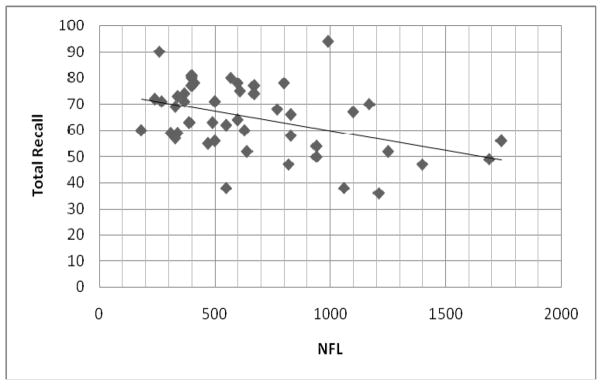
Significant reverse correlation (r = −.423; p = .003) between BSRT Total Recall performance (in number of words; y axis) and CSF NFL (in in ng/L; x axis).
4 Discussion
The quantitation of NFL proteins in CSF is an effective indirect measure of neuronal damage and higher levels of CSF NFL are commonly found in subjects with different neurological disorders, compared to controls, including AD [13]. However, so far no evidence had been reported that APOE genotype had any effect on CSF NFL concentrations, despite the APOE ε4 allele being an established genetic risk factor for AD and, to a lower degree, cerebrovascular disease. In our results, based on a sample of 47 healthy and cognitively intact elderly subjects, we confirm this finding and show that the presence of APOE ε4 alone does not affect CSF NFL levels significantly. However, importantly, our data show an interaction between the effects of APOE and of TOMM40 poly-T variants. TOMM40 is a gene in linkage disequilibrium with APOE, also found to affect AD risk; longer TOMM40 poly-T variants are associated with higher risk. Our results show that subjects with APOE ε4 have higher CSF NFL levels than non- ε4 carriers, only when they do not carry a S poly-T variant of TOMM40, which may act as protective against the effect of ε4. This set of results, if confirmed with a larger N, suggests that TOMM40 poly-T variants play a critical role in modulating APOE-dependent CSF NFL increase, thus reinforcing the notion that TOMM40 poly-T variants may be instrumental in affecting risk of AD, in conjunction with APOE genotype [e.g. 19, 20].
CSF NFL is primarily a marker of subcortical axonal degeneration. As noted, AD sufferers show early signs of subcortical pathology, which cannot be directly ascribed to the effect of APOE ε4, as this allele was not found to increase CSF levels of NFL by itself, either in previous reports [16, 17] or in our study. Our findings provide some preliminary evidence that TOMM40 poly-T variants, which we show to interact with APOE in affecting CSF NFL levels, might be involved with mechanisms that influence subcortical degeneration, in association with APOE ε4. Moreover, it has been argued [e.g. 24–26] that subcortical pathology occurs early in the process that leads from asymptomatic aging to AD; as our subjects were all cognitively intact and showed no signs of dementia, our results suggest that the genotyping for both APOE and TOMM40 may be useful for the detection of risk a long time before the appearance of symptoms of AD.
Our secondary analyses showed that, as expected, CSF NFL correlated with common CSF biomarkers of AD, such as Aβ 40, T and P tau; however, contrary to expectations, there was no correlation with Aβ 42. In addition, we observed CSF cortisol and NFL to correlate, providing direct support to the notion that higher levels of cortisol may be deleterious to neuronal substrate. Finally, the negative correlation between memory performance measures and CSF NFL provide further evidence that CSF NFL is an effective marker of subtle neuronal alterations even in cognitive intact elderly. Of note, nuclei involved in subcortical pathology, such as the nucleus basalis and locus coeruleus, all provide axonal projections to the hippocampus and other cortical areas, thus potentially affecting memory function in an indirect way.
MDD did not appear to affect levels of CSF NFL in our sample. This finding is inconsistent with the results from a previous report [31], where higher CSF NFL levels in 11 elderly women with MDD and no dementia were observed in comparison with 65 age-matched, cognitively-intact, healthy controls. A critical difference that might account for the discrepancy in the results is related to age; our groups were a few years younger (mean age ~ 67) than the subjects in [31] (mean age was ~ 74), thus suggesting MDD-related differences in neuronal degeneration might emerge later in life.
One limitation of our study was the small sample size with respect to each of the rs10524523 genotypes, which limited our analysis to testing whether the presence of a protective S poly-T influenced (specifically, in this case, decreased) CSF NFL levels and forced us to adopt an unbalanced study design, with different group sizes. In the future, studies with a larger N should overcome these limitations and examine in greater detail TOMM40 poly-T genotype sub-groups (e.g., L-L vs. L-VL), as defined by differences in poly-T length. Sample sizes should also be equal to avoid unbalanced designs, which may lead to sub-optimal interpretations of effects in ANOVAs, especially if the assumptions of normality and homogeneity of variance are not met [32]. In this respect, however, it should be noted that, once we transformed our data, these assumptions were met and the interaction between APOE and TOMM40 remained significant.
Future studies should also consider the enrollment of a majority of non-psychiatric subjects to represent the general population more accurately. In this respect, it should be noted that although we enrolled paid volunteers only, who were recruited from the local community through advertisements, which could have potentially introduced a selection bias, we believe that, if such a bias exists, it may be favorable to the scope of this study, as our enrollment strategy made it more likely for us to recruit cognitively-intact individuals.
Most evidence to date suggests that CSF levels of NFL reflect subcortical axonal injury and white matter disease, in contrast with CSF tau levels, which are more strongly linked to injurious processes in the cerebral cortex, such as AD [33]. The reason for this is that neurofilaments are expressed predominantly in large caliber, myelinated axons [6], whereas tau has a cortical expression pattern and is abundant in thin, unmyelinated axons [34]. This consideration and our results invite future studies to investigate whether TOMM40 variants should be considered as a candidate gene for subcortical dementia, in addition to AD.
5. Conclusions
The presence of the APOE ε4 allele alone does not affect CSF NFL levels significantly. However, we found that APOE and TOMM40 interact; subjects with APOE ε4 have higher CSF NFL levels than non- ε4 carriers, only when they do not carry a S poly-T variant of TOMM40. Our results confirm the importance of TOMM40 as a factor in influencing APOE ε4 related risk and support the idea that shorter poly-T variants may have a protective effect against neuronal degeneration.
Acknowledgments
This study was supported in part by an NIHM Grant (R01 MH080405) to NP. We wish to thank Drs Ann M. Saunders and Allen D. Roses, Deane Drug Discovery Institute and Department of Medicine, Duke University, Durham, NC, for supervising the TOMM40 polymorphic assays determination. We also wish to thank Dr Andrew Rutherford, Keele University, UK, for helping with the preparation of this manuscript.
Footnotes
The authors report no conflict of interest.
References
- 1.Hoffman PN, Cleveland DW, Griffin JW, Landes PW, Cowan NJ, Price DL. Neurofilament gene expression: a major determinant of axonal caliber. Proc Natl Acad Sci. 1987;84:3472–3476. doi: 10.1073/pnas.84.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zetterberg H, Hietala MA, Jonsson M, Andreasen N, Styrud E, Karlsson I, Edman A, Popa C, Rasulzada A, Blennow K, Wallin A. Neurochemical aftermath of amateur boxing. Arch Neurol. 2006;67:1277–1280. doi: 10.1001/archneur.63.9.1277. [DOI] [PubMed] [Google Scholar]
- 3.Rosengren LE, Karlsson JE, Karlsson JO, Perrson LI, Wikkelso C. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. 1996;67:2013–2018. doi: 10.1046/j.1471-4159.1996.67052013.x. [DOI] [PubMed] [Google Scholar]
- 4.Hu YY, He SS, Wang XC, Duan QH, Khatoon S, Iqbal K, Grundke-Iqbal I, Wang JZ. Elevated levels of phosphorylated neurofilament proteins in cerebrospinal fluid of Alzheimer disease patients. Neurosci Lett. 2002;320:156–160. doi: 10.1016/s0304-3940(02)00047-2. [DOI] [PubMed] [Google Scholar]
- 5.Norgren N, Rosengren L, Stigbrand T. Elevated neurofilament levels in neurological diseases. Brain Res. 2003;987:25–31. doi: 10.1016/s0006-8993(03)03219-0. [DOI] [PubMed] [Google Scholar]
- 6.Friede RL, Samorajski T. Axon caliber related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec. 1970;167:379–387. doi: 10.1002/ar.1091670402. [DOI] [PubMed] [Google Scholar]
- 7.Bjerke M, Andreasson U, Rolstad S, Nordlund A, Lind K, Zetterberg H, Edman A, Blennow K, Wallin A. Subcortical vascular dementia biomarker pattern in mild cognitive impairment. Dement Geriatr Disord. 2009;28:348–356. doi: 10.1159/000252773. [DOI] [PubMed] [Google Scholar]
- 8.de Jong D, Jansen RWMM, Pijnenburg YAL, van Geel WJA, Borm GF, Kremer HPH. CSF neurofilament proteins in the differential diagnosis of dementia. J Neurol Neurosurg Psychiatry. 2007;78:936–938. doi: 10.1136/jnnp.2006.107326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pijnenburg YAL, Janssen JC, Schoonenboom NSM, Petzold A, Mulder C, Stigbrand T, Norgren N, Heijst H, Hack CE, Scheltens P, Teunissen CE. CSF neurofilaments in frontotemporal dementia compared with early onset Alzheimer’s disease and controls. Dement Geriatr Cogn Disord. 2007;23:225–230. doi: 10.1159/000099473. [DOI] [PubMed] [Google Scholar]
- 10.Rosengren LE, Karlsson JE, Karlsson JO, Persson LI, Wikkelso C. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J Neurochem. 1996;67:2013–2018. doi: 10.1046/j.1471-4159.1996.67052013.x. [DOI] [PubMed] [Google Scholar]
- 11.Rosengren LE, Karlsson JE, Sjogren M, Blennow K, Wallin A. Neurofilament protein levels in CSF are increased in dementia. Neurol. 1999;52:1090–1093. doi: 10.1212/wnl.52.5.1090. [DOI] [PubMed] [Google Scholar]
- 12.van Eijk JJJ, van Everbroeck B, Abdo WF, Kremer BPH, Verbeek MM. CSF neurofilament proteins levels are elevated in sporadic Creutzfeldt-Jakob disease. J Alzheimers Dis. 2010;21:569–576. doi: 10.3233/JAD-2010-090649. [DOI] [PubMed] [Google Scholar]
- 13.Petzold A, Keir G, Warren J, Fox N, Rossor MN. A systematic review and meta-analysis of CSF neurofilament protein levels as biomarkers in dementia. Neurodegenerative Dis. 2007;4:185–194. doi: 10.1159/000101843. [DOI] [PubMed] [Google Scholar]
- 14.Corder EH, Saunders AM, Strittmater WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 15.Blennow K, DeLeon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 16.Sjogren M, Blomberg M, Jonsson M, Wahlund LO, Edman A, Lind K, Rosengren L, Blennow K, Wallin A. Neurofilament protein in cerebrospinal fluid: a marker of white matter changes. J Neurosci Res. 2001;66:510–516. doi: 10.1002/jnr.1242. [DOI] [PubMed] [Google Scholar]
- 17.Sjogren M, Rosengren L, Minthon L, Davidsson P, Blennow K, Wallin A. Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurol. 2000;54:1960–1964. doi: 10.1212/wnl.54.10.1960. [DOI] [PubMed] [Google Scholar]
- 18.Jones EL, Kalaria RN, Sharp SI, O’Brien JT, Francis PT, Ballard CG. Genetic associations of autopsy-confirmed vascular dementia subtypes. Dement Geriatr Cogn Disord. 2011;31:247–253. doi: 10.1159/000327171. [DOI] [PubMed] [Google Scholar]
- 19.Caselli RJ, Saunders AM, Lutz MW, Huentelman MJ, Reiman EM, Roses AD. TOMM40, APOE, and age of onset of Alzheimer’s disease. Poster session presented at the Alzheimer’s Association International Conference on Alzheimer’s Disease; Honolulu, HI. Jul, 2010. [Google Scholar]
- 20.Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2009;10:375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutz MW, Crenshaw DG, Saunders AM, Roses AD. Genetic variation at a single locus and age of onset for Alzheimer’s disease. Alzheimers Dement. 2010;6:125–131. doi: 10.1016/j.jalz.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bruno D, Nierenberg JJ, Ritchie JC, Lutz MW, Pomara N. CSF cortisol concentrations in healthy elderly are affected by both APOE and TOMM40. Psychoneuroendocrinology. doi: 10.1016/j.psyneuen.2011.07.006. Under Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pomara N, Bruno D, Nierenberg JJ, Sidtis JJ, Martiniuk FT, Mehta PD, Zetterberg H, Blennow K. TOMM40 poly-T variants and CSF amyloid beta levels in the elderly. Neurochem Res. 2011;36:1124–1128. doi: 10.1007/s11064-011-0459-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geula C, Nagykery N, Nicholas A, Wu CK. Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer’s disease. J Neuropathol Exp Neurol. 2008;67:309–318. doi: 10.1097/NEN.0b013e31816a1df3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grinberg LT, Rüb U, Ferretti REL, Farfel JM, Polichiso L, Gierga K, Jacob-Filho W, Heinsen H Brazilian Brain Bank Study Group. The dorsal raphe nucleus shows phosphor-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathology and Applied Neurobiology. 2009;35:406–416. doi: 10.1111/j.1365-2990.2009.00997.x. [DOI] [PubMed] [Google Scholar]
- 26.Mesulam M, Shaw P, Mash D, Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol. 2004;55:815–828. doi: 10.1002/ana.20100. [DOI] [PubMed] [Google Scholar]
- 27.Blennow K, Wallin A, Ågren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical diagnostic marker for axonal degeneration in Alzheimer’s disease? Mol Chem Neuropathology. 1995;26:231–245. doi: 10.1007/BF02815140. [DOI] [PubMed] [Google Scholar]
- 28.Vanmechelen E, Vanderstichele H, Davidsson P, Van Kerschaver E, Van der Perre B, Sjögren M, Andreasen N, Blennow K. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett. 2000;285:49–52. doi: 10.1016/s0304-3940(00)01036-3. [DOI] [PubMed] [Google Scholar]
- 29.Buschke H. Retrieval in human learning. Trans NY Acad Sci. 1974;36:721–729. [Google Scholar]
- 30.Constantinescu R, Zetterberg H, Holmberg B, Rosengren L. Levels of brain related proteins in cerebrospinal fluid: An aid in the differential diagnosis of parkinsonian disorders. Parkinsonism Relat Disord. 2009;15:205–212. doi: 10.1016/j.parkreldis.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Gudmundsson P, Skoog I, Waern M, Blennow K, Zetterberg H, Rosengren L, Gustafson D. Is there a CSF biomarker profile related to depression in elderly women? Psychiatry Res. 2010;176:174–178. doi: 10.1016/j.psychres.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 32.Rutherford A. ANOVA and ANCOVA: A GLM approach. Wiley; Hoboken, NJ: 2011. [Google Scholar]
- 33.Blennow K. Dementia in 2010: Paving the way for Alzheimer disease drug development. Nature Rev Neurol. 2011;7:65–66. doi: 10.1038/nrneurol.2010.214. [DOI] [PubMed] [Google Scholar]
- 34.Trojanowski JQ, Schuck T, Schmidt ML, Lee VM. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem. 1989;37:209–215. doi: 10.1177/37.2.2492045. [DOI] [PubMed] [Google Scholar]