

Abstract
Background
Haemophilus influenzae is an opportunistic bacterial pathogen that exclusively colonises humans and is associated with both acute and chronic disease. Despite its clinical significance, accurate identification of H. influenzae is a non-trivial endeavour. H. haemolyticus can be misidentified as H. influenzae from clinical specimens using selective culturing methods, reflecting both the shared environmental niche and phenotypic similarities of these species. On the molecular level, frequent genetic exchange amongst Haemophilus spp. has confounded accurate identification of H. influenzae, leading to both false-positive and false-negative results with existing speciation assays.
Results
Whole-genome single-nucleotide polymorphism data from 246 closely related global Haemophilus isolates, including 107 Australian isolate genomes generated in this study, were used to construct a whole-genome phylogeny. Based on this phylogeny, H. influenzae could be differentiated from closely related species. Next, a H. influenzae-specific locus, fucP, was identified, and a novel TaqMan real-time PCR assay targeting fucP was designed. PCR specificity screening across a panel of clinically relevant species, coupled with in silico analysis of all species within the order Pasteurellales, demonstrated that the fucP assay was 100 % specific for H. influenzae; all other examined species failed to amplify.
Conclusions
This study is the first of its kind to use large-scale comparative genomic analysis of Haemophilus spp. to accurately delineate H. influenzae and to identify a species-specific molecular signature for this species. The fucP assay outperforms existing H. influenzae targets, most of which were identified prior to the next-generation genomics era and thus lack validation across a large number of Haemophilus spp. We recommend use of the fucP assay in clinical and research laboratories for the most accurate detection and diagnosis of H. influenzae infection and colonisation.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1857-x) contains supplementary material, which is available to authorized users.
Keywords: Haemophilus influenzae, Haemophilus haemolyticus, NTHi, Genomics, fucP, PCR, Real-time PCR, Species, TaqMan, Assay
Background
The Gram-negative Haemophilus spp. bacteria comprise a diverse group containing at least 12 currently recognised species, all of which are commensal or pathogenic to humans or animals. Haemophilus influenzae is the best-known member of this genus, particularly serotybe b (Hib), the leading cause of invasive bacterial disease in children prior to the introduction of the first licensed Hib conjugate vaccine in 1987 [1]. In regions where Hib vaccination has been implemented, the spectrum of severe Hib disease is now close to eradication [2]. Other H. influenzae serotypes (a; c-f), and nonencapsulated, “nontypeable” H. influenzae (NTHi), which are not targeted by the Hib vaccine, are now recognised as important causes of primarily mucosal acute and chronic infections [3]. NTHi is a common coloniser of the upper respiratory tract in healthy individuals but can cause otitis media, conjunctivitis, sinusitis, and lower respiratory infections in children, exacerbations of chronic obstructive pulmonary disease (COPD) and cystic fibrosis (CF) in adults, and sepsis in neonates and immunocompromised adults [4]. Although far less common than H. influenzae, other Haemophilus species also have the potential to cause human disease including H. haemolyticus, H. parainfluenzae, H. aegyptius (a biogroup of H. influenzae), H. pittmaniae, H. parahaemolyticus and H. paraphrohaemolyticus [5–14].
Misidentification of near-neighbour Haemophilus species as H. influenzae has broad-ranging implications for clinical diagnosis, reported carriage rates and assessment of disease outcomes from antibiotic or vaccine clinical trials. Microbiological differentiation of H. influenzae from other species has conventionally relied upon colonial morphology, haemin and NAD (X and V factor) dependence, and for capsular strains, serotyping using various methods [15]. For NTHi, identification relies on the absence of capsule and is thus more challenging than capsulated H. influenzae. In 2007, Murphy and colleagues were the first to report the misidentification of non-haemolytic strains of H. haemolyticus as NTHi [16]. These strains are phenotypically indistinguishable from NTHi and represent the only other Haemophilus spp. for which X and V factor dependence is a diagnostic criterion.
Numerous genetic methods for discriminating H. influenzae from other species have been described [16–23]. However, accurate delineation of H. influenzae from other Haemophilus species using genetic methods has proven challenging. Recombination between H. influenzae and other Haemophilus spp., particularly H. haemolyticus [17, 22, 24], has confounded molecular speciation attempts, especially in the absence of genomic data. Binks and colleagues [22] recently assessed the ability of a number of published and novel PCR-based methods to discriminate NTHi from closely-related Haemophilus species compared with recA and 16S rRNA gene sequencing, and reported that an assay targeting sequence diversity within the hpd gene, which encodes for Protein D, was superior to other molecular signatures. Additionally, the hpd#3 assay was specific for H. influenzae when compared against a panel of common respiratory bacteria and has been used to quantify H. influenzae directly from clinical specimens [22]. However, we recently reported the absence of hpd in a proportion of H. influenzae isolates, which was only identified following whole-genome sequencing analysis of 20 NTHi isolates [25]. This finding highlights both the limitation of hpd for H. influenzae detection and the requirement for genomic data spanning a comprehensive Haemophilus dataset to identify a “gold standard” molecular signature.
Here, we describe a large-scale comparative genomics approach comprising 246 closely related, global Haemophilus spp. isolates to identify loci unique to H. influenzae. One of these loci, fucP, was used to develop a real-time PCR assay targeting H. influenzae. PCR screening of the fucP assay across 59 genome-sequenced Australian Haemophilus spp. isolates and 35 clinically relevant species demonstrated 100 % specificity towards H. influenzae.
Methods
Ethics statement
Whole-genome analysis of the isolates in this study was covered by the Human Research Ethics Committee of the Northern Territory Department of Health and Menzies School of Health Research, approval numbers 07/63 and 07/85, and the Princess Margaret Hospital for Children Ethics Committee, approval number 1295/EP.
Bacterial isolates
A total of 511 isolates were examined in this study, the majority of which were Haemophilus spp. (Table 1). Haemophilus isolates originated from a wide range of clinical sites, clinical conditions and geographic regions. Samples were obtained from nasopharyngeal swabs, sputum, bronchoalveolar lavage, throat, or blood specimens, and include isolates sourced from either healthy carriers or cases of otitis media, bronchiectasis, protracted bacterial bronchitis, chronic obstructive pulmonary disease and bacteraemia.
Table 1.
Bacterial isolates and genomic data used in this study
| Species | No. strains (no. with genome data) |
|---|---|
| Haemophilus influenzae, incl. NTHi and biogroup aegyptius a | 338 (201) |
| Haemophilus haemoglobinophilus | 1 (1) |
| Haemophilus haemolyticus b | 107 (32) |
| Haemophilus parahaemolyticus | 2 (1) |
| Haemophilus parainfluenzae | 5 (4) |
| Haemophilus paraphrohaemolyticus | 1 (1) |
| Haemophilus sp. (novel “fuzzy” species) | 17 (13) |
| Achromobacter sp. | 1 |
| Aggregatibacter (formerly Haemophilus) aphrophilus | 1 |
| Alcaligenes sp. | 1 |
| Alloiococcus otitidis | 1 |
| Bacillus sp. | 1 |
| Bacteroides fragilis | 1 |
| Burkholderia cenocepacia | 1 |
| Burkholderia cepacia | 1 |
| Burkholderia diffusa | 1 |
| Burkholderia multivorans | 1 |
| Burkholderia pseudomallei | 1 |
| Burkholderia pyrrocinia | 1 |
| Burkholderia thailandensis | 1 |
| Burkholderia vietnamiensis | 1 |
| Burkholderia spp. | 2 |
| Chromobacterium violaceum | 1 |
| Chryseobacterium sp. | 1 |
| Comamonas sp. | 1 |
| Cupriavidus spp. | 2 |
| Delftia sp. | 1 |
| Escherichia coli | 1 |
| Klebsiella pneumoniae | 1 |
| Moraxella catarrhalis | 1 |
| Neisseria meningitidis | 1 |
| Pandoraea sp. | 1 |
| Pigmentiphaga sp. | 1 |
| Proteus mirabilis | 1 |
| Pseudomonas aeruginosa | 2 |
| Ralstonia mannitolilytica | 1 |
| Staphylococcus aureus | 2 |
| Staphylococcus epidermidis | 1 |
| Stenotrophomonas sp. | 1 |
| Streptococcus mitis | 1 |
| Streptococcus pneumoniae | 1 |
| Streptococcus pyogenes | 2 |
| TOTAL | 511 (253) |
aSpecies determined according to a positive fucP assay result and/or by whole-genome sequencing
bSpecies determined by a negative fucP result, a positive H. haemolyticus hpd HRM result [27], and/or by whole-genome sequencing
Two-hundred and forty-six closely related Haemophilus isolates (H. influenzae, H. haemolyticus and a novel ‘fuzzy’ Haemophilus species) had whole-genome data available for this study (Fig. 1; Additional file 1). Our dataset included 87 unique global NTHi isolates from Brazil, China, Czech Republic, Finland, Ghana, Iceland, Malaysia, Papua New Guinea, South Africa, South Korea, Spain, Sweden, United Kingdom and USA [26] that were recently deposited into the European Nucleotide Archive database (http://www.ebi.ac.uk/ena/). The current study generated a further 107 Australian Haemophilus spp. genomes (Additional file 1). The remaining 52 genomes were downloaded from the NCBI public Nucleotide data repository (http://www.ncbi.nlm.nih.gov/nucleotide/) or the Sequence Read Archive database (http://www.ncbi.nlm.nih.gov/sra). Amongst the 246 genomes were 201 H. influenzae (comprising 186 NTHi, 11 capsulated strains, three Biogroup aegyptius strains and one strain with unspecified capsular status) and 32 H. haemolyticus isolates. Thirteen isolates from our Australian laboratories that possessed X and V factor dependence but could not be definitively speciated by whole-genome sequencing were classed as Haemophilus spp. (Additional file 1). Two-hundred and twelve additional Australian Haemophilus spp. designated as H. influenzae or H. haemolyticus using the hpd PCR high-resolution melt (HRM) assay [27], one each of H. parahaemolyticus and H. parainfluenzae, and 40 non-Haemophilus isolates (Table 1) were tested by fucP PCR only.
Fig. 1.
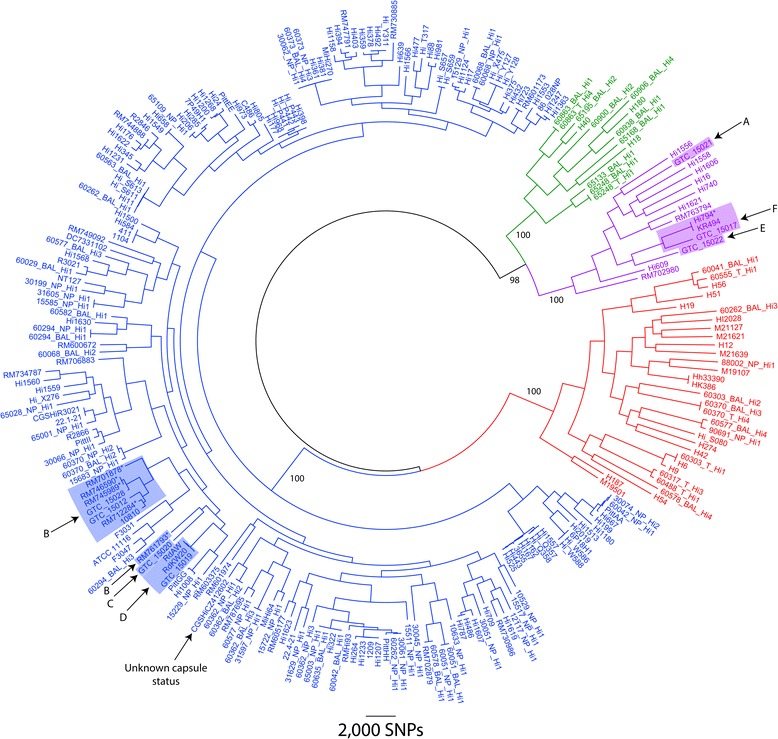
Genome-based differentiation of Haemophilus influenzae from closely-related species. A midpoint-rooted maximum parsimony phylogeny was constructed using genomic data from 246 global, closely related Haemophilus spp. isolates, 107 of which were Australian Haemophilus isolates sequenced in the present study. Phylogenetic reconstruction of 63,447 orthologous, core genome, bi-allelic single-nucleotide polymorphisms (SNPs) enabled differentiation of nontypeable and serotypeable H. influenzae (blue text) from H. haemolyticus (red text) and other “fuzzy” Haemophilus species (green text). ‘Clade I’ H. influenzae, which are genetically distinct from other H. influenzae [26], are denoted by purple text. NTHi strains encoding capsular loci that are not expressed (according to [26]) are denoted by an asterisk. The H. haemolyticus and “fuzzy” strains share the same ecological niches as H. influenzae and are indistinguishable from H. influenzae based on morphological characteristics including X and V factor dependency. Bootstrap values are shown for major branches. Consistency index = 0.14. NB. More distantly related Haemophilus species (H. haemoglobinophilus, H. parahaemolyticus, H. parainfluenzae and H. paraphrohaemolyticus) were excluded to maximise core genome size
Phenotypic selection of the Australian Haemophilus spp. isolates was undertaken prior to molecular speciation. Only clinically-derived isolates that were both X and V factor-dependent and that failed to react with capsular antisera using the Phadebact® Haemophilus coagglutination test (MKL Diagnostics, Sweden) were selected for further analysis. Of the 107 Australian Haemophilus isolates that underwent genome sequencing, 18 were not speciated by molecular methods, 15 were speciated using 16S rDNA sequencing [16], and the remainder underwent molecular speciation using hpd-based methods [22, 27].
Isolates were subcultured for purity through a minimum of three passages prior to DNA extraction. The Qiagen DNeasy kit (Qiagen, Doncaster, VIC, Australia) was used for DNA extraction according to the manufacturer’s instructions, with enzymatic pre-treatment as described previously [28]. DNA was quality-checked for purity and extraction efficiency using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Scoresby, VIC, Australia). All DNA samples were diluted 1:100 in molecular-grade H2O prior to PCR.
H. influenzae whole-genome sequencing
Paired-end genomic data for the Australian isolates were generated using the HiSeq or MiSeq platforms (Illumina, Inc., San Diego, CA, USA), and were sequenced by Macrogen Inc. (Geumcheon-gu, Seoul, Republic of Korea). The comparative genomics pipeline SPANDx v2.6 [29] was used to analyse the Haemophilus genomes (Additional file 1). Input data for SPANDx were already in paired-end Illumina format, except for publicly available reference genomes, which were first converted to synthetic paired-end Illumina reads with ART (version VanillaIceCream) [30] using quality shifts of 10. The closed NTHi 86-028NP genome [31] was used as the reference for short-read alignment mapping. Synthetic reads for 86-028NP were included as a control.
Phylogenetic analysis
Species boundaries for H. influenzae and H. haemolyticus were established a posteriori following phylogenetic reconstruction of 63,447 high-confidence orthologous, core genome, bi-allelic single-nucleotide polymorphisms (SNPs) identified across 246 closely related Haemophilus genomes. Genomes from more distantly related species (H. parainfluenzae, H. haemoglobinophilus, H. parahaemolyticus and H. paraphrohaemolyticus) were excluded from this analysis to maximise the core genome size. No additional SNP filtering (e.g. to exclude recombined regions) was performed. Maximum parsimony trees were generated using PAUP* 4.0b10 [32]; bootstrapping was based on 200 replicates. Trees were visualised using FigTree v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/).
Identification of H. influenzae-specific signatures
We deliberately chose not to pursue SNPs for H. influenzae speciation due to the high risk of eventually encountering a homoplastic event [33]. This risk is greatly increased in highly recombinogenic species like H. influenzae. Instead, we aimed to identify discrete, H. influenzae-specific genetic loci for assay design. To identify such loci, the coverageBed module of BEDTools v2.18.2 [34], which is part of the SPANDx pipeline, was applied across the 246 closely related Haemophilus genomes as described previously [35], using default settings. This tool performs presence/absence analysis of Illumina reads for all genomes against the reference genome. MS Excel 2013 was used to visualise presence/absence outputs. Candidate H. influenzae-specific loci were identified by filtering for regions with 100 % read coverage in the target species but with <50 % coverage in outgroup strains. Using this approach, three candidate loci ≥4 kb were identified (Table 2). One locus, fucP (NTHI0865 in 86-028NP), which encodes L-fucose permease, was chosen for real-time PCR assay design as this region was highly conserved amongst the 201 H. influenzae genomes. The coverageBED output was also used to assess presence/absence of the following previously reported H. influenzae-specific targets: fucK (NTHI0870 in 86-028NP [18]), hap (NTHI0354 [18]), hpd (NTHI0811 [21, 22, 27]), iga (NTHI1164 [17]), lgtC (NTHI0365 [17]), ompP2 (NTHI0225 [36]) and ompP6 (NTHI0501 [37]).
Table 2.
Haemophilus influenzae-specific loci
| Qualifiersa | Size (kb)b | Encoded genes (putative function) |
|---|---|---|
| 824000..828000c | 4 | fucP, fucA, fucU, fucK, fucI (partial) (fucose transport and degradation) |
| 829000..830000 | 1 | fucI (partial), fucR (fucose transport and degradation) |
| 1617000..1623000 | 6 | pstS, pstC, pstA, pstB, phoB, phoR (phosphate binding and regulation) |
| 1795000..1796000 | 1 | pdxS, dpxT (glutamine metabolism) |
H. influenzae real-time PCR assay
The fucP assay was used to complement our hpd results due to the recent observation that some Australian H. influenzae strains lack full-length hpd, and can therefore be erroneously genotyped with this assay [25]. Unlabelled primers fucP-F (5′-GCCGCTTCTGAGGCTGG) and fucP-R (5′-AACGACATTACCAATCCGATGG) (Sigma-Aldrich, Castle Hill, NSW, Australia) were designed to generate a 68-bp fragment. A TaqMan probe (fucP-Probe: 5′-6FAM TCCATTACTGTTTGAAATAC-MGBNFQ; Life Technologies, Grand Island, NY, USA) was included to increase specificity and to provide a “gold standard” PCR methodology for clinical specimens. Microbial discontiguous MegaBLAST analysis (http://blast.ncbi.nlm.nih.gov/; analysis performed 28-Dec-14) of the H. influenzae fucP amplicon across 3,546 complete microbial genomes, 10,247 Proteobacterial draft genomes and 1,734 complete plasmid genomes (total: 15,527 genomes) confirmed locus specificity, with a 100 % match in all H. influenzae (including biogroup aegyptius) at both primer- and probe-binding sites, and several primer and probe mismatches in the next closest species match (the avian pathogen Avibacterium paragallinarum [38]; two nucleotide mismatches in the forward primer, four mismatches in the reverse primer and one mismatch in the TaqMan probe). No other Haemophilus spp. yielded a detectable BLAST result for the fucP amplicon, indicating the absence of this locus in other species.
Real-time PCRs were performed using the RotorGene 6000 (Qiagen, Chadstone, VIC, Australia) and ABI PRISM 7900HT (Life Technologies, Mulgrave, VIC, Australia) platforms. Each reaction contained 0.25 μM of each primer, 0.1 μM of probe and 1 μL genomic DNA. For the RotorGene 6000 instrument, 1X Platinum PCR SuperMix (Life Technologies) was used to a total reaction volume of 10 μL. For the 7900HT instrument, 1X TaqMan Universal Master Mix (Life Technologies) and 384-well plates were used, enabling 5 μL reaction volumes. The 317 isolates tested by PCR in this study (Table 1; includes 46 H. influenzae and 13 H. haemolyticus that were also subjected to whole-genome sequencing) were assessed in duplicate, and all runs contained appropriate positive control and no-template control reactions. For both instruments, thermocycling was carried out as follows: 50 °C for 2 min, 95 °C for 10 min, followed by 45 cycles of 95 °C for 5 sec (15 sec for the ABI PRISM) and 60 °C for 5 sec (1 min for the ABI PRISM). The green/FAM channels were used for fluorescence detection.
Results and discussion
This study is the first to use extensive whole-genome sequence data from global Haemophilus isolates to identify and design a highly accurate molecular assay targeting H. influenzae. Based on microbiological characteristics alone, H. influenzae, and particularly NTHi, cannot always be differentiated from non-haemolytic H. haemolyticus [16] or closely related “fuzzy” Haemophilus species. Molecular methods are therefore essential for accurate identification of H. influenzae. However, assay design has conventionally been thwarted by high levels of recombination between H. influenzae and other Haemophilus species, and has even been documented between Haemophilus and Neisseria meningitidis [39]. Compounding this issue is the lack of rigorous, comparative in silico analysis of putative molecular signatures using large-scale whole-genome sequence data. These inherent obstacles with accurate H. influenzae speciation have likely led to underreporting of false-positive and false-negative results for this clinically important bacterium [16, 22].
To address this issue, we combined whole-genome data generated for 107 Australian strains by our laboratory with all closely related Haemophilus spp. genome data available in the public domain, including 87 unique NTHi genomes from De Chiara et al. [26], to identify a highly specific signature for H. influenzae. Species boundaries were first established a posteriori using phylogenetic analysis of 246 Haemophilus genomes (Fig. 1). Based on this analysis, 201 of these strains were identified as H. influenzae, 32 as H. haemolyticus and 13 as possible novel “fuzzy” Haemophilus species (Fig. 1). This phylogeny was highly similar to a recent whole-genome phylogeny constructed using 97 predominantly NTHi strains [26]. Amongst the 107 Australian Haemophilus spp. isolates subjected to whole-genome sequencing, 89 had prior speciation data by PCR-based methods [16, 22, 27]. Interestingly, the genome phylogeny reassigned two NTHi isolates that had previously been identified by 16S rDNA PCR (n = 1) [16] or hpd#3 PCR (n = 1) [22] as H. haemolyticus, and reassigned 10 NTHi, one H. haemolyticus, and two equivocal isolates as a potentially novel “fuzzy” Haemophilus species.
Following species delineation on a whole-genome SNP level, loci specific to H. influenzae were located. Core genome analysis of the 201 H. influenzae genomes found that 936 kb was conserved amongst all H. influenzae strains, represented by 100 % read coverage across all H. influenzae genomes; however, across the larger 246 Haemophilus genome dataset, only a minute fraction (12 kb) was unique to H. influenzae (Table 2). This very low prevalence of H. influenzae-specific loci exemplifies the inherent difficulties in molecular speciation of this bacterium, particularly in lieu of whole-genome data. Four loci, ranging from 1 to 6 kb in size, were identified as H. influenzae-specific (Table 2). A 4 kb locus, part of a fucose transport and degradation operon, was selected for assay design due to high sequence conservation across all H. influenzae isolates. Within this locus we targeted the L-fucose permease-encoding gene, fucP [40]. L-fucose permease is a pH-dependent major facilitator superfamily transporter that uptakes L-fucose, a substrate that can act as a sole carbon source for bacteria [41]. In the human host, the fucose operon may impart a competitive advantage and virulence potential to H. influenzae, as has been documented in Campylobacter jejuni [42].
Following its design and in silico validation (as detailed in Methods), the fucP TaqMan real-time PCR assay was screened for specificity against 35 bacterial species (comprising 40 isolates) of clinical relevance (Table 1). As expected, only H. influenzae amplified using the fucP assay. A further selection of 212 nasopharyngeal, bronchoalveolar lavage and throat isolates, previously designated H. influenzae or H. haemolyticus using the hpd PCR high-resolution melt (HRM) assay [27], were screened using the fucP assay; 124/137 (91 %) hpd-defined H. influenzae isolates and 0/75 hpd-defined H. haemolyticus isolates amplified with the fucP assay. Subsequent genomic analysis of 10 of the 14 presumptive NTHi isolates that failed fucP PCR confirmed they are neither H. influenzae nor H. haemolyticus, but rather represent a possible novel “fuzzy” Haemophilus species (Fig. 1, green text). An additional three Haemophilus isolates (H18, H40 and H180) not screened with the hpd HRM assay also grouped with this “fuzzy” Haemophilus species based on whole-genome phylogenetic analysis. The remaining four isolates were not whole-genome sequenced in this study, but we suspect that they will also group with the “fuzzy”, fucP-negative, hpd HRM H. influenzae-positive isolates. We plan to genome-sequence these strains in the future to confirm their phylogenetic placement.
Several H. influenzae-specific molecular targets have been reported previously and include fucK, hap, hpd, iga, lgtC, ompP2 and ompP6. In the current study, we compared the presence/absence profiles of these loci across our Haemophilus genome dataset. The fucP and fucK loci were best at differentiating H. influenzae from closely-related species, demonstrating 100 % specificity towards H. influenzae. The lgtC and iga loci grouped the closely related “fuzzy” Haemophilus isolates with H. influenzae, and were absent in H. haemolyticus. The hap, hpd and omp loci were least effective at differentiating H. influenzae from near-neighbour species, with potential false-positives and negatives for hpd, hap and ompP2, and false-positives for ompP6 (Fig. 2).
Fig. 2.
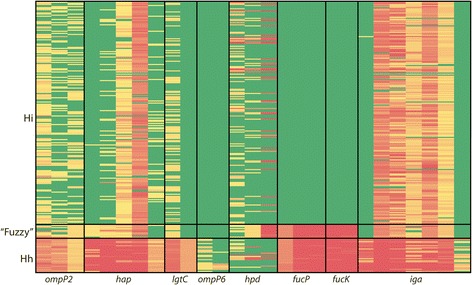
Genomic comparison of fucP with existing Haemophilus influenzae-specific targets. Red, <50 % Illumina paired-end read coverage across a 1 kb locus window; yellow, between 50 and 99 % read coverage; green, >99 % read coverage. Hi, H. influenzae; Hh, H. haemolyticus, “fuzzy”, intermediate Haemophilus species
These findings are noteworthy given that the hpd target in particular has gained popularity due to its purported ability to differentiate H. influenzae from H. haemolyticus [e.g. [27, 43, 44]]. We recently reported that the hpd “gold standard” PCR target for H. influenzae identification is absent in some NTHi strains, an observation only made possible by the provision of genomic data [25]. The non-essential hpd gene encodes Protein D immunoglobulin, the H. influenzae component of the multivalent Synflorix vaccine [45]. Therefore, targeted selection pressure towards H. influenzae may have contributed to the emergence of vaccine escape variants in recent years. In support of our previous findings, we found that 6 % of NTHi and 23 % of H. haemolyticus strains lack hpd (Fig. 2). Based on our genomic analysis, we no longer recommend the use of hpd for detection or differentiation of H. influenzae and H. haemolyticus.
The fucK locus, like fucP, appears to be an excellent target for H. influenzae speciation (Fig. 2). Other studies have identified fucose operon-negative H. influenzae strains [40, 46], a concerning finding given that fucK is one of seven loci used in the H. influenzae multilocus sequence typing scheme (http://pubmlst.org/hinfluenzae/; [47]). Our data suggest that the fucose operon may constitute an essential metabolic pathway in H. influenzae, contrary to these earlier reports. In the absence of whole-genome data, it seems likely that fucK-negative H. influenzae strains are in fact H. haemolyticus or closely-related “fuzzy” species that have been misidentified using lower-resolution genotyping methods. In such cases, the identification of fucP- or fucK-negative strains should be seen as an opportunity to investigate species designation with higher-resolving methods such as whole-genome sequencing, and should not be judged as a failure of the assays to detect H. influenzae.
Nevertheless, it cannot be reasonably expected that a single assay will accurately speciate all H. influenzae all the time. First, there is the possibility for as-yet-unobserved SNPs to eventually be encountered in the fucP primer and probe binding sites, leading to poor amplification of certain H. influenzae isolates and resultant false-negatives or ambiguous genotype calls. As a salient example, using BLAST we identified three SNPs residing within a published fucK black-hole quencher probe [37]: one SNP in H. influenzae 10810 and two SNPs in KR494. This assay, which like fucP has been designed for H. influenzae detection in real-time PCR using a fluorogenic probe, would be expected to adversely affect amplification in H. influenzae strains harbouring these SNPs. Second, amplification of H. parahaemolyticus, H. parainfluenzae and Pasteurella multocida has been observed using a probeless fucK PCR [22] due to fucK orthologues in these species, leading to false-positive calls. Third, high rates of lateral gene transfer within and amongst Haemophilus species, combined with insufficient diversity in certain loci (e.g. 16S rDNA), will eventually lead to false-positive results and erroneous species assignments, especially when based on a single gene or locus. All of these possibilities demonstrate that large-scale genome datasets, extensive in silico validation efforts and the inclusion of fluorogenic probes targeting highly conserved regions are important considerations when designing H. influenzae-specific PCR assays to maximise specificity. Others have used the strategy of interrogating multiple genetic loci to improve species determination [5, 17]. Similarly, in instances where 100 % H. influenzae detection is essential, we recommend that two or more independent and well-validated assays, or ideally whole-genome sequencing coupled with phylogenetic analysis, should be used to verify species assignment.
One recognised limitation of this study is that, despite analysing whole-genome sequencing data, the delineation of Haemophilus species boundaries remains somewhat arbitrary. In particular, the exclusion of the “fuzzy” clade from the H. influenzae group is potentially contentious given that these isolates share a node with ‘Clade I’ H. influenzae [26], and thus may in fact represent a novel H. influenzae clade rather than a distinct species. However, for the purposes of this study, these “fuzzy” isolates were classified as distinct from H. influenzae due to their relatively high dissimilarity on the nucleotide level (only 94–98 % sequence identity in orthologous regions) and their unclear clinical relevance. In the absence of extensive transcriptional, metabolic and DNA hybridisation analysis of these isolates, we have chosen not to classify these isolates as H. influenzae. This approach does not diminish the value of the fucP assay for H. influenzae identification; rather, it highlights a deficit in our current understanding of Haemophilus diversity and the need for greater genomic, transcriptomic and metabolic studies within this genus. Interestingly, the hpd HRM assay grouped these “fuzzy” isolates with H. influenzae on 100 % of occasions, suggesting close relatedness of these species, although it remains unclear whether hpd HRM genotypes are variable in “fuzzy” isolates as has been observed in NTHi. On a crude presence/absence level, the iga or lgtC loci also provided good detection of H. influenzae and “fuzzy” isolates to the exclusion of H. haemolyticus (Fig. 2). Closer investigation of conserved regions within these or similar loci will enable detection of the “fuzzy” species clade and H. influenzae as a single group. We are currently investigating suitable targets for this purpose.
Conclusions
We have used a large-scale genomic approach to characterise the highly recombinogenic Haemophilus spp., including 107 new isolates from Australia. This approach enabled accurate delineation of H. influenzae from morphologically identical near-neighbour species. Using extensive genomic data, we next designed and validated a real-time PCR assay targeting the fucP locus in H. influenzae, which provided 100 % specificity for this bacterium both in silico and across a diverse bacterial DNA panel. The fucP TaqMan assay format enables rapid testing of clinical specimens, leading to faster and more accurate diagnosis of this bacterium without the requirement for culture.
Availability of supporting data
Eight Australian reference genomes (comprising two H. influenzae, three H. haemolyticus and three Haemophilus sp.) that support the results of this article are available in the NCBI SRA repository under BioProject ID PRJNA292146 (http://www.ncbi.nlm.nih.gov/bioproject/292146).
Acknowledgements
This project was funded by the Channel 7 Children’s Research Foundation (13699) and the Australian National Health and Medical Research Council (grant no. 1023781). We are grateful to the Rebecca L. Cooper Medical Research Foundation for provision of the NanoDrop 2000 spectrophotometer. HSV and L-AK are supported by NHMRC Career Development Fellowships 1024175 and 1061428, and RLM is supported by NHMRC Frank Fenner Early Career Fellowship 1034703.
We would like to thank the families who participated in these studies and for their continued support of our research. We wish to acknowledge Daniel J. Morton, University of Oklahoma Health Sciences Center, for the provision of a blood-derived H. haemolyticus isolate. We would also like to thank the Menzies Respiratory, Ear Health Research and Child Health Laboratory Teams and particularly Professor Amanda Leach for clinical swabs, clinical data, and laboratory support.
Abbreviations
- NTHi
non-typeable Haemophilus influenzae
- PCR
polymerase chain reaction
Additional files
Haemophilus spp. genomes used in this study. (XLS 57 kb)
Footnotes
Competing interests
The author(s) declare that they have no competing interests.
Authors’ contributions
EPP wrote the manuscript, designed the experiments, and conducted genomic and PCR analyses. DSS, EN, RLM, JP, ABC and HCS-V reviewed and revised the manuscript. DSS assisted with genomic pipeline development and bioinformatic analyses. EN and JB performed genomic DNA extractions and PCRs. RLM, JP, L-ASK, ADK, ABC and HCS-V provided clinical specimens or isolates, or provided microbiological or database assistance. All authors read and approved the final manuscript.
Contributor Information
Erin P. Price, Phone: +61 8-8946-8577, Email: erin.price@menzies.edu.au
Derek S. Sarovich, Email: derek.sarovich@menzies.edu.au
Elizabeth Nosworthy, Email: elizabeth.nosworthy@menzies.edu.au.
Jemima Beissbarth, Email: jemima.beissbarth@menzies.edu.au.
Robyn L. Marsh, Email: robyn.marsh@menzies.edu.au
Janessa Pickering, Email: janessa.pickering@uwa.edu.au.
Lea-Ann S. Kirkham, Email: lea-ann.kirkham@uwa.edu.au
Anthony D. Keil, Email: anthony.keil@health.wa.gov.au
Anne B. Chang, Email: anne.chang@menzies.edu.au
Heidi C. Smith-Vaughan, Email: heidi.smith-vaughan@menzies.edu.au
References
- 1.US Centers for Disease Control and Prevention. Haemophilus influenzae Type b vaccine. MMWR Morb Mortal Wkly Rep. 1988;36:832. [PubMed]
- 2.Bisgard KM, Kao A, Leake J, Strebel PM, Perkins BA, Wharton M. Haemophilus influenzae invasive disease in the United States, 1994–1995: near disappearance of a vaccine-preventable childhood disease. Emerg Infect Dis. 1998;4:229–37. doi: 10.3201/eid0402.980210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Eldere J, Slack MP, Ladhani S, Cripps AW. Non-typeable Haemophilus influenzae, an under-recognised pathogen. Lancet Infect Dis. 2014;14:1281–92. doi: 10.1016/S1473-3099(14)70734-0. [DOI] [PubMed] [Google Scholar]
- 4.Foxwell AR, Kyd JM, Cripps AW. Nontypeable Haemophilus influenzae: pathogenesis and prevention. Microbiol Mol Biol Rev. 1998;62:294–308. doi: 10.1128/mmbr.62.2.294-308.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson R, Wang X, Briere EC, Katz LS, Cohn AC, Clark TA, et al. Haemophilus haemolyticus isolates causing clinical disease. J Clin Microbiol. 2012;50:2462–5. doi: 10.1128/JCM.06575-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morton DJ, Hempel RJ, Whitby PW, Seale TW, Stull TL. An invasive Haemophilus haemolyticus isolate. J Clin Microbiol. 2012;50:1502–3. doi: 10.1128/JCM.06688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynn DJ, Kane JG, Parker RH. Haemophilus parainfluenzae and influenzae endocarditis: a review of forty cases. Medicine (Baltimore) 1977;56:115–28. doi: 10.1097/00005792-197703000-00003. [DOI] [PubMed] [Google Scholar]
- 8.US Centers for Disease Control and Prevention. Brazilian purpuric fever: Haemophilus aegyptius bacteremia complicating purulent conjunctivitis. MMWR Morb Mortal Wkly Rep. 1986;35:553–4. [PubMed]
- 9.Boucher MB, Bedotto M, Couderc C, Gomez C, Reynaud-Gaubert M, Drancourt M. Haemophilus pittmaniae respiratory infection in a patient with siderosis: a case report. J Med Case Rep. 2012;6:120. doi: 10.1186/1752-1947-6-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Floch AS, Cassir N, Hraiech S, Guervilly C, Papazian L, Rolain JM. Haemophilus parahaemolyticus septic shock after aspiration pneumonia, France. Emerg Infect Dis. 2013;19:1694–5. doi: 10.3201/eid1910.130608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parsons M, Faris I. Empyema of the gallbladder due to Haemophilus parahaemolyticus, with a brief review of its role as a pathogen. J Clin Pathol. 1973;26:604–5. doi: 10.1136/jcp.26.8.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CC, Wang SJ, Fuh JL. Isolation of Haemophilus parahaemolyticus in a patient with cryptogenic brain abscess. Scand J Infect Dis. 2001;33:385–6. doi: 10.1080/003655401750174129. [DOI] [PubMed] [Google Scholar]
- 13.Douglas GW, Buck LL, Rosen C. Liver abscess caused by Haemophilus paraphrohaemolyticus. J Clin Microbiol. 1979;9:299–300. doi: 10.1128/jcm.9.2.299-300.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jordan IK, Conley AB, Antonov IV, Arthur RA, Cook ED, Cooper GP, et al. Genome sequences for five strains of the emerging pathogen Haemophilus haemolyticus. J Bacteriol. 2011;193:5879–80. doi: 10.1128/JB.05863-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winn Jr. WC, Allen SD, Janda WJ, Koneman EW, Procop G, Schreckenberger PC. Chapter 9: Miscellaneous fastidious Gram-negative bacilli, p. 448. In Koneman's colour atlas and textbook of diagnostic microbiology, 6th ed. Lippincott Williams & Wilkins, Baltimore, MD. 2005
- 16.Murphy TF, Brauer AL, Sethi S, Kilian M, Cai X, Lesse AJ. Haemophilus haemolyticus: a human respiratory tract commensal to be distinguished from Haemophilus influenzae. J Infect Dis. 2007;195:81–9. doi: 10.1086/509824. [DOI] [PubMed] [Google Scholar]
- 17.McCrea KW, Xie J, LaCross N, Patel M, Mukundan D, Murphy TF, et al. Relationships of nontypeable Haemophilus influenzae strains to hemolytic and nonhemolytic Haemophilus haemolyticus strains. J Clin Microbiol. 2008;46:406–16. doi: 10.1128/JCM.01832-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nørskov-Lauritsen N. Detection of cryptic genospecies misidentified as Haemophilus influenzae in routine clinical samples by assessment of marker genes fucK, hap, and sodC. J Clin Microbiol. 2009;47:2590–2. doi: 10.1128/JCM.00013-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nørskov-Lauritsen N, Overballe MD, Kilian M. Delineation of the species Haemophilus influenzae by phenotype, multilocus sequence phylogeny, and detection of marker genes. J Bacteriol. 2009;191:822–31. doi: 10.1128/JB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prymula R, Kriz P, Kaliskova E, Pascal T, Poolman J, Schuerman L. Effect of vaccination with pneumococcal capsular polysaccharides conjugated to Haemophilus influenzae-derived protein D on nasopharyngeal carriage of Streptococcus pneumoniae and H. influenzae in children under 2 years of age. Vaccine. 2009;28:71–8. doi: 10.1016/j.vaccine.2009.09.113. [DOI] [PubMed] [Google Scholar]
- 21.Wang X, Mair R, Hatcher C, Theodore MJ, Edmond K, Wu HM, et al. Detection of bacterial pathogens in Mongolia meningitis surveillance with a new real-time PCR assay to detect Haemophilus influenzae. Int J Med Microbiol. 2011;301:303–9. doi: 10.1016/j.ijmm.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Binks MJ, Temple B, Kirkham LA, Wiertsema SP, Dunne EM, Richmond PC, et al. Molecular surveillance of true nontypeable Haemophilus influenzae: an evaluation of PCR screening assays. PLoS One. 2012;7 doi: 10.1371/journal.pone.0034083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pickering J, Richmond PC, Kirkham LA. Molecular tools for differentiation of non-typeable Haemophilus influenzae from Haemophilus haemolyticus. Front Microbiol. 2014;5:664. doi: 10.3389/fmicb.2014.00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witherden EA, Bajanca-Lavado MP, Tristram SG, Nunes A. Role of inter-species recombination of the ftsI gene in the dissemination of altered penicillin-binding-protein-3-mediated resistance in Haemophilus influenzae and Haemophilus haemolyticus. J Antimicrob Chemother. 2014;69:1501–9. doi: 10.1093/jac/dku022. [DOI] [PubMed] [Google Scholar]
- 25.Smith-Vaughan HC, Chang AB, Sarovich DS, Marsh RL, Grimwood K, Leach AJ, et al. Absence of an important vaccine and diagnostic target in carriage- and disease-related nontypeable Haemophilus influenzae. Clin Vaccine Immunol. 2014;21:250–2. doi: 10.1128/CVI.00632-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Chiara M, Hood D, Muzzi A, Pickard DJ, Perkins T, Pizza M, et al. Genome sequencing of disease and carriage isolates of nontypeable Haemophilus influenzae identifies discrete population structure. Proc Natl Acad Sci U S A. 2014;111:5439–44. doi: 10.1073/pnas.1403353111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pickering J, Binks MJ, Beissbarth J, Hare KM, Kirkham LA, Smith-Vaughan H. A PCR-high-resolution melt assay for rapid differentiation of nontypeable Haemophilus influenzae and Haemophilus haemolyticus. J Clin Microbiol. 2014;52:663–7. doi: 10.1128/JCM.02191-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith-Vaughan H, Byun R, Nadkarni M, Jacques NA, Hunter N, Halpin S, et al. Measuring nasal bacterial load and its association with otitis media. BMC Ear Nose Throat Disord. 2006;6:10. doi: 10.1186/1472-6815-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarovich DS, Price EP. SPANDx: a genomics pipeline for comparative analysis of large haploid whole genome re-sequencing datasets. BMC Res Notes. 2014;7:618. [DOI] [PMC free article] [PubMed]
- 30.Huang W, Li L, Myers JR, Marth GT. ART: a next-generation sequencing read simulator. Bioinformatics. 2012;28:593–4. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison A, Dyer DW, Gillaspy A, Ray WC, Mungur R, Carson MB, et al. Genomic sequence of an otitis media isolate of nontypeable Haemophilus influenzae: comparative study with H. influenzae serotype d, strain KW20. J Bacteriol. 2005;187:4627–36. doi: 10.1128/JB.187.13.4627-4636.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Sunderland, MA: Sinauer Associates [Version 4]. 2003.
- 33.Price EP, Dale JL, Cook JM, Sarovich DS, Seymour ML, Ginther JL, et al. Development and validation of Burkholderia pseudomallei-specific real-time PCR assays for clinical, environmental or forensic detection applications. PLoS One. 2012;7 doi: 10.1371/journal.pone.0037723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price EP, Sarovich DS, Webb JR, Ginther JL, Mayo M, Cook JM, et al. Accurate and rapid identification of the Burkholderia pseudomallei near-neighbour, Burkholderia ubonensis, using real-time PCR. PLoS One. 2013;8 doi: 10.1371/journal.pone.0071647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maaroufi Y, De Bruyne JM, Heymans C, Crokaert F. Real-time PCR for determining capsular serotypes of Haemophilus influenzae. J Clin Microbiol. 2007;45:2305–8. doi: 10.1128/JCM.00102-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdeldaim GM, Stralin K, Kirsebom LA, Olcen P, Blomberg J, Herrmann B. Detection of Haemophilus influenzae in respiratory secretions from pneumonia patients by quantitative real-time polymerase chain reaction. Diagn Microbiol Infect Dis. 2009;64:366–73. doi: 10.1016/j.diagmicrobio.2009.03.030. [DOI] [PubMed] [Google Scholar]
- 38.Blackall PJ, Christensen H, Beckenham T, Blackall LL, Bisgaard M. Reclassification of Pasteurella gallinarum, [Haemophilus] paragallinarum, Pasteurella avium and Pasteurella volantium as Avibacterium gallinarum gen. nov., comb. nov., Avibacterium paragallinarum comb. nov., Avibacterium avium comb. nov. and Avibacterium volantium comb. nov. Int J Syst Evol Microbiol. 2005;55:353–62. doi: 10.1099/ijs.0.63357-0. [DOI] [PubMed] [Google Scholar]
- 39.Kroll JS, Wilks KE, Farrant JL, Langford PR. Natural genetic exchange between Haemophilus and Neisseria: intergeneric transfer of chromosomal genes between major human pathogens. Proc Natl Acad Sci U S A. 1998;95:12381–5. doi: 10.1073/pnas.95.21.12381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ridderberg W, Fenger MG, Norskov-Lauritsen N. Haemophilus influenzae may be untypable by the multilocus sequence typing scheme due to a complete deletion of the fucose operon. J Med Microbiol. 2010;59:740–2. doi: 10.1099/jmm.0.018424-0. [DOI] [PubMed] [Google Scholar]
- 41.Dang S, Sun L, Huang Y, Lu F, Liu Y, Gong H, et al. Structure of a fucose transporter in an outward-open conformation. Nature. 2010;467:734–8. doi: 10.1038/nature09406. [DOI] [PubMed] [Google Scholar]
- 42.Stahl M, Friis LM, Nothaft H, Liu X, Li J, Szymanski CM, et al. L-fucose utilization provides Campylobacter jejuni with a competitive advantage. Proc Natl Acad Sci U S A. 2011;108:7194–9. doi: 10.1073/pnas.1014125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hare KM, Binks MJ, Grimwood K, Chang AB, Leach AJ, Smith-Vaughan H. Culture and PCR detection of Haemophilus influenzae and Haemophilus haemolyticus in Australian Indigenous children with bronchiectasis. J Clin Microbiol. 2012;50:2444–5. doi: 10.1128/JCM.00566-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Theodore MJ, Anderson RD, Wang X, Katz LS, Vuong JT, Bell ME, et al. Evaluation of new biomarker genes for differentiating Haemophilus influenzae from Haemophilus haemolyticus. J Clin Microbiol. 2012;50:1422–4. doi: 10.1128/JCM.06702-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Croxtall JD, Keating GM. Pneumococcal polysaccharide protein D-conjugate vaccine (Synflorix; PHiD-CV) Paediatr Drugs. 2009;11:349–57. doi: 10.2165/11202760-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 46.Norskov-Lauritsen N, Overballe MD, Kilian M. Delineation of the species Haemophilus influenzae by phenotype, multilocus sequence phylogeny, and detection of marker genes. J Bacteriol. 2009;191:822–31. doi: 10.1128/JB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meats E, Feil EJ, Stringer S, Cody AJ, Goldstein R, Kroll JS, et al. Characterization of encapsulated and noncapsulated Haemophilus influenzae and determination of phylogenetic relationships by multilocus sequence typing. J Clin Microbiol. 2003;41:1623–36. doi: 10.1128/JCM.41.4.1623-1636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]