

Abstract
Quantitative analysis of gene expression is a fundamental experimental approach in many fields of plant biology, but it requires the use of internal controls representing constitutively expressed genes for reliable transcript quantification. In this study, we identified fifteen putative reference genes from an A. angustifolia transcriptome database. Variation in transcript levels was first evaluated in silico by comparing read counts and then by quantitative real-time PCR (qRT-PCR), resulting in the identification of six candidate genes. The consistency of transcript abundance was also calculated applying geNorm and NormFinder software packages followed by a validation approach using four target genes. The results presented here indicate that a diverse set of samples should ideally be used in order to identify constitutively expressed genes, and that the use of any two reference genes in combination, of the six tested genes, is sufficient for effective expression normalization. Finally, in agreement with the in silico prediction, a comprehensive analysis of the qRT-PCR data combined with validation analysis revealed that AaEIF4B-L and AaPP2A are the most suitable reference genes for comparative studies of A. angustifolia gene expression.
Introduction
Quantitative analysis of gene expression is important for many fields of biological research and in this regard, quantitative real-time PCR (qRT-PCR) is a popular method for mRNA detection and quantification due to its high sensitivity, reproducibility and high throughput capability [1–3], as well as its wide dynamic range [4–9]. For an accurate and reliable analysis, internal controls must be used to eliminate the experimental noise generated by variations in mRNA amount, reverse transcription efficiency and the co-purification of inhibitory compounds that affect the amplification efficiency [10–13].
Genes involved in basic cellular processes, such as cell structure maintenance or primary metabolism, are often used as internal controls to normalize gene expression between samples [14]. Commonly used examples include 18S ribosomal RNA (18S), actin (ACT), tubulin (TUB), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), polyubiquitin (UBQ) and elongation factor 1-α (EF 1-α) [8,15–17]. However, several reports have described the characterization of other reference genes for use in particular species [8,14,16,18–23]. A reliable constitutively expressed control for qRT-PCR analysis should, by definition, exhibit constant levels of transcript abundance between the cells of different tissues and under different experimental conditions [21]; however, the identification of such control genes can be laborious, especially with species for whom a comprehensive genome sequence is not yet available, such as the Brazilian pine (Araucaria angustifolia) [24–25]. This native conifer is currently classified as a critically endangered species [26] and the seeds are recalcitrant to storage, since they maintain high levels of water and active metabolic rates at the mature stage, resulting in a rapid loss of viability, so conservation strategies are restricted [27]. The establishment of a successful somatic embryogenesis system as an alternative propagation approach for conservation requires extensive knowledge of the expression profiles of genes related to morphogenesis in the zygotic counterpart. Recently, a comparative transcriptome analysis of Brazilian pine early somatic embryo formation and seed development was reported, providing a foundation for further gene expression studies [25].
This current study describes a survey of the A. angustifolia transcriptome data set [25], the identification of fifteen potential qRT-PCR constitutively expressed reference genes, and an assessment of their suitability as controls for transcript profiling of a large set of biological samples representing different seed developmental stages, vegetative tissues and embryogenic cell lines.
Materials and Methods
Plant material
Two developmental stages of the zygotic embryo (Fig 1A–1C), aciculas (Fig 1D) and three embryogenic cultures of A. angustifolia, a critically endangered species [26], were analyzed (Fig 1E–1G). The globular zygotic embryos with the corresponding megagametophyte (GZE), cotyledonal zygotic embryos (CZE), megagametophytes of the cotyledonal embryos (CZE MG) and aciculas were harvested from five trees located in the Parque Estadual de Campos do Jordão (22° 41.792' south; 045° 29.393' west, 1.529 m) (authorization by Secretaria do Meio Ambiente, Instituto Florestal, in accordance with CARTA COTEC n° 066/2014 D139/2013 AP), Campos do Jordão, São Paulo, Brazil. Two of the embryogenic cell lines, SE1 and SE6, are abscisic acid (ABA)-responsive and non-responsive, respectively, while the third embryogenic culture was derived from the maturation of embryos generated by SE1 (S1M) (Fig 1). For our studies, the SE1 and SE6 cultures were allowed to proliferate for 21 days on basic MS medium [28] supplemented with 1.46 g dm-3 L-glutamine (MSG) [29] before harvesting. The S1M embryogenic culture was developed by growing the SE1 culture for 90 days on maturation medium (MSG medium supplemented with abscisic acid (ABA), maltose and PEG 4000) and sub-culturing every 30 days [25]. For each zygotic embryo developmental stage, we collected two or three seed cones per mother tree, mixed the seeds and divided them in three sub-samples containing 70 seeds. Each sub-sample was considered a biological replicate. For somatic sample, we collected five callus with 500 mg each (collected from three Petri dishes), mixed, divided in three biological replicates and stored at—80°C until further processing.
Fig 1. Araucaria angustifolia tissues/organs used in this study.

Globular zygotic embryos (left, scale bar = 1 mm, and the megagametophyte (right, scale bar = 10 mm) (A); late cotyledonal zygotic embryo (B) and the corresponding megagametophyte (C); aciculas (D); abscisic acid (ABA)-responsive (E), ABA non-responsive (F) and mature ABA-responsive (G) embryogenic cell lines. Arrows indicate globular somatic embryos. Scale bars for panels b-g = 10 mm, 10 mm, 100 mm, 10 mm, 10 mm and 10 mm, respectively.
RNA extraction, DNAse treatment and cDNA synthesis
Plant material was homogenized in liquid nitrogen with a pestle and mortar and RNA samples were extracted using three different protocols, depending on the tissue: Chang et al. [30] for zygotic samples, Salzmann et al. [31] for aciculas and Trizol reagent (Invitrogen Life Technologies—Burlington, ON, Canada) for the cultured cell lines. The quantity and purity of the RNA samples were assessed using a Nanodrop ND-100 spectrophotometer (Thermo Fisher Scientific) and, samples with 260/280 nm and 260/230 nm ratios between 1.8–2.2 and 1.6–2.2, respectively, were considered to be of sufficient purity. The integrity of the samples was confirmed by electrophoresis on a 1% (w/v) agarose gel and stained with ethidium bromide. Approximately 2 μg of each RNA sample was subjected to DNAse I (Life Technologies, Inc) treatment and reverse transcribed with random primers using SuperScript III Reverse Transcriptase (Life Technologies, Inc). To confirm that the reverse transcription reaction had worked and to confirm the absence of genomic DNA contamination, samples were subjected to PCR analysis with a pair of intron-flanking primers for Ubiquitin (UBI- accession number GW924714.1), Forward: 5’-CCTCGTGTCGATTTACGTC-3’, Reverse: 5’-GGGCGGCTTCTGGATTTG-3’). The PCR reactions were conducted in a total volume of 25 μl containing 1 μl cDNA (1:10), 0.2 μl Taq polymerase (Invitrogen, Carlsbad, CA, USA), 0.2 mM each dNTP, 0.2 μM each primer, 5 μl 10x PCR buffer (Invitrogen, Carlsbad, CA, USA) and 1.5 mM MgCl2. The amplification conditions were: 94°C for 3 min, 35 cycles of 94°C for 30 s, 60°C for 30 s, and then 72°C for 1 min. cDNA samples were diluted 1:10 to a final concentration of 5 ng reverse-transcribed RNA/μl.
Candidate gene selection and primer design
A total of fifteen candidate reference genes were selected, based on the reports of Brunner et al. [32], Iskandar et al. [33], Czechowski et al. [16], Expósito-Rodríguez et al. [18], Hu et al. [34], Cruz et al. [20], Artico et al. [21], Narsai et al. [35], Mafra et al. [36]; de Vega-Bartol et al. [22] and Perini et al. [14] (Table 1). The A. angustifolia transcriptome database [25] was surveyed using the BLAST program [37] and the corresponding Arabidopsis thaliana protein sequences as query sequences. The abundance of the candidate gene transcripts was assessed using the RPKM values (number of reads that map per kilobase of exon model per million mapped reads for each gene, for each tissue or sample), and pairwise and global comparisons between samples and/or tissues, except aciculas (sample not included in the Brazilian pine transcriptome analysis), were performed using the nonparametric Kruskal-Wallis test (p < 0.05) (R software, version 2.8.0) [38]. Specific primers for qRT-PCR were designed according to the MIQE guidelines (see S1 Table) [39] using the program Oligo Perfect 3.1 (http://tools.lifetechnologies.com) based on the sequences listed in Table 2. Primers were tested in PCR reactions using a pool of all cDNA samples as described above.
Table 1. Candidate reference genes and their annotated functions.
| Gene abbreviation | Gene name | Araucaria angustifolia unigene a | Arabidopsis thaliana locus | Protein function b | References |
|---|---|---|---|---|---|
| 60S | 60S ribosomal protein L18A-1 | comp44885_c0_seq2 | At1g29970 | Structural constituent of ribosome. | Saccharum spp. (Iskandar et al. 2004). |
| ARP7 | Actin-Related Protein 7 | comp43799_c0_seq2 | At3g60830 | Cell division. | P. trichocarpa (Brunner et al. 2004); Saccharum spp. (Iskandar et al. 2004); G. hirsutum (Artico et al. 2010); P. abies and P. pinaster (de Vega-Bartol et al. 2013); M. domestica (Perini et al. 2014). |
| CYP | Cyclophilin | comp39853_c0_seq1 | At2g21130 | Peptidyl-prolyl cis-trans isomerase activity, involved in protein folding. | P. trichocarpa (Brunner et al. 2004); G. max (Hu et al. 2009); C. sinensis (Mafra et al. 2012). |
| EF-1α | Elongation Factor 1α | comp52960_c0_seq8 | At5g60390 | Calmodulin binding protein involved in translational elongation. | Arabidopsis thaliana (Czechowski et al. 2005); Solanum lycopersicum (Expósito-Rodríguez et al. 2008); Gossypium hirsutum (Artico et al. 2010); Oryza sativa (Narsai et al. 2010); Citrus sinensis (Mafra et al. 2012); Picea abies and Pinus pinaster (de Vega-Bartol et al. 2013); M. domestica (Perini et al. 2014). |
| EIF4B-L | Translational initiation factor 4B | comp50365_c0_seq1 | At4g38710 | Protein transduction initiation. | P. trichocarpa (Brunner et al. 2004); O. sativa (Narsai et al. 2010). |
| FBOX | F-BOX family protein | comp48365_c0_seq1 | At5g15710 | Cell cycle regulation. | A. thaliana (Czechowski et al. 2005); G. hirsutum (Artico et al. 2010); C. sinensis (Mafra et al. 2012). |
| PP2A | Protein phosphatase 2A | comp39762_c0_seq1 | At1g59830 | Catalytic subunit of protein phosphatase 2A. | A. thaliana (Czechowski et al. 2005); C. arabica (Cruz et al. 2009); G. hirsutum (Artico et al. 2010); M. domestica (Perini et al. 2014). |
| PSAB | D1 subunit | comp50019_c0_seq1 | ATCG00340 | Component of the photosystem I and II reaction centers | Coffea arabica (Cruz et al. 2009). |
| S24 | S24 ribosomal protein S24 | comp54446_c0_seq1 | At3g04920 | Structural constituent of ribosome. | C. arabica (Cruz et al. 2009). |
| SAM | S-adenosyl-L-methionine-dependent methyltransferase | comp43283_c0_seq3 | At2g32170 | Methylation and nucleotide biosynthetic process. | C. sinensis (Mafra et al. 2012). |
| TUA5 | Tubulin α3 / α5 chain | comp14778_c0_seq1 | At5g19780 | Structural constituent of cytoskeleton. | Populus trichocarpa (Brunner et al. 2004); Glycine max (Hu et al. 2009); Malus domestica (Perini et al. 2014). |
| UBC21 | Ubiquitin-conjugating enzyme 21 | comp42656_c0_seq3 | At5g25760 | Fatty acid beta-oxidation, peroxisome organization and phosphatidylinositol biosynthetic process. | C. sinensis (Mafra et al. 2012); P. abies and P. pinaster (de Vega-Bartol et al. 2013). |
| UBC9 | Ubiquitin conjugating enzyme | comp52968_c0_seq1 | At4g27960 | Ubiquitin-dependent protein catabolic process. | Saccharum spp. (Iskandar et al. 2004); C. sinensis (Mafra et al. 2012). |
| UBQ7 | Ubiquitin 7 | comp51531_c0_seq6 | At2g35635 | Ubiquitin-dependent protein catabolic process. | P. trichocarpa (Brunner et al. 2004); C. sinensis (Mafra et al. 2012). |
| UBQ10 | Polyubiquitin | comp51531_c0_seq24 | At4g05320 | Ubiquitin-dependent protein catabolic process. | P. trichocarpa (Brunner et al. 2004); Saccharum spp. (Iskandar et al. 2004); A. thaliana (Czechowski et al. 2005); G. max (Hu et al. 2009); G. hirsutum (Artico et al. 2010); O. sativa (Narsai et al. 2010); M. domestica (Perini et al. 2014). |
a Araucaria angustifolia transcriptome database (Elbl et al. 2015).
b Encoded-protein function according to TAIR database (http://www.arabidopsis.org/).
Table 2. Primers used for gene amplification.
| Gene | Primer sequences (forward/reverse primer) | Expected amplicon size (bp) |
|---|---|---|
| Aa60S | 5'-CCTATGTGTGCTTAGATGACC-3' | 214 |
| 5'-CCTATTGTTTCCTCTCCTCTCC-3' | ||
| AaADC a | 5’-GGTGGAGGGCTTGGCATC-3’ | 199 |
| 5’-CGAAAACGAGGAGGGAATGG-3’ | ||
| AaARP7 | 5'-CGGTGTTTTCCAGAAGTTGTCGC-3' | 220 |
| 5'-CAGATTGCCTATGAAGAGACGC-3' | ||
| AaCAT a | 5’-GCTTTTGGAGGACTATCACC-3’ | 192 |
| 5’-GAGAATCGCACAATAACGGG-3’ | ||
| AaCYP | 5'-GAAAGTTGTTGTTGAAGATTGCGGC-3' | 153 |
| 5'-CGTAAACCCTCACAGTAGAAAACC-3' | ||
| AaEF-1α | 5'-GATGACGATGATGAGGTTTTACTG-3' | 164 |
| 5'-CGGCATAATGATTCCACAGC-3' | ||
| AaEIF4B-L | 5'-CAGTCGCCTCCTGTCTTG-3' | 233 |
| 5'-CCGTCGTCTGGTGAAAATG-3' | ||
| AaFBOX | 5'-CGTCCCCAAATCTTCTCTTCC-3' | 196 |
| 5'-GCAAAAGCGAGTTGTTATCTGATG-3' | ||
| AaPP2A | 5'-GATGAAGGTCAATGTAGAGGG-3' | 178 |
| 5'-GGTGGGGCTTATTTTGCTTTG-3' | ||
| AaPSAB | 5'-CCTCCTCATCTCTTTAGTTTTC-3' | 228 |
| 5'-CCCTTCCTTGTCCTGAATC-3' | ||
| AaS24 | 5'-CCCCAGACCATATTTGTTTTCGGC-3' | 185 |
| 5'-CTGTTCTTCCTTTCCTTCATTTGC-3' | ||
| AaSAM | 5'-CACCTCAACAAAGTCCCC-3' | 172 |
| 5'-GAACCAAACTCAAGCACCC-3' | ||
| AaTPS3 a | 5’-CGATGAATGTAGCCCTCACTATGC-3’ | 178 |
| 5’-CTCAATCCAAATCCAATACCCCAGC-3’ | ||
| AaTUA5 | 5'-CGTGAGGTGATGTTAGAGAGAG-3' | 213 |
| 5'-CGAATGAAGAAGGCGTTTGC-3' | ||
| AaUBC21 | 5'-CTCTGGTGATAATCGTGGG-3' | 185 |
| 5'-CACTGGCAGCAAATGGTTG-3' | ||
| AaUBC9 | 5'-CTCTTGAACTGTAACCCCATTCG-3' | 221 |
| 5'-GAAGCCTGCCACCTATGAGC-3' | ||
| AaUBQ7 | 5'-CCAATCCCGAGCCCTTTCAG-3' | 232 |
| 5'-CCAGCGAATATAAGCCTCTGC-3' | ||
| AaUBQ10 | 5'-CCAATCCCGAGCCCTTTCAG-3' | 232 |
| 5'-CCAGCGAATATAAGCCTCTGC-3' | ||
| AaUGP a | 5’-GAAGTTGTGGTTCCCTATC-3’ | 214 |
| 5’-CTCTGCTATTGTATTTGTCGTTGAG-3’ |
a Genes used for reference gene validation.
qRT-PCR analysis
Transcript abundance was assessed by qRT-PCR analysis using a 7500 Real-Time PCR system (Applied Biosystems by Life Technologies, NY, USA) (S1 Table). The PCR reactions were performed with 4 μl of cDNA (1:10), 10 μl 2X SYBR Green Master Mix (Applied Biosystems) and the following cycling conditions: 95°C for 10 min, 40 cycles of 95°C for 15 s, 60°C for 30 s and 72°C for 30 s. All reactions were performed in duplicate for each of three biological replicates. The amplification of single products was confirmed by melting curve analysis. After testing primer concentrations of 200 nM, 400 nM and 800 nM, we selected 400 nM as the optimal concentration based on the lowest quantification cycle (Cq) values and the absence of primer dimers. The Cq values and the efficiency of the reaction with each primer were determined using LinRegPCR software [40] and only the genes with transcripts yielding Cq values ≤ 35 were included in subsequent analyses (S1 Table).
Statistical analysis of gene expression consistency
The consistency of expression of the candidate reference genes was evaluated according to Expósito-Rodríguez et al. [18], by applying two different statistical approaches: using geNorm v.3.5 software (http://medgen.ugent.be/~jvdesomp/genorm/) [41] and NormFinder software (http://www.mdl.dk/publicationsnormfinder.htm) [42].
Validation of the selected reference genes
To confirm our procedure for the selection of control genes, the relative expression level of AaADC, AaCAT, AaTPS3 and AaUGP were evaluated using five combinations of reference genes and the results were confronted with Araucaria transcriptome dataset [25]. The primers (Table 2) and qRT-PCR reactions were conducted as described in qRT-PCR section. The relative expression was quantified in comparison with the average expression, normalized against the aciculas samples, and the Cq values of target genes were normalized against the geometric average of a combination of reference genes, followed by ANOVA analysis. The co-variation between the qRT-PCR genes and the transcriptome profile were calculated as Euclidean distances by the *omeSOM software (version v2.27.17, available in http://sourcesinc.sourceforge.net/omesom/). For this analysis, data were normalized as previously described [43]. A 3 X 3 map was selected to group co-expresssed genes showing direct expression patterns using group neighbor neurons with a visualization neighborhoods equal 1 (Vn = 1).
Results
Identification of putative Araucaria angustifolia reference genes
Using the sequences of previously reported A. thaliana orthologs, fifteen putative reference genes were retrieved from the A. angustifolia transcriptome database. In order to identify suitable constitutively expressed reference genes for a number of distinct tissues, genes from a range of functional categories, such as ‘cytoskeleton and cell division’, ‘protein/lipid/nucleotide metabolism’, ‘photosynthesis’ and ‘gene expression regulation’ were chosen (Table 1). An in silico analysis of the A. angustifolia transcriptome data set indicated that of the candidate genes, only AaPSAB, AaPP2A and AaEIF4B-L showed consistent transcript levels amongst the samples represented in the transcriptome database (Table 3), suggesting that these represented the most promising candidates to be used as constitutively expressed reference genes.
Table 3. In silico analysis of RPMK average ± standard deviation values of the candidate reference genes retrieved from the Araucaria angustifolia transcriptome database (Elbl et al. 2015).
| Sample a | |||||||
|---|---|---|---|---|---|---|---|
| Gene | GZE | CZE | CZE MG | S1M | SE1 | SE6 | Kruskal-Wallis (p < 0.05) b |
| AaTUA5 | 1,353.0 ± 762.0 | 956.2 ± 28.3 | 1,052.9 ± 98.0 | 149.0 ± 54.7 | 240.8 ± 126.1 | 240.2 ± 14.1 | 0.01 |
| AaPSAB | 8.7 ± 12.8 | 1.0 ± 0.7 | 3.9 ± 0.8 | 1.4 ± 0.8 | 3.7 ± 5.4 | 1.6 ± 0.3 | 0.27 |
| AaEF-1α | 9.0 ± 1.1 | 16.2 ± 0.5 | 9.8 ± 0.1 | 0.6 ± 0.4 | 3.9 ± 2.1 | 2.4 ± 0.6 | 0.01 |
| AaS24 | 25.7 ± 8.1 | 30.7 ± 0.8 | 29.8 ± 11.9 | 9.9 ± 6.9 | 72.6 ± 34.5 | 94.1 ± 15.1 | 0.02 |
| AaPP2A | 27.7 ± 3.9 | 27.6 ± 1.3 | 33.4 ± 6.1 | 6.2 ± 4.3 | 53.4 ± 27.0 | 35.5 ± 4.5 | 0.07 |
| AaCYP | 135.2 ± 52.9 | 180.6 ± 11.7 | 121.4 ± 25.2 | 42.0 ± 11.4 | 222.9 ± 104.9 | 1,008.4 ± 198.7 | 0.02 |
| AaUBC21 | 8.7 ± 2.0 | 6.8 ± 0.9 | 10.0 ± 0.4 | 2.3 ± 2.1 | 26.7 ± 7.4 | 17.8 ± 2.6 | 0.01 |
| AaSAM | 2.5 ± 0.4 | 4.6 ± 0.6 | 2.5 ± 0.4 | 0.8 ± 0.4 | 8.0 ± 4.5 | 4.2 ± 1.1 | 0.01 |
| Aa60S | 179.9 ± 33.6 | 187.7 ± 15.7 | 195.4 ± 53.4 | 13.5 ± 8.2 | 87.2 ± 37.8 | 135.2 ± 13.3 | 0.02 |
| AaUBC9 | 241.2 ± 63.1 | 307.4 ± 2.6 | 314.2 ± 61.4 | 796.3 ± 432.1 | 1,377.9 ± 1,281.5 | 1,807.3 ± 212.4 | 0.01 |
| AaUBQ10 | 76.6 ± 22.7 | 155.9 ± 10.0 | 98.4 ± 26.7 | 63.0 ± 12.7 | 171.6 ± 60.4 | 219.7 ± 29.0 | 0.01 |
| AaARP7 | 3.9 ± 1.9 | 7.4 ± 1.1 | 3.6 ± 0.2 | 0.8 ± 1.0 | 4.9 ± 2.2 | 2.6 ± 0.9 | 0.04 |
| AaFBOX | 1.3 ± 0.2 | 3.7 ± 0.7 | 2.0 ± 0.3 | 0.6 ± 0.4 | 4.8 ± 2.5 | 2.9 ± 0.3 | 0.02 |
| AaEIF4B-L | 51.0 ± 37.3 | 83.0 ± 4.2 | 49.1 ± 10.2 | 28.0 ± 12.4 | 51.8 ± 26.3 | 83.3 ± 18.5 | 0.10 |
| AaUBQ7 | 93.3 ± 21.0 | 182.0 ± 12.7 | 103.2 ± 26.2 | 57.2 ± 11.7 | 251.5 ± 114.2 | 390.3 ± 145.3 | 0.01 |
a Samples according to Fig 1.
b Genes with significant differential expression among samples are highlighted in bold.
cDNA quality and test of primers
cDNA samples were derived from RNA extracted from the globular zygotic embryo plus megagametophyte (GZE), the late cotyledonal zygotic embryo (CZE) and the corresponding megagametophyte (CZE MG), the ABA-responsive (SE1) embryogenic cell lines, the ABA-non responsive (SE6) embryogenic cell lines, the maturated SE1 (S1M) embryogenic cell lines and aciculas. All samples were extracted as biological triplicates. The effectiveness of the DNAse treatment was tested by PCR using specific UBI intron-flanking primers (Fig 2A).
Fig 2. cDNA quality (a) and primer (b) test.

Amplification products of PCR analyses using genomic DNA (gDNA) or a pool of all cDNA samples (Fig 1) and UBI intron-flanking specific primers (A). Amplicons obtained by PCR using a pool of all cDNA samples and specific primers for the reference genes (B). bp = base pairs.
Out of the fifteen genes initially selected, only nine were successfully amplified from a pool of cDNA (Fig 2B). For AaS24, AaSAM, AaUBC9, AaARP7 and AaUBQ7 no amplification products were obtained, while for AaUBQ10 the amplicon was larger than expected. The might be probably due to errors in transcriptome assembly. The efficiency and specificity of the primer pairs were tested in qRT-PCR. AaTUA5, and Aa60S showed Cq values ≥ 35 cycles and the reactions did not reach the plateau phase, while the primers for AaPSAB resulted in dimer formation. Thus, six genes (AaFBOX, AaEF-1α, AaPP2A, AaUBC21, AaEIF4B-L and AaCYP) were selected for further analyses.
qRT-PCR amplification
Analysis of the Cq values showed that the selected reference genes had different levels of expression among the analyzed samples, with AaCYP (24.30 ± 1.36) and AaFBOX (33.27 ± 1.92) being the most highly and least abundantly expressed, respectively. EF-1α, AaPP2A, AaEIF4B-L and AaUBC21 showed intermediate levels of expression, with average Cq values from 25 to 29 cycles. The intra-assay variation was evaluated by correlation analysis and demonstrated a reasonably good fit (r2 = 0.992, S1 Fig). The efficiency for all six primer pairs was ≥ 1.88 for the entire experimental set of samples (Fig 3).
Fig 3. Box plot of the Cq value distribution of candidate reference genes in all Araucaria angustifolia samples (Fig 1).
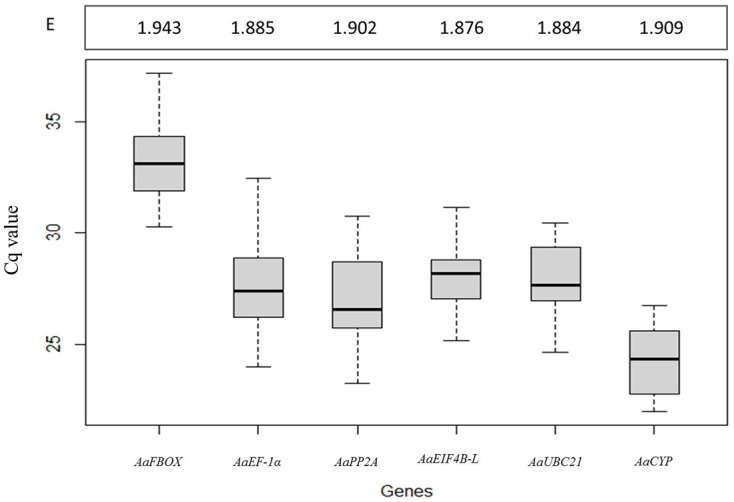
The median is indicated by a thick horizontal line. Gray boxes and vertical lines indicate interquartile range and the variance between Cq values for each gene, respectively. E: primer efficiency.
Analysis of reference gene expression using geNorm and NormFinder software
The consistency of gene expression was analyzed using geNorm (Table 4) and NormFinder (Table 5) software with data derived from the entire set of samples (ALL), or from four subsets: zygotic (GZE, CZE and CZE MG), cell lines (SE1, SE6 and S1M), zygotic/somatic embryos (GZE and S1M) and aciculas. The geNorm analysis scored all six genes as suitable ‘normalizers’ according to their stability value, although the relative ranking of the genes varied depending on the sample. AaPP2A, AaEIF4B-L and AaCYP exhibited the most consistent expression among the different samples except for the “cell line” subset. The optimal number of reference genes was calculated by the pairwise variation value (V) and it was found that for a cut-off threshold of 0.15, two reference genes were sufficient for an accurate normalization in all the experimental data sets evaluated (Table 4). NormFinder software also indicated a distinct ranking of genes but the best pair of normalizers for the five samples were different from those suggested by geNorm. The NormFinder analysis further suggested the best combination of genes using a pairwise comparison approach and for only two subsets of data, zygotic and zygotic/somatic embryos, the selected pair was in agreement with the stability ranking, while for the other three the chosen genes were AaFBOX and AaCYP (Table 5). The best reference gene, as indicated by both geNorm and NormFinder, was different for each subset of samples; however, when the sample diversity was larger (i.e. by including all the data), the results indicated that AaEIF4B-L represents the best reference gene for A. angustifolia.
Table 4. Ranking of Araucaria angustifolia candidate reference genes based on GeNorm analysis.
| ALL | ZYGOTIC | CELL LINES | ZYGOTIC/SOMATIC EMBRYOS | ACICULAS c | |||||
|---|---|---|---|---|---|---|---|---|---|
| Ranking | M a | Ranking | M a | Ranking | M a | Ranking | M a | Ranking | M a |
| AaEIF4B-L/AaPP2A | 0.284 | AaPP2A/AaCYP | 0.280 | AaFBOX/ AaEF-1α | 0.150 | AaPP2A/AaEIF4B-L | 0.205 | AaUBC21/AaCYP | 0.198 |
| AaEF-1α | 0.310 | AaEIF4B-L | 0.308 | AaEIF4B-L | 0.225 | AaEF-1α | 0.282 | AaEF-1α | 0.427 |
| AaUBC21 | 0.380 | AaEF-1α | 0.336 | AaPP2A | 0.274 | AaFBOX | 0.306 | AaEIF4B-L | 0.399 |
| AaCYP | 0.399 | AaFBOX | 0.344 | AaUBC21 | 0.393 | AaCYP | 0.311 | AaFBOX | 0.476 |
| AaFBOX | 0.424 | AaUBC21 | 0.360 | AaCYP | 0.450 | AaUBC21 | 0.341 | AaPP2A | 0.346 |
| V b | V b | V b | V b | V b | |||||
| V2/3 | 0.097 | V2/3 | 0.097 | V2/3 | 0.085 | V2/3 | 0.101 | V2/3 | 0.136 |
| V3/4 | 0.104 | V3/4 | 0.072 | V3/4 | 0.075 | V3/4 | 0.064 | V3/4 | 0.104 |
| V4/5 | 0.073 | V4/5 | 0.061 | V4/5 | 0.110 | V4/5 | 0.060 | V4/5 | 0.083 |
| V5/6 | 0.070 | V5/6 | 0.059 | V5/6 | 0.086 | V5/6 | 0.062 | V5/6 | 0.089 |
a Stability coefficient is the mean of the variation of two internal control genes between an individual and all other tested genes. The most stable gene has the lowest M value (cut-off < 1.5).
b Pairwise variation values Vn/n+1 < 0.15 mean that use of the two most stable genes is sufficient to normalize the expression of a test gene in the corresponding set of samples. n: number of genes.
Table 5. Ranking of Araucaria angustifolia reference genes calculated using NormFinder.
| ALL | ZYGOTIC | CELL LINES | ZYGOTIC/SOMATIC EMBRYOS | ACICULAS | |||||
|---|---|---|---|---|---|---|---|---|---|
| Ranking | Stability value a | Ranking | Stability value a | Ranking | Stability value a | Ranking | Stability value a | Ranking | Stability value a |
| AaEIF4B-L | 0.730 | AaFBOX | 0.576 | AaPP2A | 0.697 | AaCYP | 0.170 | AaEIF4B-L | 0.639 |
| AaEF-1α | 0.741 | AaPP2A | 0.624 | AaEF-1α | 0.731 | AaFBOX | 0.260 | AaUBC21 | 0.732 |
| AaPP2A | 0.749 | AaUBC21 | 0.663 | AaEIF4B-L | 0.785 | AaEF-1α | 0.371 | AaEF-1α | 0.823 |
| AaUBC21 | 0.807 | AaCYP | 0.671 | AaFBOX | 0.942 | AaPP2A | 0.404 | AaPP2A | 0.846 |
| AaCYP | 0.919 | AaEF-1α | 0.698 | AaUBC21 | 1.016 | AaUBC21 | 0.428 | AaCYP | 0.885 |
| AaFBOX | 1.093 | AaEIF4B-L | 0.727 | AaCYP | 1.164 | AaEIF4B-L | 0.586 | AaFBOX | 1.269 |
| Best Combination b | Best Combination b | Best Combination b | Best Combination b | Best Combination b | |||||
| AaFBOX and AaCYP | 0.390 | AaFBOX and AaPP2A | 0.364 | AaFBOX and AaCYP | 0.253 | AaFBOX and AaCYP | 0.165 | AaFBOX and AaCYP | 0.346 |
a Genes are ranked according to minimal estimated intra- and intergroup variation. Genes with the lowest stability value have the most stable expression.
b The pair of genes with the highest degree of similarity in their expression profiles.
Validation of the selected reference genes
The relative expression levels of AaADC, AaCAT, AaTPS3 and AaUGP were used as target genes to demonstrate the usefulness of the candidate reference genes in qRT-PCR. Five combinations of the most stable reference genes established by geNorm and NormFinder were applied for normalization (Fig 4A). In all four situations, when only one normalizer was used (EIF4B-L) the relative expression was different than the observed when two or more reference genes were used, except to sample CZE. Likewise, CYP+FBOX showed discrepant values when compared to EIF4B-L+PP2A or EIF4B-L+PP2A+EF and similar expression values was observed only for AaADC in SE1 and AaCAT in GZE. The combinations EIF4B-L+PP2A and EIF4B-L+PP2A+EF did not exhibited significant difference in expression values, except in SE1 for AaCAT, AaTPS3 and AaUGP genes (Fig 4A). The addition of more reference genes did not improve the analysis of target and reference genes.
Fig 4. Validation of the most stable reference genes.
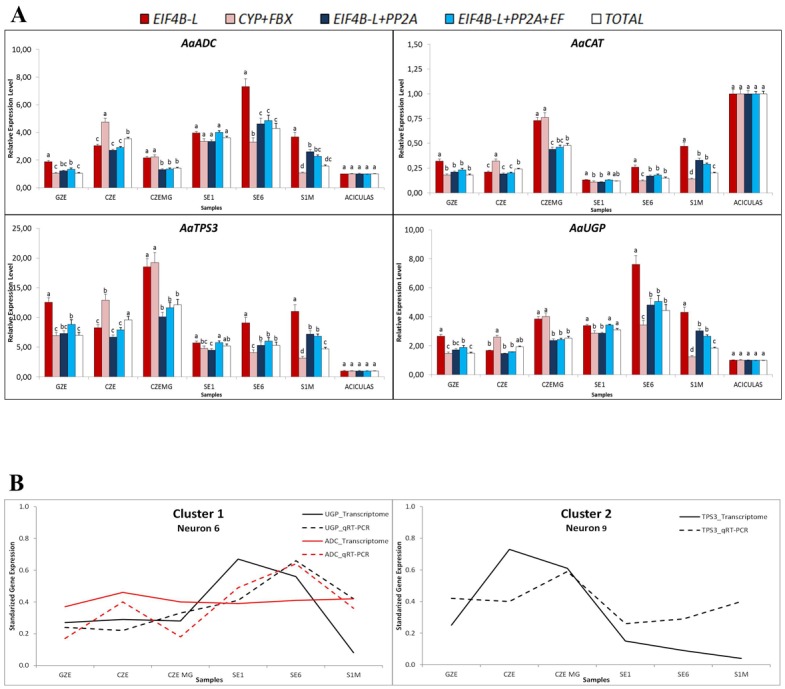
(A) Relative expression of AaADC, AaCAT, AaTPS3 and AaUGP in the samples used in this study, normalized with different combinations of reference genes. (B) Co-variation patterns by neural network analysis, performed by applying the *omeSOM software [43].
For comparison between the qRT-PCR and transcriptome profiles we used a neural clustering model through a self-organizing map (*omeSOM) [43], using the qRT-PCR results obtained with the EIF4B-L+PP2A pair. Among all the genes analyzed AaADC and AaUGP (Cluster 1) and AaTPS3 (Cluster 2) showed a correlation between qRT-PCR and transcriptome profile (Fig 4B). The results suggest that normalizers, mainly EIF4B-L and PP2A, were satisfactory for accurate qRT-PCR assay, since the majority of target genes tested showed relative expression levels correlating with the transcriptome expression profile.
Discussion
Gene expression analysis is central to developing a mechanistic understanding of physiological and developmental processes and qRT-PCR is currently the most widely used method for quantifying transcript expression [6–7,9,23]. Consequently, the identification of suitable reference genes for use as internal controls to standardize expression levels is an important goal. This is particularly challenging for non-model species that do not have a reference genome sequence [8,14,19–21,23]. The Brazilian pine (A. angustifolia) is an example of a non-model species for which there is a considerable research invested in studying aspects of growth, development and environmental responses, and thus for which the identification of suitable reference genes would be of considerable value. For example, several studies have investigated the molecular mechanisms that control embryogenesis in A. angustifolia [44–46] and recently a substantial collection of transcriptome sequences was generated and annotated [25]. Large datasets, such as those associated with microarrays, expressed sequence tags and cDNA libraries, can provide valuable resources to identify genes showing minimal variation in expression [14,24,35]. Accordingly, we used published information derived from several plant species to identify candidate constitutively expressed reference genes in an in silico analysis of the A. angustifolia transcriptome dataset, which contains genes expressed during early somatic embryo and seed development (Table 1). This search suggested fifteen such candidate reference genes and a subsequent non-parametric statistical test highlighted AaPSAB, AaPP2A and AaEIF4B-L as those with invariable expression among the sets of samples that comprise the A. angustifolia transcriptome dataset (Table 3).
Of the fifteen genes, six were successfully amplified by PCR and showed satisfactory qRT-PCR quality parameters, so their consistency of expression was further evaluated using geNorm and NormFinder software (Tables 4 and 5). According to the former, all the identified genes were deemed sufficiently reliable such that only two should be necessary for an accurate normalization of qRT-PCR data from all the tested subsets of the A. angustifolia samples.
In accordance with similar studies of gene expression in Glycine max [34], Citrus spp. [36] and Pyrus spp. [47], the best pair of reference genes for A. angustifolia varied within subsets of samples, suggesting that the genes showing the most constant expression profile varied between tissues and/or samples. It is noteworthy that the two programs suggested different optimal pairs of reference genes for a given subset of samples. However, when all the samples (ALL) were examined together, both algorithms indicated the same three genes as being the most effective for normalizing A. angustifolia expression data: AaEIF4B-L, AaPP2A and AaEF-1α, which is in agreement with the initial in silico analysis. Thus, it appears that the wider and more diverse the sample pool, the more accurate the selection of constitutively expressed genes. We also note that EF-1α has been extensively tested as a reference gene and has been ranked as highly effective for use in gene expression studies with Picea abies and Pinus pinaster, two other gymnosperm species [22]. EIF4B-L has not been widely used to normalize gene expression in plants and was reported to be somewhat effective as a reference gene in studies of both vegetative and reproductive organs of Populus trichocarpa [32]. Finally, PP2A has been shown to have consistent expression levels in Gossypium hirsutum and Malus domestica leaves, buds, flowers and fruits [14,21]. Thus, for most plant species and samples, there appears to be a suitable group of “universal” candidate reference genes that are constitutively expressed and that encode proteins involved in basic cellular processes (Table 1).
The reference gene approach was validated by quantifying relative expression of A. angustifolia AaADC, AaCAT, AaTSP3 and AaUGP genes. In a previous study, it was observed that transcripts involved in A. angustifolia embryogenesis related to polyamines, oxidative and carbohydrate metabolism were differentially expressed [25]. ADC encodes an enzyme that controls the polyamine flux in seeds of conifers by the putrescine (Put) biosynthesis [48]. Put is one of the main polyamines found in plants and is normally associated to embryogenesis and abiotic stress response [49–50]. Related to stress response, CAT plays important roles in plant antioxidative and detoxification processes that are closely related with reactive oxygen species generation [51]. In carbohydrate metabolism, TPS and UGP have a participation in biosynthesis of plant trehalose (α-D-glucopyranosyl α-D-glucopyranoside) a non-reduced disaccharide which plays important roles in protecting plants from heat, cold, osmotic and dehydration stress [52]. These genes encode enzymes related to sugar sensing, energy metabolism and seem involved in embryogenic potential of A. angustifolia cell lines (Navarro et al. unpublished data).
For all target genes and samples, similar expression values were observed when normalized with the combinations EIF4B-L+PP2A, EIF4B-L+PP2A+EF and TOTAL, showing that the use of three or more reference genes is unnecessary (Fig 4A). Interestingly, when we used the EIF4B-L+PP2A pair in the neural network analysis the result was in accordance with the Araucaria dataset expression profiles (Fig 4B). Therefore, the proposed reference genes are reliable for obtaining accurate expression profiles in different target genes and samples during A. angustifolia embryogenesis.
In conclusion, this report describes an efficient pipeline for the identification of constitutively expressed reference genes for subsequent gene expression analyses. We also demonstrate that large databases of gene expression can provide valuable resources for identifying genes that show little variation in expression and, in this regard, data derived from a diverse set of samples are recommended for the selection of such reference genes.
Supporting Information
Ct values of the replicates were plotted against each other.
(TIF)
(PDF)
Acknowledgments
The authors thank the PlantScribe (www.plantscribe.com) for editing this manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (02437/09-0), Fundação de Amparo à Pesquisa do Estado de São Paulo (2011/51659-7) and Conselho Nacional de Desenvolvimento Científico e Tecnológico(307238/2013-0).
References
- 1. Czechowski T, Bari RP, Stitt M, Scheible WR, Udvardi MK (2004) Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root-and shoot-specific genes. Plant Journal 38: 366–379. [DOI] [PubMed] [Google Scholar]
- 2. Morrison T, Hurley J, Garcia J, Yoder K, Katz A, Roberts D, et al. (2006) Nanoliter high throughput quantitative PCR. Nucleic Acids Research 34: e123–e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quadrana L, Almeida J, Otaiza SN, Duffy T, da Silva JVC, de Godoy F, et al. (2013). Transcriptional regulation of tocopherol biosynthesis in tomato. Plant Molecular Biology 81: 309–325. 10.1007/s11103-012-0001-4 [DOI] [PubMed] [Google Scholar]
- 4. Bustin AS, Nolan T (2004) Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. Journal of Biomolecular Techniques 15: 155–166. [PMC free article] [PubMed] [Google Scholar]
- 5. Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonák J, Lind K, et al. (2006) The real-time polymerase chain reaction. Molecular Aspects of Medicine 27: 95–125. [DOI] [PubMed] [Google Scholar]
- 6. Gachon CMA, Charrier B (2004) Real-time PCR: what relevance to plant studies? Journal of Experimental Botany 55:1445–1454. [DOI] [PubMed] [Google Scholar]
- 7. Derveaux S, Vandesompele J, Hellemens J (2010) How to do successful gene expression analysis using real-time PCR. Methods 50: 227–230. 10.1016/j.ymeth.2009.11.001 [DOI] [PubMed] [Google Scholar]
- 8. Chi X, Hu R, Yang Q, Zhang X, Pan L, Chen N, et al. (2012) Validation of reference genes for gene expression studies in peanut by quantitative real-time RT-PCR. Molecular Genetics and Genomics 287: 167–176. 10.1007/s00438-011-0665-5 [DOI] [PubMed] [Google Scholar]
- 9. Huang L, Yan H, Jiang X, Zhang X, Zhang Y, Huang X, et al. (2014) Evaluation of candidate reference genes for normalization of quantitative RT-PCR in switchgrass under various abiotic stress conditions. BioEnergy Research 7: 1201–1211. [Google Scholar]
- 10. de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, et al. (2005) Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Laboratory investigation 85: 154–159. [DOI] [PubMed] [Google Scholar]
- 11. Dheda K, Huggett JF, Bustin SA, Johnson MA, Rook G, Zumla A (2004) Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 37: 112–119. [DOI] [PubMed] [Google Scholar]
- 12. Nolan T, Hands RE, Bustin SA (2006) Quantification of mRNA using real-time RT-PCR. Nature protocols 1: 1559–1582. [DOI] [PubMed] [Google Scholar]
- 13. Guénin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, Gutierrez L (2009) Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. Journal Experimental Botany 60: 487–493. [DOI] [PubMed] [Google Scholar]
- 14. Perini P, Pasquali G, Margis-Pinheiro M, de Oliviera PRD, Revers LF (2014) Reference genes for transcriptional analysis of flowering and fruit ripening stages in apple (Malus × domestica Borkh.). Molecular Breeding, 34:829–842. [Google Scholar]
- 15. Bustin SA (2002) Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. Journal of Molecular Endocrinology 29: 23–39. [DOI] [PubMed] [Google Scholar]
- 16. Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiology 139: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gutierrez L, Mauriat M, Guénin S, Pelloux J, Lefebvre JF, Louvet R, et al. (2008) The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnology Journal 6: 609–618. 10.1111/j.1467-7652.2008.00346.x [DOI] [PubMed] [Google Scholar]
- 18. Expósito-Rodríguez M, Borges AA, Borges-Pérez A, Pérez JA (2008) Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biology 8: 131 10.1186/1471-2229-8-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Remans T, Smeets K, Opdenakker K, Mathijsen D, Vangronsveld J, Cuypers A (2008) Normalisation of real-time RT-PCR gene expression measurements in Arabidopsis thaliana exposed to increased metal concentrations. Planta 227: 1343–1349. 10.1007/s00425-008-0706-4 [DOI] [PubMed] [Google Scholar]
- 20. Cruz F, Kalaoun S, Nobile P, Colombo C, Almeida J, Barros LM, et al. (2009) Evaluation of coffee reference genes for relative expression studies by quantitative real-time RT-PCR. Molecular Breeding 23: 607–616. [Google Scholar]
- 21. Artico S, Nardeli SM, Brilhante O, Grossi-de-Sa MF, Alves-Ferreira M (2010) Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biology 10: 49 10.1186/1471-2229-10-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Vega-Bartol JJ, Santos RR, Simões M, Miguel CM (2013) Normalizing gene expression by quantitative PCR during somatic embryogenesis in two representative conifer species: Pinus pinaster and Picea abies . Plant Cell Reports 32: 715–729. 10.1007/s00299-013-1407-4 [DOI] [PubMed] [Google Scholar]
- 23. Galli V, Borowski JM, Perin EC, da Silva Messias R, Labonde J, dos Santos Pereira I, et al. (2015) Validation of reference genes for accurate normalization of gene expression for real time-quantitative PCR in strawberry fruits using different cultivars and osmotic stresses. Gene 554: 205–214. 10.1016/j.gene.2014.10.049 [DOI] [PubMed] [Google Scholar]
- 24. Faccioli P, Ciceri GP, Provero P, Stanca AM, Morcia C, Terzi V (2007) A combined strategy of “in silico” transcriptome analysis and web search engine optimization allows an agile identification of reference genes suitable for normalization in gene expression studies. Plant Molecular Biology 63: 679–688. [DOI] [PubMed] [Google Scholar]
- 25. Elbl P, Campos RA, Lira BS, Andrade SCS, Jo L, dos Santos ALW, et al. (2015) Comparative transcriptome analysis of early somatic embryo formation and seed development in Brazilian pine, Araucaria angustifolia (Bertol.) Kuntze. Plant Cell Tissue and Organ Culture 120: 903–915. [Google Scholar]
- 26.International Union of Conservation of Nature Red List of Threatened Species (2014). http://www.iucnredlist.org/details/32975/0. Accessed 03 December 2014.
- 27. Steiner N, Santa-Catarina C, Andrade JBR, Balbuena TS, Guerra MP, Handro W, et al. (2008) Araucaria angustifolia biotechnology. Functional Plant Science and Biotechnology 2: 20–28. [Google Scholar]
- 28. Murashige T, Skoog F (1962) A revised medium for rapid growth and bio-assays with tobacco tissue cultures. Physiologia Plantarum 15: 473–497. [Google Scholar]
- 29. Becwar MR, Noland TL, Wyckoff JL (1989) Maturation, germination, and conversion of Norway spruce (Picea abies L.) somatic embryos to plant. In Vitro Cellular and Developmental Biology 26: 575–580. [Google Scholar]
- 30. Chang S, Puryear J, Cairney J (1993) A simple and efficient method for isolating RNA from Pine trees. Plant Molecular Biology Reporter 11: 113–116. [Google Scholar]
- 31. Salzman RA, Fujita T, Zhu-Salzman K, Hasegawa PM, Bressan RA (1999) An improved RNA isolation method for plant tissues containing high levels of phenolic compounds or carbohydrates. Plant Molecular Biology Reporter 17: 11–17. [Google Scholar]
- 32. Brunner AM, Yakovlev IA, Strauss SH (2004) Validating internal controls for quantitative plant gene expression studies. BMC Plant Biology 4:14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iskandar HM, Simpson RS, Casu RE, Bonnett GD, Maclean DJ, Manners JM (2004) Comparison of reference genes for quantitative real-time polymerase chain reaction analysis of gene expression in sugarcane. Plant Molecular Biology Reporter 22: 325–337. [Google Scholar]
- 34. Hu R, Fan C, Li H, Zhang Q, Fu YF (2009) Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Molecular Biology 10: 93 10.1186/1471-2199-10-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Narsai R, Ivanova A, Ng S, Whelan J (2010). Defining reference genes in Oryza sativa using organ, development, biotic and abiotic transcriptome datasets. BMC Plant Biology 10: 56 10.1186/1471-2229-10-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mafra V, Kubo KS, Alves-Ferreira M, Ribeiro-Alves M, Stuart RM, Boava LP, et al. (2012) Reference genes for accurate transcript normalization in citrus genotypes under different experimental conditions. PloS One 7: e31263 10.1371/journal.pone.0031263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. Journal Molecular Biology 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 38. R Development Core Team. R: A language and environment for statistical computing: reference index version 2.8.0. Vienna foundation for statistical computing, 2011. <http://www.r-project.org.> 02 Jan. 2015 [Google Scholar]
- 39. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry 55: 611–622. 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
- 40. Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, Van den Hoff MJB, et al. (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Research 37: e45 10.1093/nar/gkp045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vandesompele J, de Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology. 3(7):research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Andersen CL, Jensen JL, Ørntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Research 64: 5245–5250. [DOI] [PubMed] [Google Scholar]
- 43. Milone DH, Stegmayer GS, Kamenetzky L, López M, Lee JM, Giovannoni JJ, et al. (2010) * omeSOM: a software for clustering and visualization of transcriptional and metabolite data mined from interspecific crosses of crop plants. BMC Bioinformatics 11: 438 10.1186/1471-2105-11-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steiner N, Santa-Catarina C, Guerra MP, Cutri L, Dornelas MC, Floh EIS (2012) A gymnosperm homolog of SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASE-1 (SERK1) is expressed during somatic embryogenesis. Plant Cell, Tissue and Organ Culture 109: 41–50. [Google Scholar]
- 45. Schlögl PS, dos Santos ALW, Vieira LN, Floh EIS, Guerra MP (2012a) Cloning and expression of embryogenesis-regulating genes in Araucaria angustifolia (Bert.) O. kuntze (Brazilian pine). Genetics and Molecular Biology 35(1):172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schlögl PS, dos Santos ALW, Vieira LN, Floh EIS, Guerra MP (2012b) Gene expression during early somatic embryogenesis in Brazilian pine (Araucaria angustifolia (Bert) O. Ktze). Plant Cell Tissue and Organ Culture 108: 173–180. [Google Scholar]
- 47. Wu T, Zhang R, Gu C, Wu J, Wan H, Zhang S, et al. (2012) Evaluation of candidate reference genes for real time quantitative PCR normalization in pear fruit. African Journal of Agricultural Research 7: 3701–3704. [Google Scholar]
- 48. Minocha R, Majumdar R, Minocha SC (2014) Polyamines and abiotic stress in plants: a complex relationship. Frontiers in Plant Science 5: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Majumdar R, Shao L, Minocha R, Long S, Minocha SC (2013) Ornithine: the overlooked molecule in the regulation of polyamine metabolism. Plant Cell Physiology. pct053. [DOI] [PubMed] [Google Scholar]
- 50. Jo L, dos Santos ALW, Bueno CA, Barbosa H.R. and Floh E.I.S. (2014) Proteomic analysis and polyamines, ethylene and reactive oxygen species levels of Araucaria angustifolia (Brazilian pine) embryogenic cultures with different embryogenic potential. Tree Physiology 34: 94–104 10.1093/treephys/tpt102 [DOI] [PubMed] [Google Scholar]
- 51. Mhamdi A, Noctor G, and Baker A (2012) Plant catalases: peroxisomal redox guardians. Archives of Biochemistry and Biophysics 525: 181–194. 10.1016/j.abb.2012.04.015 [DOI] [PubMed] [Google Scholar]
- 52. Yang HL, Liu YJ, Wang CL, Zeng QY (2012) Molecular evolution of trehalose-6-phosphate synthase (TPS) gene family in Populus, Arabidopsis and rice. PloS One, 7(8): e42438 10.1371/journal.pone.0042438 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ct values of the replicates were plotted against each other.
(TIF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.