

Significance
Calcium ions play a central role in controlling contraction in heart muscle cells. We have investigated the function of a calcium ion signaling protein found in the heart called STIM1 (stromal interaction molecule 1) that has a known function in many other cells. We discovered, however, that in the heart STIM1 works completely differently. Instead of enabling Ca2+ entry into the cell, STIM1 enhances the rate and amount of Ca2+ stored and released from intracellular organelles in the steady state. Hence, more STIM1 expression increases the magnitude of the calcium signal, and this increases contraction. STIM1 thus may play an important and novel role in ventricular myocytes under physiological conditions by regulating the [Ca2+]i transient and contraction.
Keywords: STIM1, phospholamban, store-operated calcium entry, diffusion coefficient, heart
Abstract
In ventricular myocytes, the physiological function of stromal interaction molecule 1 (STIM1), an endo/sarcoplasmic reticulum (ER/SR) Ca2+ sensor, is unclear with respect to its cellular localization, its Ca2+-dependent mobilization, and its action on Ca2+ signaling. Confocal microscopy was used to measure Ca2+ signaling and to track the cellular movement of STIM1 with mCherry and immunofluorescence in freshly isolated adult rat ventricular myocytes and those in short-term primary culture. We found that endogenous STIM1 was expressed at low but measureable levels along the Z-disk, in a pattern of puncta and linear segments consistent with the STIM1 localizing to the junctional SR (jSR). Depleting SR Ca2+ using thapsigargin (2–10 µM) changed neither the STIM1 distribution pattern nor its mobilization rate, evaluated by diffusion coefficient measurements using fluorescence recovery after photobleaching. Two-dimensional blue native polyacrylamide gel electrophoresis and coimmunoprecipitation showed that STIM1 in the heart exists mainly as a large protein complex, possibly a multimer, which is not altered by SR Ca2+ depletion. Additionally, we found no store-operated Ca2+ entry in control or STIM1 overexpressing ventricular myocytes. Nevertheless, STIM1 overexpressing cells show increased SR Ca2+ content and increased SR Ca2+ leak. These changes in Ca2+ signaling in the SR appear to be due to STIM1 binding to phospholamban and thereby indirectly activating SERCA2a (Sarco/endoplasmic reticulum Ca2+ ATPase). We conclude that STIM1 binding to phospholamban contributes to the regulation of SERCA2a activity in the steady state and rate of SR Ca2+ leak and that these actions are independent of store-operated Ca2+ entry, a process that is absent in normal heart cells.
Store-operated Ca2+ entry (SOCE) is a cellular mechanism to ensure that sufficient levels of Ca2+ are present in the intracellular Ca2+ stores to enable robust signaling (1). SOCE depends on the presence and interaction of two proteins, STIM1 (stromal interaction molecule 1) and Orai1 (a low conductance plasma/sarcolemmal Ca2+ channel), or their equivalents (2–5). STIM1 is an endo/sarcoplasmic reticulum (ER/SR) Ca2+-sensitive protein that interacts with Orai1 to activate the channel function of Orai1, a Ca2+ selective channel, and thus permit Ca2+ entry. SOCE is clearly present in nonexcitable cells such as T lymphocytes and some excitable cells including skeletal muscle cells (4, 6–13). STIM1 is a membrane-spanning ER/SR protein with a single transmembrane domain and a luminal Ca2+ ([Ca2+]ER/SR)-sensing domain. When luminal Ca2+ is low (i.e., [Ca2+]ER/SR drops to less than 300 µM), then STIM1 self-aggregates and associates with Orai1 to activate it, producing a SOCE current (ISOCE) (2, 14–16) and Ca2+ entry (with a reversal potential ESOCE ∼ +50 mV or more) (17, 18). Then, as [Ca2+]ER/SR increases in response to the Ca2+ influx, the process reverses.
In adult skeletal muscle cells, Ca2+ influx is normally low, and it has been suggested that SOCE is needed for maintaining an appropriate level of [Ca2+]ER/SR and correct Ca2+ signaling (6, 7, 9, 19). In skeletal muscle, it has been hypothesized that STIM1 is prelocalized in the SR terminal cisternae (6, 20) and hence can more rapidly respond to changes in [Ca2+]ER/SR. The putative importance of SOCE in skeletal muscle was further supported by the observation that the skeletal muscle dysfunction is significant in STIM1-null mice where 91% (30/33) of the animals died in the perinatal period from a skeletal myopathy (6). Furthermore, in humans, STIM1 mutations were identified as a genetic cause of tubular aggregate myopathy (21).
Despite the clarity of the SOCE paradigm, the canonical SOCE activation process described above does not apply to all conditions in which STIM1 and Orai1 interact. For example, in T lymphocytes, STIM1 clustering is necessary and sufficient to activate SOCE, regardless of whether [Ca2+]ER/SR is low (4). When present, the STIM1 EF hand mutation causes STIM1 oligomerization and constitutive Ca2+ influx across the plasma membrane into cells with full Ca2+ stores (4). Although this is consistent with the use of STIM1 clusters and puncta to measure the activation of Orai1 (15, 16, 22, 23), it does not necessarily reflect the state of [Ca2+]ER/SR. Furthermore, several small-molecule bioactive reagents, such as 2-APB and FCCP, neither of which causes [Ca2+]ER/SR depletion, induce STIM1 clustering (24). Thus, STIM1 may have actions that are more complicated than simple [Ca2+]ER/SR sensing and Orai1 signaling.
Cardiomyocytes have been reported to have SOCE (8, 13, 25, 26) but are very different from many of the cells noted above that exhibit significant [Ca2+]ER/SR depletion-sensitive Ca2+ entry through the Ca2+-selective Orai1. Cardiac ventricular myocytes are different from the other cells in that they have large, regular, and dynamic changes in [Ca2+]i and robust influx and extrusion pathways across the sarcolemmal membrane. For example, it is not unusual for investigators to measure a 10–20 nA calcium current (ICa,L) in single cardiac ventricular myocytes that is readily extruded by the sarcolemmal Na+/Ca2+ exchanger. Because of these large fluxes, adult ventricular myocytes have no “need” for SOCE and the same logic applies to neonatal cardiomyocytes. Nevertheless, reports of SOCE in neonatal cardiac myocytes are clear (10, 12, 13). Against this background, we have attempted to determine if STIM1 is present in adult cardiomyocytes and, if so, where the protein is located, how it is mobilized, and how it may interact with other Ca2+ signal proteins. In the work presented here, we show that STIM1 is present but that its function in heart is distinct from the canonical SOCE behavior and does not contribute to Ca2+ influx through ISOCE. Instead we show that STIM1 binds phospholamban (PLN), an endogenous SERCA2a inhibitor in the heart (27), and by doing so reduces the PLN-dependent inhibition of SERCA2a and thereby indirectly activates SERCA2a.
Results
Experiments were designed to investigate whether or not STIM1 is present in adult rat cardiac ventricular myocytes and, if so, what the function of STIM1 may be. The experiments were carried out using freshly isolated single ventricular myocytes from Sprague–Dawley rats (Materials and Methods).
STIM1 in Heart.
Using rabbit polyclonal anti-STIM1 antibody (Sigma-Aldrich) to probe a Western blot at a distance around 84 kDa, we identified STIM1 protein from liver, from a ventricle homogenate, and from a single cardiac myocyte preparation, as shown in Fig. 1A. These bands were absent in STIM1-null mice for samples taken from single cardiac myocytes and from mouse flexor digitalis brevis (FDB) skeletal muscle (SI Appendix, Fig. S1A). Our observations thus suggest that there is STIM1 protein in heart muscle cells. Similar results were seen with other STIM1 antibodies from BD Biosciences and Santa Cruz Biotechnology. To determine where the STIM1 in rat ventricular myocytes was located, we carried out confocal immunofluorescence imaging, as shown in Fig. 1B. The Left panel (red) shows the image of STIM1 immunofluorescence localization that resembles the distribution of the junctional SR (jSR) along the Z-disk. The position of the STIM1 within the sarcomere is confirmed by the simultaneous immunostaining of α-actinin (green). The antibody specificity is excellent, as shown in SI Appendix, Fig. S1B. To improve further the signal of the immunofluorescence localization of STIM1 in rat ventricular myocytes, we used preabsorbed antibodies (with STIM1 KO cells; Materials and Methods) to reduce nonspecific signals (Fig. 1C). The jSR staining pattern is better seen in Fig. 1C with a mixture of puncta and Z-line signals.
Fig. 1.

Expression and localization of STIM1 in rat ventricular myocytes. (A) The STIM1 protein is found in isolated ventricular myocytes (Left), in ventricular tissue (Middle), and in liver tissue (Right) by Western blot to be the same size. The total protein loaded for each lane is indicated at the bottom. (B) Immunofluorescence imaging shows the cellular distribution of STIM1 (Left) and α-actinin (Middle) in a freshly isolated ventricular myocyte. Superimposed images (Right) indicate that the STIM1 immunofluorescence is at the Z-disk, identified by α-actinin. The insets (Bottom) correspond to the area marked with the dashed white box in the upper left of each image. STIM1 appears to form puncta and line segments along the Z-disk, consistent with it being located in clusters at the jSR. (C) STIM1 immunofluorescence using anti-STIM1 antibody that was preabsorbed using STIM1-null myocytes shows that the longitudinal streaks of “STIM1 fluorescence” shown in the Left panel of B was nonspecific binding. The Inset better reveals the jSR pattern of immunofluorescence for STIM1.
Effect of SR Ca2+ Depletion on STIM1 Distribution Within a Ventricular Myocyte.
Because the fundamental feature of SOCE is that it is activated by the depletion of Ca2+ from the SR/ER store, we carried out experiments to deplete the SR/ER Ca2+ stores. We used the “standard method,” which involves applying thapsigargin (Tg; 2 and 10 μM) to block the SR/ER Ca2+ ATPase (SERCA or the SR Ca2+ pump). In the presence of prolonged Tg (10 min) exposure, the SR/ER stores become completely depleted of Ca2+ because the leak of Ca2+ out of the SR is no longer balanced by the SERCA-dependent Ca2+ influx (SI Appendix, Fig. S2 A and B). The time course of this loss (SI Appendix, Fig. S2 C and D) is rapid and complete. In the absence of a functioning SERCA, the relative contributions to the [Ca2+]i transient of L-type Ca2+ entry and triggered SR Ca2+ release is clear (SI Appendix, Fig. S2 C–F) and shown to be about 25% in rat ventricular myocytes under these conditions. This is an upper limit because the jSR Ca2+-dependent inactivation of ICa,L is removed under these conditions, and this leads to a larger Ca2+ influx through ICa,L. When the canonical SOCE in nonexcitable cells is activated following Ca2+ store depletion, STIM1 proteins aggregate to form clusters of STIM1 proteins, also called STIM1 puncta (14, 28, 29). The organization of endogenous STIM1 proteins in ventricular myocytes is shown before Ca2+ store depletion in the heart (Figs. 1 B and C and 2A) and after depletion (Fig. 2 A, Middle and Right). There is no apparent reorganization of the endogenous STIM1 proteins after the SR is depleted of Ca2+ in rat adult ventricular myocytes. The consistent results were obtained from STIM1 overexpressing myocytes (SI Appendix, Fig. S3), where STIM1-mCherry and STIM1-eYFP were cotransfected. What is clear is that the proteins appear to exist as a mixture of puncta and linear segments at the Z-disk in the presence and absence of [Ca2+]SR/ER (SI Appendix, Fig. S4). In contrast, the fairly uniform distribution of STIM1 in the ER/SR in nonexcitable cells when [Ca2+]SR/ER is high changes to a pattern of STIM1 puncta (clusters of STIM1 proteins) upon ER/SR Ca2+ depletion due to the Tg treatment (4). These STIM1 clusters interact with the plasmalemmal Ca2+ channel Orai1 and signal them to open and thereby produce SOCE.
Fig. 2.

STIM1 properties in intact and SR Ca2+-depleted rat ventricular myocytes. (A) Immunofluorescence imaging shows the cellular distribution of endogenous STIM1 in intact (Left) and Tg (2 and 10 μM)-treated myocytes (Middle and Right). (Lower panel) Magnified images of the boxed region in the Upper panel. (B) A representative myocyte shows the infection of STIM1-mCherry in ventricular myocyte cultured for 48 h. (C) Zoom-in image of the framed region in B. Dashed white box shows a 6 × 6 µm area subject to photobleaching. (D) Western blot shows the overexpression of STIM1-mCherry (upper band) in rat ventricular myocytes. (E) STIM1-mCherry FRAP in the absence and presence of Tg. (F) Summary data of the diffusion coefficient of STIM1-mCherry in the absence and presence of Tg (2 µM; n = 21 and 15 myocytes in control and Tg, respectively).
Although STIM1 organization in adult cardiac ventricular myocytes seems unresponsive to [Ca2+]SR/ER, the mobility of these proteins could change. To measure the STIM1 mobility, adenovirus carrying STIM1-mCherry was transfected into a primary culture of adult rat ventricular myocytes. Fig. 2 B–D shows that STIM1-mCherry is clearly expressed in rat ventricular myocytes and the spatial distribution is similar to that of WT. Fluorescence recovery after photobleaching (FRAP) experiments were performed to evaluate the mobility of STIM1-mCherry before and after [Ca2+]SR/ER depletion (Fig. 2 B, C, E, and F). Photobleaching was carried out in a 6 × 6 µm square (avoiding the nucleus) using high illumination intensity (543 nm and 488 nm laser). The fluorescence imaging during the recovery was monitored continuously at 0.5 Hz (Fig. 2E). Our results show that STIM1-mCherry mobilizes slowly within the SR membrane. The diffusion coefficient, estimated using SimFRAP (written by Levi A. Gheber’s Lab at Ben Gurion University of the Negev, Israel, Plugin in ImageJ, NIH), is ∼0.012 µm2/s (Fig. 2F), much slower than in HeLa or RBL (rat basophilic leukemia) cells (22), but similar to some membrane proteins such as water channel protein aquaporin-3 (0.0104 µm2/s) and delayed rectifier K+ channel (Kv2.1; 0.03–0.06 µm2/s) (30, 31). The diffusion coefficient of a protein may vary and depend on many factors. For example, the diffusion coefficient of BKα (Ca2+-activated K+ channel α subunit) in vascular smooth muscle cells is ∼20 times slower than in HEK293 cells and 10,000 times slower than in bilayer (32). Therefore, it is not surprising that the diffusion coefficient of STIM1 in ventricular myocytes is slower than in HeLa cells, particularly in light of the possible STIM1 clustering in heart cells (see results below). In the nonexcitable cells, ER Ca2+ depletion with ionomycin clearly slowed diffusion by a factor of 2 (from 0.1 to 0.05 μm2/s), and this slowing was attributed to oligomerization of the STIM1 (22). In contrast to the FRAP behavior of STIM1 in adult ventricular myocytes, in cultured neonatal cardiac myocytes the STIM1-GFP diffusion coefficient is ∼0.025 μm2/s (SI Appendix, Fig. S5), about 2 times faster than that in adult. The FRAP recovery time and STIM1 diffusion coefficients are largely unaffected in both adult and neonatal rat heart cells by [Ca2+]SR/ER depletion following Tg treatment. Taken together, these results suggest that STIM1 in adult ventricular myocytes diffuses more slowly within the SR/ER structure than the STIM1 protein does in neonatal cardiac myocytes, and neither diffusion rate constants are sensitive to changes in [Ca2+]SR/ER. Furthermore, the slow rate of diffusion raises the possibility that the STIM1 may be part of a large protein complex or may be oligomerized or both.
The very slow movement of STIM1 in adult heart cells may reflect the complex multimeric assembly of the STIM1 proteins, as suggested by Liou et al. (2007) (22). Fig. 3A shows that STIM1, displayed in a native gel, forms high molecular weight (MW) protein complexes in ventricular myocytes (see band ∼550 kDa). The slow mobility of STIM1 seen in ventricular myocytes (Fig. 2) is likely due in part to this high MW protein complex. There are no monomeric bands of STIM1 present in Fig. 3A, but there is a band at ∼240 kDa that is consistent with a STIM1 dimer. The composition of the dominant high MW band (∼550 kDa) is not fully known but may contain STIM1 multimers as well as STIM1 monomers or multimers associated with other proteins components. Although the dominant high MW band is large enough to be consistent with a hexameric STIM1 cluster or a smaller multimer with other proteins, additional experiments are required to determine the composition of the high MW band. Importantly, this dominant band is seen under all conditions and is significantly enhanced when STIM1-mCherry is expressed. These data suggest that a STIM1 dimer certainly exists, according to the results obtained from 2D blue native polyacrylamide gel electrophoresis (BN–PAGE) (Fig. 3 B and C), a finding consistent with earlier studies (22, 33). We observe direct evidence of STIM1/STIM1-mCherry and STIM1-mCherry/STIM1-eYFP dimers in our 2D BN-PAGE and coimmunoprecipitation (co-IP) data (Fig. 3 B–D), under conditions when all three STIM1 proteins are expressed. The STIM1 clusters and the high MW bands in Fig. 3A under all conditions of [Ca2+]SR/ER in adult ventricular myocytes are consistent with our observation that STIM1 is organized into both puncta and linear structures in the Z-disk, as shown in Figs. 1–3 (also see SI Appendix, Figs. S3 and S4).
Fig. 3.

Multimeric properties of STIM1 in rat ventricular myocytes. (A) BN-PAGE shows that endogenous and overexpressed STIM1 forms multimers or protein complex in ventricular myocytes. (B) 2D BN-PAGE and SDS/PAGE show the separation of STIM1 multimer or protein complex using anti-mCherry (to detect STIM1-mCherry, Left panel) and anti-STIM1 (to detect STIM1 and STIM1-mCherry, Right panel). (C) Amplification of the corresponding area marked with the dashed white boxes in B (Upper panel). Band distribution profiles corresponding to the dashed color boxes in the Upper panels show overlap of STIM1 and STIM1-mCherry (Lower panel). Values are presented as arbitrary units (AUs) of the intensity, with background subtracted. (D) Representative (of two independent tests) co-IP shows physical association between STIM1, STIM1-mCherry, and STIM1-eYFP in the absence and presence of Tg (10 µM, 30 min). The lysates from the myocytes overexpressing mCherry and STIM1-eYFP (left lane) or STIM1-mCherry and STIM1-eYFP (middle and right lanes) were immunoprecipitated with anti-mCherry and probed with anti-YFP, anti-mCherry, and anti-STIM1.
Is There Evidence for SOCE in Rat Cardiac Ventricular Myocytes?
That STIM1 is preassembled in puncta at the Z-disk suggests that it may be poised to interact with Orai1 (or the equivalent) to enable SOCE when [Ca2+]SR/ER is very low. To test this possibility, a whole-cell patch-clamp approach was used to measure current components related to [Ca2+]SR/ER depletion (see Materials and Methods for details). From a holding potential of –80 mV, the membrane potential was depolarized to +50 mV and repolarized in a linear ramp to –100 mV over 1 s, and the measured current is shown in Fig. 4 A and B. Multiple recordings were carried out with 20 s between measurements. Cells were superfused with Tyrode’s solution supplemented with verapamil (10 µM) and TEA (tetraethylammonium, 10 mM) to block L-type Ca2+ channels and K+ channels. Tg (10 µM) was applied after 10–20 recordings when a stable current was obtained. A clear Tg (10 μM)-sensitive current (–218 ± 19 pA, at –80 mV, n = 14 cells) is observed, but this current is not blocked by a specific SOCE blocker (34) (BTP-2, 20 μM) nor by La3+ (20 µM) (see green and purple lines in Fig. 4B). Fig. 4C shows the time course of measurements of membrane current at –80 mV before and after Tg (10 μM) and the absence of any change produced by La3+ (20 μM). The apparent absence of any SOCE-dependent current is consistent with the observed reversal potential (Erev,∼–15 mV) of the Tg-sensitive current that we have seen (Fig. 4 A–D). Although the Erev shown in Fig. 4B is consistent with a nonselective ion channel and is similar to the data presented by Hulot et al. (2011), they report it as a SOCE current (8). Note that ISOCE, when measured in cells where SOCE has been validated, has been reported to reverse close to +50 mV or more (17, 18), as it is highly selective for Ca2+. Finally, measurements of [Ca2+]i under physiological conditions in the presence and absence of Tg (2 μM) shown in Fig. 4 E and F show no significant difference. Consistent with these results, neither change of diastolic membrane potential nor ICa,L was observed in STIM1 overexpressing myocytes or in the myocytes from STIM1-null mice (Fig. 4G and SI Appendix, Fig. S6). Taken together, these data, along with the undetectable Orai1 in rat ventricular myocytes (SI Appendix, Fig. S7A), indicate that there is no SOCE in normal adult ventricular myocytes.
Fig. 4.

Whole-cell patch-clamp recording and Ca2+ imaging of store-operated Ca2+ current/entry in rat ventricular myocytes. (A) Whole-cell patch-clamp current recorded in the absence and presence of Tg (10 μM), La3+ (nonselective SOCE blocker), and/or BTP-2 (20 μM, selective SOCE blocker). (B) Current–voltage relationships show Tg, BTP-2, and La3+-sensitive currents obtained from A by subtracting the corresponding controls. (C) A representative recording (–80 mV) shows a Tg-induced current that is not La2+-sensitive. (D) Mean data show a Tg-sensitive current in ventricular myocytes (n = 14, 14, 8, and 6 cells from left to right columns). *P < 0.05 vs. control. (E) Traces show fluo 4 intensity (cytosolic Ca2+) in control (Upper) and STIM1 overexpressing (Lower) myocytes. Cells were initially exposed to physiological saline solution, followed by Ca2+-free solution, and then by Ca2+-free solution with Tg (2 μM). After ∼5 min, Ca2+ was reintroduced into the bath solution. The increase of cytosolic Ca2+ upon Ca2+ readdition was analyzed and compared between control and STIM1 overexpressing myocytes, as shown in F. (F) Summary data of fluo 4 intensity at physiological Ca2+ after SR Ca2+ depletion. n = 20 and 21 cells for control and STIM1, respectively. (G) Summary of the diastolic membrane potential in control and STIM1 overexpressing rat ventricular myocytes. n = 6 cells for the control and STIM1 overexpressing groups.
What Does STIM1 Do in Ventricular Myocytes?
The presence of STIM1 protein as puncta and other Z-disk structures suggests the protein serves a function. A clue to a possible important role comes from adult ventricular myocytes overexpressing STIM1. These cells were observed to have a relatively large number of propagating Ca2+ waves, compared with rare Ca2+ waves found in control cells (Fig. 5). The control cells expressed mCherry rather than STIM1-mCherry (Fig. 5 A and B). Although 27% (n = 40 cells) of control cells had observable Ca2+ waves, 93% (n = 58 cells) of the STIM1overexpressing cells displayed Ca2+ waves. Fig. 5C shows the consistent and persistent Ca2+ waves from the STIM1 overexpressing cells. Fig. 5D reports the histogram comparing Ca2+ wave frequency in control versus STIM1 overexpressing cells, and these data are summarized in Fig. 5E. Consistent with STIM1 overexpressing results, STIM1 knockout mouse ventricular myocytes display reduced spontaneous Ca2+ waves (SI Appendix, Fig. S8D). That these Ca2+ waves in STIM1 cells depend on [Ca2+]SR and not SOCE is revealed because Tg (2 μM) blocked them but Gd3+ (1 μM) does not, as shown in Fig. 5 F and G. Supporting this conclusion is the observation that ryanodine (10 μM) also abolished the Ca2+ waves. These data demonstrated that this STIM1-induced Ca2+ wave generation is nonrelated to SOCE but dependent on SR Ca2+ uptake and release. Indeed, no increased frequency of Ca2+ waves was observed in myocytes even when Orai1 is overexpressed (SI Appendix, Fig. S7 B and C).
Fig. 5.

The change of SR Ca2+ leak and STIM1 overexpression. (A) XY time-series images of [Ca2+]i (fluo 4) in control (Upper) and STIM1 (Lower) overexpressing myocytes. (B) Line scan images of fluo 4 in control and STIM1 overexpressing myocytes (lines are shown in A). (C) An example of Ca2+ wave profile (white box in A) in STIM1 overexpressing myocytes. (D) Histogram shows the cell count and Ca2+ wave frequency in control and STIM1 overexpressing myocytes. (E) Summary data show the change of Ca2+ wave frequency in STIM1 overexpressing myocytes (n = 40 and 58 cells in control and STIM1, respectively). (F) Representative Ca2+ wave profiles show the effect of GdCl3 (1 μM), KB-R 7943 (10 μM), and Tg (2 μM). (G) Summary data show the effect of GdCl3 and KB-R 7943 on spontaneous Ca2+ wave frequency in STIM1 overexpressing myocytes (n = 7 and 5 cells in the Gd3+ and KB-R groups, respectively). *P < 0.05 vs. control.
How Does STIM1 Affect [Ca2+]SR?
SR Ca2+ content was measured in STIM1 and control cells as previously described (35) (SI Appendix, Fig. S9). These data show that SR Ca2+ content is increased by 44% in STIM1 overexpressing cells compared with control. The SR Ca2+ content in control cells was 67.5 ± 8.6 µM/L cytosol (n = 7 cells) versus 97.4 ± 10.6 µM/L cytosol in STIM1 overexpressing cells (n = 5 cells) (see Fig. 6 A and B for measurements and Fig. 6C for statistics). That the SR Ca2+ content is significantly different suggested to us that we should examine the [Ca2+]i transients in STIM1 and control cells. Fig. 7 A and B shows that the peak of the [Ca2+]i transients was significantly higher in STIM1 overexpressing cells. The kinetics of decay of the [Ca2+]i transients were notably faster in the STIM1 overexpressing rat ventricular myocytes, whereas the time to peak is not changed (Fig. 7 A and C), suggesting faster Ca2+ removal from cytosol. A result consistent with this observation is seen in STIM1 knockout mouse ventricular myocytes (SI Appendix, Fig. S8 A–C). Knocking out STIM1 reduced the peak of the [Ca2+]i transient and slowed the rate of decline. This raises the question of how STIM1 can affect [Ca2+]SR and the kinetics of decay of the [Ca2+]i transient. The array of changes observed in the STIM1overexpressing cells is consistent with an increase in the turnover rate of SERCA. Changes in protein expression that could account for these observations include an increase in phosphorylated PLN (p-PLN) expression, an increase of SERCA2a expression, or a decrease of PLN expression. However, none of these changes is observed, as shown in Fig. 8 A and D. Nevertheless, there is an alternative possibility if STIM1 were to bind to the non–p-PLN and thereby to decrease the amount of PLN that could bind to SERCA2a and inhibit it. To investigate this possibility, interactions between STIM1 and PLN were examined in the experiments shown in Fig. 8B. When pulling down STIM1, PLN was detected in the protein complex extracted from isolated ventricular myocytes (Fig. 8B). SERCA2a was also probed but demonstrated much less binding to STIM1 than PLN did (Fig. 8B). In the myocytes where SR Ca2+ was depleted by Tg (10 μM, 20 min), more PLN (∼117% of control) was detected compared with that in control myocytes (Fig. 8 B and E), suggesting that SR Ca2+ depletion promotes STIM1–PLN interaction (see Discussion for a more complete explanation). However, application of isoproterenol (0.1 and 1 μM), which causes SR Ca2+ increase and PLN phosphorylation, decreased the interaction between STIM1 and PLN (Fig. 8 C and E), suggesting that STIM1 favors binding to non–p-PLN. To explore this possibility, the ability of STIM1 to bind PLN and p-PLN was examined, as shown in Fig. 8 F and G and SI Appendix, Fig. S10. In Fig. 8G, PLN was identified as a STIM1-interacting protein in a T7 phage display screen using STIM1 c terminus (STIM1c) as bait. This interaction was verified in vitro by ELISA (Fig. 8 F and G). In SI Appendix, Fig. S10, the relative ability of PLN to bind STIM1c was compared with the ability of p-PLN to bind STIM1c and was shown to be about 20-fold greater. To control for the possibility that the amount of p-PLN limited the sensitivity of the assay, the same test was repeated but following 10 nM treatment with isoproterenol, which increases p-PLN. As shown in SI Appendix, Fig. S10B, the binding of PLN to STIM1c remained about 20-fold greater than p-PLN. When these data are placed in a physiological context, it would appear that when STIM1 expression increased in rodent hearts, STIM1 expression leads to SERCA2a activation. The thinking is that if the STIM1–PLN interaction were to lead to a reduction in the amount of PLN that binds to SERCA2a, then SERCA2a function and turnover rate would be increased by the presence of STIM1.
Fig. 6.

SR Ca2+ content and STIM1. (A) Representative traces show the NCX currents in control and STIM1 overexpressing myocytes upon application of caffeine (10 mM). (B) SR Ca2+ content demonstrated by the integral of NCX current in A. (C) Summary data show SR Ca2+ content in control and STIM1 overexpressing myocytes. *P < 0.05 vs. control. n = 7 and 5 cells in the control and STIM1 groups, respectively.
Fig. 7.

The effect of STIM1 on SR Ca2+ release and Ca2+ reuptake in ventricular myocytes. (A) Representative Ca2+ transients induced by field stimulation (0.5 Hz) in primary cultured control (mCherry) and STIM1 overexpressing rat ventricular myocytes. (B) Summary data show the change of [Ca2+]i transient amplitude in control and STIM1 overexpressing myocytes. (C) Summary data show the time to peak and 90% decay of [Ca2+]i transient. *P < 0.05 vs. control. n = 10 cells in the control and STIM1 groups, respectively.
Fig. 8.
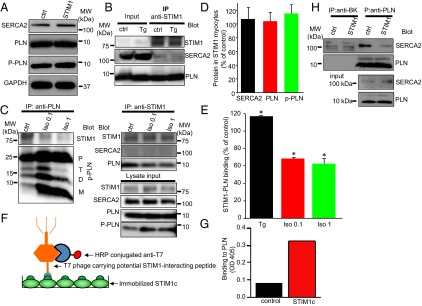
STIM1 binds PLN. (A) Western blot shows the protein level of SERCA2a, PLN, and p-PLN in control and STIM1 overexpressing myocytes. (B) Co-IP shows that STIM1 in ventricular myocytes strongly binds to PLN but weakly binds to SERCA2a. (C) Co-IP shows the effect of isoproterenol stimulation on STIM1–PLN binding. D, dimer; M, monomer; P, pentamer; p-PLN, phosphorylated PLN; T, tetramer. (D) Summary data show protein changes in STIM1 overexpressing myocytes for SERCA2a, PLN, and p-PLN (n = 6, 5, and 4, respectively). (E) Summary data show the change of STIM1–PLN binding in isolated myocytes treated with Tg (10 μM, n = 3) or isoproterenol (Iso; 0.1 and 1 μM, n = 4). Data were normalized to their corresponding control. *P < 0.05 vs. control. (F) Diagram of ELISA to verify the STIM1c interacting protein following T7 phage display screen. (G) PLN expressing T7 phage was identified as one of the STIM1c interacting phages. The binding intensity presented in the figure reflects colorimetric readings at 405 nm. (H) Representative (of two independent tests) co-IP shows reduction of SERCA–PLN binding in STIM1 overexpressing myocytes. Anti-BK (Ca2+-activated K+ channel α subunit) served as the negative control.
To test the hypothesis that STIM1 binds to PLN and thereby disinhibits SERCA, PLN was immunoprecipitated in control and STIM1 overexpressing myocytes (Fig. 8H). The immunoprecipitate was then probed for SERCA2a. The relative reduction of SERCA2a detected in the immunoprecipitate in the STIM1overexpressing cells is consistent with the hypothesis. Importantly, the amount of PLN and the amount of SERCA2a in the “input” was the same for both control and STIM1 overexpressing cells (Fig. 8 A and H). In this scenario, the effect of STIM1 overexpression in PLN-null myocytes would be barely seen. SI Appendix, Fig. S11 indicates that absence of PLN causes increased Ca2+ leak, similar to our finding in rat ventricular myocytes that overexpressed STIM1. STIM1 overexpression does not further increase Ca2+ leak in PLN-null myocytes (SI Appendix, Fig. S11), suggesting that the presence of PLN is necessary for the effects of STIM1 to be seen, in adult rodent heart, at least. Taken together, these data suggest that STIM1 binding to PLN leads to the activation of SERCA2a.
Discussion
STIM1 is found in normal cardiac ventricular myocytes, but the abundance of the protein is low. Nevertheless, the STIM1 that is present is found in highly organized puncta and linear segments at the Z-disks in both normal and low [Ca2+]SR. In this manner, STIM1 appears to be “frozen” in the organizational state found in developing cells with low levels of Ca2+ in the SR/ER stores. Consistent with this is the observed mobility of STIM1 in intracellular membranes at the jSR in that it diffuses slowly whether or not there is Ca2+ in the SR/ER lumen. The STIM1 phenotype is thus similar to what we would expect to observe in Ca2+-depleted SR/ER. Nevertheless, STIM1 does appear to influence SR Ca2+ in normal ventricular myocytes. When STIM1 is overexpressed, SR Ca2+ overload is produced and Ca2+ instability is observed in the form of propagating Ca2+ waves. For this to happen, it would appear to bind to the SERCA2a modulator PLN. If by doing so STIM1 could bind PLN and remove it from interacting with SERCA2a (and inhibiting function), then the net effect of this process would be to increase SR Ca2+ content. Given that there is no SOCE in normal rat ventricular myocytes, the mechanism of action of STIM1 in heart is, in effect, like that of revving up SERCA2a (see discussion below) and similar to knocking down PLN. The level of STIM1 expression thus appears to “set” the background SERCA Ca2+ pump rate, with the moment-to-moment rate regulated by PLN phosphorylation. These provocative observations raise questions that focus our attention on what STIM1 may do when it does not produce SOCE.
PLN and SERCA2a.
PLN is one of the primary regulators of SERCA2a (27). When PLN binds to SERCA2a, it inhibits the Ca2+ pumping function and thus reduces the ability of the SR to acquire and store Ca2+. The affinity of PLN for SERCA2a decreases when it is phosphorylated by PKA or Ca2+-calmodulin–dependent protein kinase. As PLN is converted into p-PLN, SERCA2a is disinhibited (i.e., activated). In addition to PLN, there are other modulators of SERCA2a (27). The mechanism by which STIM1 interacts with SERCA2a (i.e., by binding the un–p-PLN) to indirectly activate SERCA2a is thus consistent with the sweep of other SERCA2a regulatory factors.
SR Ca2+ Leak.
STIM1 overexpression also increases SR Ca2+ leak. However, there is no evidence to date that it does this by interacting directly with the SR Ca2+ release channel, the ryanodine receptor. Instead, we speculate that the increase in [Ca2+]SR due to the indirect activation of SERCA2a by STIM1 leads to enhanced SR Ca2+ efflux due to the luminal dependence of the ryanodine receptor on [Ca2+]SR (36, 37) and also by mass action. The higher the [Ca2+]SR, the higher the SR Ca2+ leak flux when RyR2s are open.
What Does the Continuing Presence of STIM1 Puncta Mean in Cardiac Ventricular Myocytes?
That STIM1 forms puncta in cardiac jSR without requiring low [Ca2+]SR/ER suggests that its function does not depend on a dynamic and active clustering of STIM1 proteins. Perhaps the interaction of STIM1 with PLN alone is sufficient to induce the clustering. If true, it does raise the speculation that the abundance of STIM1 per se may be the mechanism by which STIM1 influences Ca2+ in cardiac SR. This would suggest that the Ca2+-sensing EF hand motif in STIM1 does not control the behavior of STIM1 or its function in heart. Instead, the STIM1 function would appear to depend on its ability to stimulate SERCA by “tying up” PLN and thereby disinhibiting SERCA. This is consistent with the physiological observations presented here in Fig. 5 and those made by Hulot and Luo, who show that expression of STIM1 goes up in pressure overload hypertrophy (8, 13). Nevertheless, additional experiments are needed to better understand how STIM1 overexpression leads to elevated [Ca2+]SR.
STIM1 and Its Protein Partners.
The discovery and characterization of SOCE in nonexcitable cells revealed an important partnership between STIM1 and Orai1 with separate [Ca2+]SR/ER sensing (STIM1) and separate Ca2+ channel (Orai1) responsibilities. That a similar system occurs in many cell types including skeletal muscle has informed our investigation in the heart. In skeletal muscle, both Orai1 and STIM1 are found to be involved in SOCE (38–40). However, during development in skeletal muscle, there appears to be a reciprocal relationship between SOCE and the presence of a PLN-like SERCA1a modulator, sarcolipin (SLN). As SOCE increases during skeletal muscle development, SLN decreases (41). Thus, the high level of PLN and absence of Orai1 and SOCE in adult heart echoes the protein and functional relationships in skeletal muscle. In adult heart, we found no consistent clear evidence that Orai1 is present, whereas there is some evidence for Orai1 and SOCE during development (12). As noted earlier, the absence of Orai1 and SOCE did not surprise us, as huge Ca2+ entry occurs with each heartbeat in the adult ventricular myocyte. The function served by STIM1, based on our work, suggests that it can bind to PLN, the cardiac modulator of SERCA2a, and possibly tune the SERCA2a function by disinhibiting it. In Fig. 8 B and E, we observed that SR Ca2+ depletion by Tg enhances STIM1–PLN binding. We do not have a definitive explanation for this observation, but it could arise if the relative amount of free PLN were to increase due to the Tg treatment. An increase in the amount of unbound PLN could occur if the binding of Tg to SERCA (which inhibits its function) were also to decrease the binding affinity of PLN for SERCA, as recently suggested (42). If so, the increased availability of PLN (from the PLN freed from the SERCA) would then increase the amount of PLN bound to STIM1. In contrast, the activation of PKA by isoproterenol (0.1 and 1 μM) stimulation decreases the binding of STIM1–PLN presumably by changes in PLN following its phosphorylation (i.e., p-PLN). These two observations suggest that STIM1 does not readily bind to p-PLN (SI Appendix, Fig. S10). As a consequence, STIM1 competes with SERCA2a for binding to PLN. These suggestions are consistent with our observations that STIM1 overexpression activates SERCA2a, presumably by mass action, and the more STIM1 there is, the more SERCA2a is disinhibited by the reduction of PLN bound to it. If true, STIM1 trafficking and expression may work as a “long-term” regulatory mechanism serving to modulate [Ca2+]SR/ER.
Physiological Versus Pathophysiological Ca2+ Signaling in the Heart and STIM1.
The absence of SOCE and ISOCE in healthy heart cells is clearly demonstrated in our article. Nevertheless, in studies of de-differentiated or pathological cardiac myocytes and studies of cells in long-term culture, there is support for the presence of Orai1 (or the equivalent). Because Orai1 is not detectable in adult ventricular myocytes by us and it is a necessary component of SOCE, our findings of the absence of SOCE and Orai1 are consistent. However, SOCE may be present during development or during the time when a cardiac pathology produces Orai1 (12). Importantly, in normal adult ventricular myocytes, there is no convincing evidence of ISOCE. Ca2+ entry upon SR Ca2+ depletion has been reported in adult ventricular myocytes (26), but no compelling data have demonstrated a current through the Ca2+-selective Orai1 channel. Briefly, ISOCE that is dependent on SR Ca2+ depletion and shows very high Ca2+ selectivity has not been identified in normal adult ventricular myocytes. However, in a study of pressure overload cardiac hypertrophy, evidence was presented for the development of a nonselective cation channel (8) whose reversal potential is far negative to ECa. This nonselective cation channel could be a member of the TRPC channel family, as suggested by some studies in cardiac myocytes (10, 43–45).
SOCE has been suggested to play an essential role in early heart development. This finding is supported by the expression of both STIM1 and Orai1 early in development that declines with increasing maturity (13). Thus, it seems reasonable to suggest that SOCE may exist in early development, yet more work is needed to characterize the magnitude of ISOCE and its function(s) and to account for its roles in developmental physiology and biology. Our work here in adult rat heart shows low expression of STIM1 and the absence of Orai1. The question is thus raised about what STIM1 may be doing in adult ventricular myocytes under physiological conditions. A clue comes from our discovery that elevated expression of STIM1 in 2-day primary cultures of adult ventricular myocytes leads to [Ca2+]SR overload and the development of arrhythmogenic Ca2+ waves. These findings suggest that STIM1 can regulate SR Ca2+ content. The additional investigations here indicate that this regulation arises from a novel mechanism: STIM1 binds to un–p-PLN and, by this means, STIM1 increases SERCA2a-mediated Ca2+ uptake into the SR. With the lower expression of STIM1 as seen under physiological conditions, these data suggest that STIM1 moderates SERCA2a-dependent [Ca2+]SR. This function is probably important, as the long-term absence of STIM1 in cardiac-specific STIM1 knockout animals underlies serious disease (46). These animals develop a progressive dilated cardiomyopathy starting around 20 wk of age and die prematurely starting around week 40 (46). Although in young STIM1 cardiac-specific knockout mice there is no significant dysfunction, ER/SR stress increases in these animals starting around 12 wk (46). It is thus plausible that this ER/SR stress might arise from the decreases in SR Ca2+ content (a common marker of ER/SR stress) (47) due to the mechanisms presented here. STIM1 may thus play an important and novel role in ventricular myocytes under physiological conditions by regulating [Ca2+]SR and hence the [Ca2+]i transient and contraction.
Materials and Methods
Cell Preparation and Transfection.
Male Sprague–Dawley rats were anesthetized by a lethal i.p. injection of pentobarbital sodium. Ventricular myocytes were isolated as previously described (48), and the freshly isolated ventricular myocytes were used within 6 h of isolation or in short-term primary culture [M-199 medium supplemented with 0.1% ITS (insulin–transferrin–selenium) and 10 µM blebbistatin]. The cultured cells were used to examine overexpression of STIM1-mCherry or STIM1-eYFP using an adenovirus construct or mCherry alone as a control. Experiments were performed 48 h after transfection with an MOI (multiplicity of infection) of 50 or 100 for STIM1-mCherry, STIM1-eYFP, or mCherry alone. The transfection efficiency is >∼80%. For the imaging experiments, the measurements were restricted to those showing mCherry and eYFP fluorescence. For the co-IP and Western blot (including BN-PAGE), the cells were transfected with 50 or 100 MOI, and the fluorescence was not examined.
Clones and Construction of STIM1-mCherry and STIM1-eYFP Adenovirus.
A rat STIM1 plasmid was the kind gift of Shenyuan Zhang (Texas A&M University, College Station, TX) (4). We fused STIM1 to the fluorescent protein mCherry following the modification of STIM1c to remove the stop codon and incorporating a Not I site at the 3′ end using PCR. mCherry was then fused in frame to the STIM1c by PCR using primers containing Not I and Xho I sites at the 5′ and 3′ ends, respectively. The STIM1 fusion construct was ligated into pShuttle CMV (Agilent Tech Inc.) using the Not I and Xho I sites in the vector. An adenovirus expressing the STIM1 fusion protein was produced according to the manufacturer’s protocol and purified by banding through a CsCl density gradient (49). Human STIM1-eYFP adenovirus was provided by Vector BioLabs.
Co-IP.
Freshly isolated or short-term cultured (48 h) rat ventricular myocytes were placed in ice-cold lysis buffer [in mM, 150 NaCl, 25 Tris, 1 EDTA, 1% Nonidet P-40, and 5% (vol/vol) glycerol at pH 7.4, supplemented with 1% protease inhibitor mixture] (Sigma-Aldrich). After centrifugation at 10,000 × g for 10 min, the supernatant was collected and the amount of protein was measured. Co-IP experiments were performed as previously described (50). In brief, 1–5 µg of the relevant antibody (anti-PLN, Abcam; anti-STIM1, Sigma-Aldrich; anti-mCherry, Abcam) was incubated with 50 µL of coupling resin or control agarose resin in a coupling buffer (10 mM sodium phosphate, 150 mM NaCl, pH 7.2) for 2 h at room temperature. The same amount of polyclonal (Alomone Laboratories) or monoclonal (NeuroMab, University of California, Davis/NIH) antibody to the BK channel (large conductance Ca2+-activated K+ channel) α subunit was incubated with coupling resin and used as a negative control. The antibody–resin buffer mixture was then centrifuged and washed three times with coupling buffer and one time with quenching buffer (1 M Tris∙HCl). Protein lysate (500 µg to 1 mg) was added to the antibody–resin pellet and shaken overnight at 4 °C and then washed using the lysis buffer until the supernatant was protein-free. The antigen was eluted with a low pH elution buffer containing primary amines provided in the kit (Pierce Biotech). The protein was then separated and probed following a routine Western blot protocol described previously (50, 51). The HRP/chemiluminescence signal was detected using ImageQuant LAS 4000 (GE Healthcare Biosciences). Quantitative analysis was carried out using the digital image output from the LAS 4000 and ImageJ software (NIH), with the background subtracted. For quantification of co-IP results, the band intensity was first normalized to its untreated control and then to the immunoprecipitated control. Statistics were performed only for those for which more than three independent tests have been carried out. Otherwise, only representative results were shown.
First-Dimensional BN-PAGE.
For native gel protein electrophoresis, a native PAGE sample prep kit (Life Tech., 50 mM Bis∙Tris, 16 mM HCl, 50 mM NaCl, 10% glycerol, and 0.001% Ponceau S, pH 7.2) was used to produce the lysate from cardiac myocytes. A 4–12% or 4–16% native PAGE gel (Life Tech.) was used to separate proteins.
Second-Dimensional SDS/PAGE and Western Blotting.
For second-dimensional SDS/PAGE, a 4–20% one-well gel was used. Gel strips derived from the first-dimensional BN-PAGE were equilibrated in Tris–Glycine SDS sample buffer (Life Tech.) supplemented with 5% β-mercaptoethanol for 30 min at 37 °C. The strip was then placed onto the second-dimensional gel for electrophoresis. For Western blotting, proteins were extracted from cells or tissues using either a RIPA (radio-immunoprecipitation assay) lysis buffer (Sigma-Aldrich) or a SDS Tris–Glycine SDS buffer. The following antibodies were used in the blotting: anti-STIM1 (Sigma, 1:1,000), anti-SERCA2a (Thermo or Santa Cruz, 1:500), anti-YFP (abm, 1:1,000), anti-mCherry (Abcam, 1:2,000), and anti-PLN (Abcam, 1:2,000). For quantitative purposes, GAPDH was reprobed in some of the membranes. Relative protein level was quantified using ImageJ 1.44p software (NIH), with background subtracted.
Immunochemistry.
Freshly isolated adult rat ventricular myocytes were fixed using 3.7% paraformaldehyde in PBS for 15 min. After three washes with PBS, cells were permeabilized and blocked in PBS containing 5% goat serum and 0.3% triton X-100 for 1 h. Cells were then incubated overnight at 4 °C with primary antibodies: polyclonal anti-STIM1, or mouse monoclonal anti–α-actinin, at a dilution of 1:100 (anti-STIM1) or 1:1,000 (anti–α-actinin) in PBS containing 0.5% BSA and 0.3% triton X-100. After three washes in PBS, cells were then incubated for 1 h at room temperature with secondary antibodies: Alexa Fluor 488-conjugated goat anti-mouse for α-actinin and Alexa Fluor 633-conjugated goat anti-rabbit for STIM1. Fluorescence images were acquired using a Zeiss LSM-510 laser scanning confocal microscopy. Alexa Fluor 488 and 633 were excited at 488 and 633 nm and emissions collected at 505–550 and >650 nm, respectively. Negative controls were prepared by omitting primary antibodies.
T7 Phage Display Screen and ELISA.
Biotin-labeled STIM1c (238–685 aa) was expressed in sf9 cells and affinity-purified by Neutravidin column (Pierce). Purified STIM1c protein was then immobilized to avidin-coated 96-well plates and incubated with a T7 phage display peptide library (at a titer of 5.7 × 108 pfu per well) for 1 h at room temperature. The biopanning procedures were performed as directed (Novagen T7Select System). After three rounds of biopanning, the pool of enriched STIM1c-interacting phages were plated on LB plates, and single plaques were selected and amplified for individual ELISA to further verify STIM1c interaction. Specifically, individual phages (0.1 mL, 108 pfu/mL) were added into the STIM1c-coated wells and incubated for 2 h followed by five washes with PBS-T. Then anti-T7 phage antibody (Novagen) was added at a dilution of 1:2,000 and incubated for 1 h at room temperature. After washing, HRP-conjugated secondary antibody (1:5,000) was added and incubated for 1 h at room temperature. After five washes and a 30-min incubation in ABTS (2,2′-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt, 0.1 mL per well) with 0.05% H2O2 at room temperature, the plate was read at 405 nm with a plate reader. Only those phages that show notable ELISA signal to STIM1c (twofold over milk) were selected for PCR amplification with T7Select up and T7Select down primers (Novagen). The PCR products were purified and sequenced. STIM1c interacting proteins were deduced from the sequencing results and NCBI database blasting (Fig. 7F).
Preabsorption of Anti-STIM1.
To minimize the nonspecific staining by the anti-STIM1 antibody, the antibody was preabsorbed using STIM1-null (STIM1−/−) mouse ventricular myocytes. Briefly, anti-STIM1 antibody (diluted 1:5 in PBS containing 5% goat serum) was first incubated with permeabilized ventricular myocytes from a mouse (STIM1−/−) for 2 h at room temperature. The cells were then centrifuged at 1,000 × g for 5 min, and the supernatant (containing the anti-STIM1 antibody) was collected and used as a primary antibody for the immunostaining of STIM1 protein in rat ventricular myocytes.
Ca2+ Imaging and Patch-Clamp Electrophysiology.
Isolated ventricular myocytes or primary cultures of rat ventricular myocytes were loaded with fluo 4 (using 5 μM fluo 4-AM for 20 min at room temperature) and placed in a normal Tyrode’s solution containing (in mM) 140 NaCl, 10 Hepes, 0.5 MgCl2, 0.33 NaHPO4, 5.5 Glucose, 1.8 CaCl2, and 5 KCl (pH 7.4 with NaOH). Confocal imaging (Zeiss LSM-510) was used to monitor Ca2+ signals using either a line scan method (at 1.92 ms per line) or an XY image time series (at 780 ms per image). Data were analyzed offline using either ImageJ (1.44p) or IDL 6.3 with programs written by us or members of the laboratory. A conventional whole-cell patch-clamp configuration (Axopatch 200A, Clampex 8.2, Axon) was used to acquire the whole-cell patch-clamp data. The pipette solution contains (in mM) 130 CsCl, 1 MgCl2, 10 TEACl, 10 Hepes, 5 Mg-ATP, and 10 EGTA for Tg-sensitive current recording and 60 CsCl, 0.91 MgCl2, 80 TEACl, 10 Hepes, 10 NaCl, 5 Mg-ATP, 0.3 Na2GTP, and 4 KCl for Na+–Ca2+ exchanger current (Incx) recording (pH 7.2 with CsOH). The resistance of the pipettes used was 1.5–2 MΩ when filled with pipette solution. The bath solution contained (in mM) 140 NaCl, 10 Hepes, 0.5 MgCl2, 0.33 NaHPO4, 5.5 Glucose, 1.8 CaCl2, and 5 KCl (pH 7.4 with NaOH). For SR Ca2+ content measurement, Incx was recorded and integrated with cells at a holding potential of –80 mV. For Tg-sensitive current recordings, TEACl (10 mM) and verapamil (10 μM) were added to the superfusion solution. A 1-s ramp protocol from +50 mV to –100 mV was run every 20 s and then returned to a holding potential of –80 mV. After 10–20 recordings (when the current is stable), the superfusion containing Tg (10 µM) was applied. After around 10 min of Tg application, BTP-2 (20 µM) or La3+ (20 µM) was applied on top of Tg. For diastolic membrane potential recordings, current clamp was applied. Normal Tyrode solution was used in bath solution. The pipette solutions contained (in mM) 11 KCl, 120 K Aspartic acid, 5 Mg-ATP, 5 NaCl, 0.5 MgCl2, and 10 Hepes (pH 7.2 with KOH). Diastolic membrane potential was measured when no current was injected. Data were analyzed offline using Clampfit 10.4.
FRAP.
A 6 × 6 µm area (avoiding the nucleus) was used for the photobleaching experiments using high illumination intensity (488 nm and 543 nm laser) in STIM1-mCherry–infected rat ventricular myocytes after 48 h in primary culture. The fluorescence recovery in the bleached area was monitored at 2 s per frame. The intensity ratio of the fluorescence was calculated from the ratio of the intensities of the bleached area and another region (3–5 µm apart from the bleached region). The ratio derived from the image before photobleaching was set to 1, and the ratio immediately after photobleaching was set to 0. Diffusion coefficients were calculated using ImageJ (1.44p) plugin software SimFRAP, based on the two-dimensional diffusion formulation: d = , where d is the distance (µm), D is the diffusion coefficient (µm2/s), and t is the time (s).
Reagents, Data Analysis, and Statistics.
Unless otherwise specified, all reagents were purchased from Sigma-Aldrich. Data are presented as mean ± SEM. All data statistics are conducted using OriginPro 8.5.0. Where appropriate, Student’s t test or ANOVA was used to test for significance. P values less than 0.05 are considered significant.
Supplementary Material
Acknowledgments
The research leading to these results has received funding from the European Community’s Seventh Framework Program FP7/2007-2013 under Grant HEALTH-F2-2009-241526, EUTrigTreat, National Heart, Lung, and Blood Institute (NHLBI) Grants RO1 HL105239 and RO1 HL106056 (to W.J.L.) and RO1 HL093470 (to P.B.R.), and National Scientist Development Grant 10SDG4030042 (to G.Z.) from the American Heart Association.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1423295112/-/DCSupplemental.
References
- 1.Lewis RS. Store-operated calcium channels: New perspectives on mechanism and function. Cold Spring Harb Perspect Biol. 2011;3(12):a003970. doi: 10.1101/cshperspect.a003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prakriya M, et al. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443(7108):230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 3.Zeng W, et al. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32(3):439–448. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang SL, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437(7060):902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan JP, et al. TRPC channels as STIM1-regulated SOCs. Channels (Austin) 2009;3(4):221–225. doi: 10.4161/chan.3.4.9198. [DOI] [PubMed] [Google Scholar]
- 6.Stiber J, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10(6):688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dirksen RT. Checking your SOCCs and feet: The molecular mechanisms of Ca2+ entry in skeletal muscle. J Physiol. 2009;587(Pt 13):3139–3147. doi: 10.1113/jphysiol.2009.172148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hulot JS, et al. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation. 2011;124(7):796–805. doi: 10.1161/CIRCULATIONAHA.111.031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li T, et al. STIM1-Ca(2+) signaling is required for the hypertrophic growth of skeletal muscle in mice. Mol Cell Biol. 2012;32(15):3009–3017. doi: 10.1128/MCB.06599-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohba T, et al. Essential role of STIM1 in the development of cardiomyocyte hypertrophy. Biochem Biophys Res Commun. 2009;389(1):172–176. doi: 10.1016/j.bbrc.2009.08.117. [DOI] [PubMed] [Google Scholar]
- 11.Trebak M. STIM/Orai signalling complexes in vascular smooth muscle. J Physiol. 2012;590(Pt 17):4201–4208. doi: 10.1113/jphysiol.2012.233353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voelkers M, et al. Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J Mol Cell Cardiol. 2010;48(6):1329–1334. doi: 10.1016/j.yjmcc.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo X, et al. STIM1-dependent store-operated Ca²⁺ entry is required for pathological cardiac hypertrophy. J Mol Cell Cardiol. 2012;52(1):136–147. doi: 10.1016/j.yjmcc.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174(6):803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park CY, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136(5):876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454(7203):538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeromin AV, et al. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443(7108):226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355(6358):353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 19.Kurebayashi N, Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J Physiol. 2001;533(Pt 1):185–199. doi: 10.1111/j.1469-7793.2001.0185b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darbellay B, Arnaudeau S, Bader CR, Konig S, Bernheim L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J Cell Biol. 2011;194(2):335–346. doi: 10.1083/jcb.201012157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Böhm J, et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am J Hum Genet. 2013;92(2):271–278. doi: 10.1016/j.ajhg.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104(22):9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng B, Chen GL, Xu SZ. Store-independent pathways for cytosolic STIM1 clustering in the regulation of store-operated Ca(2+) influx. Biochem Pharmacol. 2012;84(8):1024–1035. doi: 10.1016/j.bcp.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Hunton DL, et al. Capacitative calcium entry contributes to nuclear factor of activated T-cells nuclear translocation and hypertrophy in cardiomyocytes. J Biol Chem. 2002;277(16):14266–14273. doi: 10.1074/jbc.M107167200. [DOI] [PubMed] [Google Scholar]
- 26.Hunton DL, Zou L, Pang Y, Marchase RB. Adult rat cardiomyocytes exhibit capacitative calcium entry. Am J Physiol Heart Circ Physiol. 2004;286(3):H1124–H1132. doi: 10.1152/ajpheart.00162.2003. [DOI] [PubMed] [Google Scholar]
- 27.Haghighi K, Bidwell P, Kranias EG. Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J Mol Cell Cardiol. 2014;77:160–167. doi: 10.1016/j.yjmcc.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15(13):1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174(6):815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnspang EC, Koffman JS, Marlar S, Wiseman PW, Nejsum LN. Easy measurement of diffusion coefficients of EGFP-tagged plasma membrane proteins using k-Space Image Correlation Spectroscopy. J Vis Exp. 2014;10:87. doi: 10.3791/51074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamkun MM, O’connell KM, Rolig AS. A cytoskeletal-based perimeter fence selectively corrals a sub-population of cell surface Kv2.1 channels. J Cell Sci. 2007;120(Pt 14):2413–2423. doi: 10.1242/jcs.007351. [DOI] [PubMed] [Google Scholar]
- 32.Yamamura H, Ikeda C, Suzuki Y, Ohya S, Imaizumi Y. Molecular assembly and dynamics of fluorescent protein-tagged single KCa1.1 channel in expression system and vascular smooth muscle cells. Am J Physiol Cell Physiol. 2012;302(8):C1257–C1268. doi: 10.1152/ajpcell.00191.2011. [DOI] [PubMed] [Google Scholar]
- 33.Covington ED, Wu MM, Lewis RS. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol Biol Cell. 2010;21(11):1897–1907. doi: 10.1091/mbc.E10-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zitt C, et al. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J Biol Chem. 2004;279(13):12427–12437. doi: 10.1074/jbc.M309297200. [DOI] [PubMed] [Google Scholar]
- 35.Walden AP, Dibb KM, Trafford AW. Differences in intracellular calcium homeostasis between atrial and ventricular myocytes. J Mol Cell Cardiol. 2009;46(4):463–473. doi: 10.1016/j.yjmcc.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Williams GS, et al. Dynamics of calcium sparks and calcium leak in the heart. Biophys J. 2011;101(6):1287–1296. doi: 10.1016/j.bpj.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walker MA, et al. Superresolution modeling of calcium release in the heart. Biophys J. 2014;107(12):3018–3029. doi: 10.1016/j.bpj.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goonasekera SA, et al. Enhanced Ca²⁺ influx from STIM1-Orai1 induces muscle pathology in mouse models of muscular dystrophy. Hum Mol Genet. 2014;23(14):3706–3715. doi: 10.1093/hmg/ddu079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyfenko AD, Dirksen RT. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J Physiol. 2008;586(Pt 20):4815–4824. doi: 10.1113/jphysiol.2008.160481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei-Lapierre L, Carrell EM, Boncompagni S, Protasi F, Dirksen RT. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat Commun. 2013;4:2805. doi: 10.1038/ncomms3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seth M, et al. Dynamic regulation of sarcoplasmic reticulum Ca(2+) stores by stromal interaction molecule 1 and sarcolipin during muscle differentiation. Dev Dyn. 2012;241(4):639–647. doi: 10.1002/dvdy.23760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bidwell P, Blackwell DJ, Hou Z, Zima AV, Robia SL. Phospholamban binds with differential affinity to calcium pump conformers. J Biol Chem. 2011;286(40):35044–35050. doi: 10.1074/jbc.M111.266759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohba T, et al. Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol Cell Cardiol. 2007;42(3):498–507. doi: 10.1016/j.yjmcc.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 44.Wu X, Eder P, Chang B, Molkentin JD. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci USA. 2010;107(15):7000–7005. doi: 10.1073/pnas.1001825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seth M, et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res. 2009;105(10):1023–1030. doi: 10.1161/CIRCRESAHA.109.206581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins HE, et al. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am J Physiol Heart Circ Physiol. 2014;306(8):H1231–H1239. doi: 10.1152/ajpheart.00075.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berridge MJ. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium. 2002;32(5-6):235–249. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- 48.Song LS, Sham JS, Stern MD, Lakatta EG, Cheng H. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J Physiol. 1998;512(Pt 3):677–691. doi: 10.1111/j.1469-7793.1998.677bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graham FL, Prevec L. Methods for construction of adenovirus vectors. Mol Biotechnol. 1995;3(3):207–220. doi: 10.1007/BF02789331. [DOI] [PubMed] [Google Scholar]
- 50.Zhao G, et al. Type 1 IP3 receptors activate BKCa channels via local molecular coupling in arterial smooth muscle cells. J Gen Physiol. 2010;136(3):283–291. doi: 10.1085/jgp.201010453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adebiyi A, et al. Isoform-selective physical coupling of TRPC3 channels to IP3 receptors in smooth muscle cells regulates arterial contractility. Circ Res. 2010;106(10):1603–1612. doi: 10.1161/CIRCRESAHA.110.216804. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.