

Abstract
Over the past 10 years, polyvalent DNA–gold nanoparticle (DNA–GNP) conjugate has been demonstrated as an efficient, universal nanocarrier for drug and gene delivery with high uptake by over 50 different types of primary and cancer cell lines. A barrier limiting its in vivo effectiveness is limited resistance to nuclease degradation and nonspecific interaction with blood serum contents. Herein we show that terminal PEGylation of the complementary DNA strand hybridized to a polyvalent DNA–GNP conjugate can eliminate nonspecific adsorption of serum proteins and greatly increases its resistance against DNase I-based degradation. The PEGylated DNA–GNP conjugate still retains a high cell uptake property, making it an attractive intracellular delivery nanocarrier for DNA binding reagents. We show that it can be used for successful intracellular delivery of doxorubicin, a widely used clinical cancer chemotherapeutic drug. Moreover, it can be used for efficient delivery of some cell-membrane-impermeable reagents such as propidium iodide (a DNA intercalating fluorescent dye currently limited to the use of staining dead cells only) and a diruthenium complex (a DNA groove binder), for successful staining of live cells.
Keywords: DNA−gold nanoparticle conjugate, PEGylation, drug delivery, DNase resistance, DNA intercalation reagent
Introduction
The polyvalent oligonucleotide–gold nanoparticle (DNA–GNP) conjugate, first developed by Mirkin et al.,1 has been demonstrated to be a wonder material for nanotechnology,1 biosensing,2−8 materials science, and medicine over the past two decades.9−13 It exhibited a number of highly attractive properties such as low/noncytotoxicity, excellent biocompatibility, good stability in high salt biological buffers, improved resistance against nuclease degradation, and universally high cell uptake via scavenger receptor-mediated endocytosis pathways. Such properties made it extremely attractive for multimodal bioimaging and drug/gene delivery. For example, the DNA–GNPs have been used for intracellular gene regulation and siRNA delivery,14−17 displaying impressive gene silencing efficiencies which are better than some widely used gene transfection reagents (e.g., lipofectmine).14 More recently, a RNA-GNP conjugate has shown to be capable of in vivo RNAi therapy of brain cancer with a mouse model.18,19 The DNA–GNP system has also been exploited for intracellular delivery of small chemotherapeutic drugs.20−24 We have found recently that a pH-responsive (PR) DNA, which exhibits a highly reversible, pH-triggered conformational switch between a four-stranded i-motif and a random coil,25−27 can be combined with GNP to develop an effective nanocarrier for doxorubicin (DOX), a widely used clinical cancer chemotherapy drug. It allows for effective treatment of cancer at the cellular level.12 The PR-DNA–GNP displays numerous features of an “ideal drug nanocarrier” outlined by Langer et al.28 It can effectively exploit the gradually acidified local pH of the natural endo/lysosomal maturation/trafficking process to achieve effective, pH-triggered intracellular drug release.
Despite significant studies, most of the DNA–GNP systems reported so far have been based on unmodified DNAs. The inherent strong negative-charge of the DNA phosphate backbone can lead to nonspecific interactions with serum proteins, altering their particle size, charge, and pharmacokinetic properties.29,30 This can lead to strong recognition by the reticuloendothelial system (RES), resulting in rapid removal from blood circulation. As a result, this can limit its ability to exploit the enhanced permeation and retention (EPR) effect, a characteristic pathological property of cancer tumor,28 to achieve tumor-targeted accumulation and hence compromising its therapeutic efficacy in vivo. Additionally, although the stability of DNA against nuclease degradation can be improved by ∼3 fold after GNP conjugation,31 this may still not be not good enough to satisfy the challenging in vivo conditions because of the extensive exposure to various nucleases.
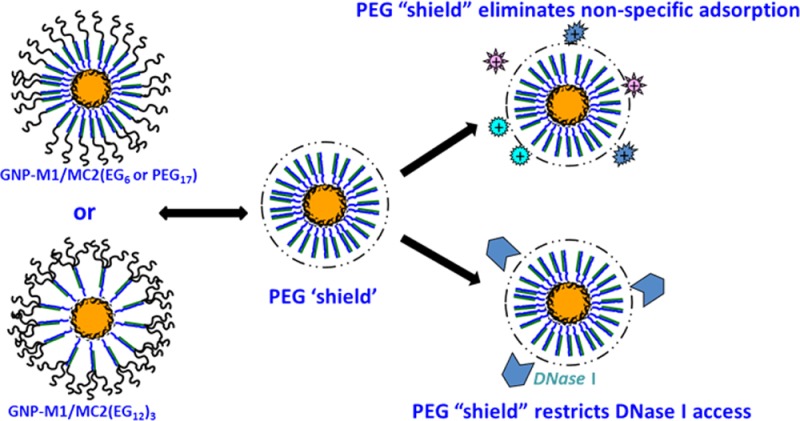
To address the problem of serum protein nonspecific adsorption, the Mirkin group used a post-treatment of the formed DNA–GNP with a thiolated poly(ethylene glycol) (PEG). Despite success, a drawback was a reduced DNA/RNA loading on the GNP, due to competitive displacement of the thiolated nucleic acid strands on the GNP surface by the thiolated PEG passivation molecules. As a result, the number of functional DNA/RNA strands on each ∼14 nm GNP was found to be only ∼35,18 a considerable reduction from the typical ≥100 strands found for nontreated DNA–GNPs.1−12 Herein we report a new PEGylation strategy for the DNA–GNP via terminal PEGylation of the complementary strand (MC2). The specific hybridization between the PR-DNA–GNP and MC2(PEG) then completes the carrier PEGylation (Figure 1A). An advantage of this strategy over the post-thiolated PEG treatment is that it yields more functional DNA strands per GNP (ca. 110 vs 35), making it potentially a more effective drug or gene nanocarrier. We show that our PEGylation approach offers complete resistance to nonspecific adsorption of serum proteins in cell culture media and provides >10 times higher resistance to DNase I-mediated enzymatic digestion. Moreover, the PEGylated DNA–GNP nanocarrier still retains high cell uptake which can be exploited for efficient delivery of both chemotherapeutic drugs (ca. DOX) and some cell membrane-impermeable reagents to live cells.
Figure 1.
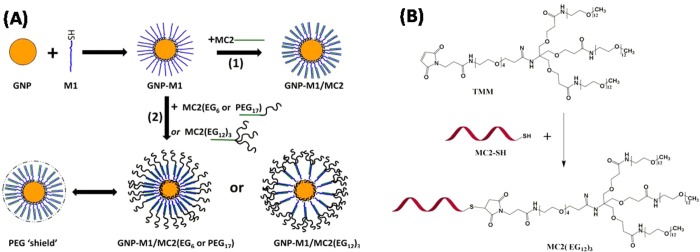
(A) Schematic procedures of our approach to PEGylated DNA–GNP drug nanocarriers. Thiolated PR-DNA (denoted as M1) was first loaded onto a citrate-stabilized 14 nm GNP via gold–thiol self-assembly to form GNP–M1, which was then hybridized to complementary MC2 (unmodified, route 1) or PEG-modified MC2s (route 2) to form the GNP–M1/MC2(PEG) carriers. (B) Schematic of MC2(EG12)3 preparation via the Michael addition between the maleimide-modified three-chain oligo(ethylene glycol) and the MC2-free sulfhydryl group, forming a stable covalently linked MC2(EG12)3.
Results and Discussion
Table 1 summarizes the DNA sequences used in this study. EGm represents uniform, single-length oligo(ethylene glycol, EG) containing m EG units, while PEGn represents poly(ethylene glycol) with mixed length PEGs containing an average number of n EG repeats. The thiolated pH-responsive (PR) DNA strand (M1) contains an i-motif domain consisting of four stretches of cytosine-rich sequences. The i-motif domain is separated by a 10-consecutive thymine (T10) linker from the 5′-thiol modification to minimize any possible nonspecific interactions with the GNP after conjugation.12 The MC2 sequence is fully complementary to the M1 i-motif domain except for two designed mismatches to stop it forming a stable G-quadruplex. The mismatches are also used to tune the stability of the resulting double-stranded (ds) DNA structure, ensuring the ability to form a stable i-motif triggered by the acidic pH environment of intracellular compartment and to release the intercalated drugs/reagents as described previously.12 The GC-rich base pairs in the M1/MC2 duplex also allow for convenient loading of doxorubicin (DOX), a widely used clinical cancer chemotherapeutic drug, via its preferred GC base pair intercalation.24
Table 1. DNA Abbreviations and Their Sequences Used in This Papera.
| DNA nameb | sequence (5′ → 3′) |
|---|---|
| M1 (PR-DNA) | HS(CH2)6-TTT TTT TTT TCC CTA ACC CTA ACC CTA ACC C |
| MC2 | GTG TTA GGT TTA GGG TTA GGG |
| MC2(EG6) | EG6-GTG TTA GGT TTA GGG TTA GGG |
| MC2(PEG17) | PEG17-GTG TTA GGT TTA GGG TTA GGG |
| MC2(EG12)3 | (EG12)3- GTG TTA GGT TTA GGG TTA GGG |
The two designed mismatched bases between MC2 and M1 are highlighted in red.
EGm: single-length oligo(ethylene glycol) containing m EG repeat. PEGn: a mixed length poly(ethylene glycol) with an average number of n EG repeats.
The MC2 modified with a 5′-terminal six EG unit, MC2(EG6), is purchased commercially from IBA GmbH (Germany). The synthesis and characterization of the MC2(PEG17), MC2 with a 5′-terminal modification of PEG with an average of 17 repeat EG units, has been reported in our previous publication.12 MC2(EG12)3 is synthesized in house by reaction of a 5′-thiol-modified MC2 with a maleimide-modified, branched three-chain PEG each containing 12 EG units [(Methyl-EG12)3-EG4-Maleimide (TMM)] as shown schematically in Figure 1B. Details of the MC2(EG12)3 characterization are given in the Supporting Information (SI).
GNP–M1 conjugates with the average M1 strand loading per GNP of 60, 85, and 110 respectively are prepared by incubating citrate stabilized GNP (∼14 nm in diameter, see SI, Figure S1) with 100, 200, and 300 mol equiv of thiolated M1s followed by salt aging as described previously.12 The resulting GNP–M1 conjugates are then hybridized to the MC2, MC2(EG6), MC2(PEG17) or MC2(EG12)3 at a fixed M1:MC2 molar ratio of 1:1 in a 2-N-morpholino ethanesulfonic acid (MES) buffer (50 mM MES, 150 mM NaCl, pH 7.4) to complete the carrier assembly. Effects of the EG (or PEG) chain length and number and the GNP surface M1 density on the carrier’s resistance to nonspecific serum protein adsorption and DNase I digestion are investigated.
PEGylation Eliminates Nonspecific Adsorption of Serum Proteins on the DNA–GNP Carrier
The size and surface properties of a drug carrier are critical to its stability, pharmacokinetics, and biodistribution in vivo, which in turn strongly affect its cancer targeting ability and efficacy. For effective cancer targeting via the EPR effect, a characteristic pathological condition of many solid tumors, an ideal carrier size should be greater than the renal clearance threshold (∼8 nm, ensuring long blood half-time)32,33 but smaller than the average gap of leaky blood vessels of solid tumors (∼100 nm).28,34 The carrier should also minimize the capture by fixed macrophages in the liver and spleen,35 and have the right surface properties to avoid being recognized and cleared out of the body during systemic circulation before reaching the target tumor.36,37 The carrier should not interact strongly with blood components to alter its size and surface properties. In this regard, PEGylation has been shown to be one of the most effective and widely used strategies.38,39 PEGylation can provide a flexible, hydrophilic shield to minimize the nonspecific uptake and removal by macrophages. Indeed, PEGylation has shown to be effective at resisting nonspecific adsorption of biomolecules on both flat and curved nanoparticle (e.g., magnetic nanoparticle, quantum dot) surfaces.40−43 Therefore, the hydrodynamic diameter (Dh) of the DNA–GNPs (with ∼110 M1 strands per GNP) in MES buffer and in Dulbecco’s Modified Eagle Medium (DMEM) cell culture media with 10% fetal bovine serum (FBS) is measured by dynamic light scattering (DLS), and the results are shown in Figure 2.
Figure 2.
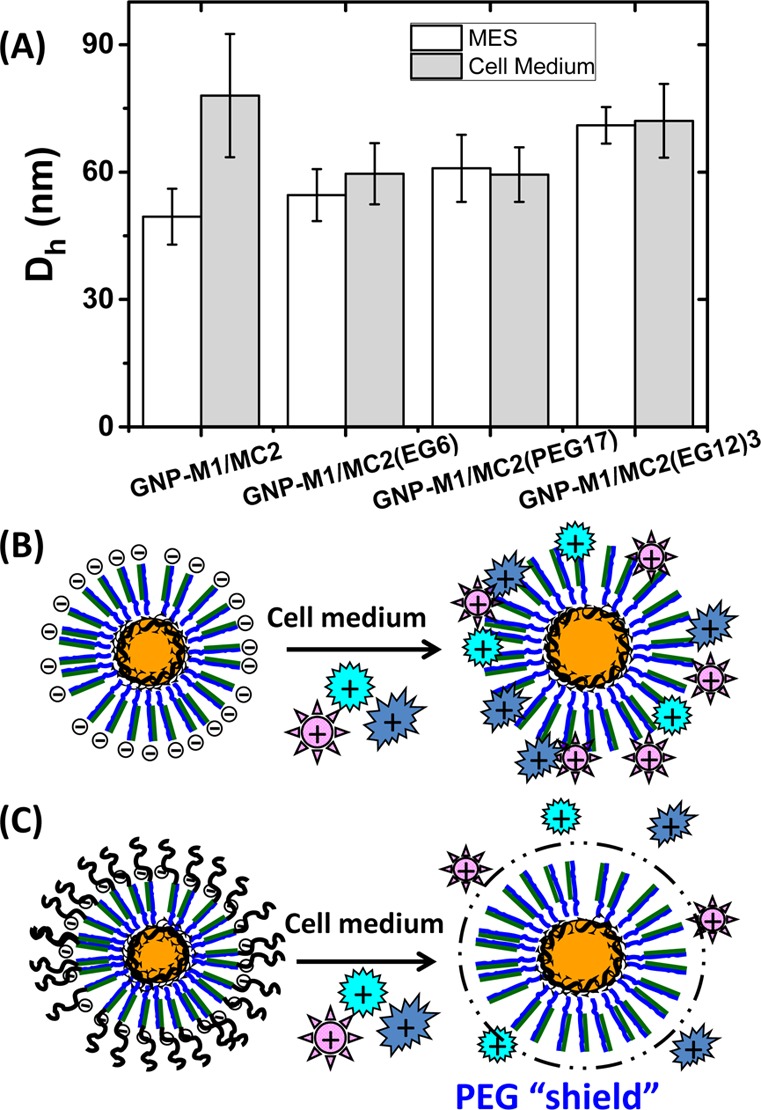
(A) Comparison of the hydrodynamic diameter (Dh) of different GNP–M1/MC2 systems in MES buffer (white bars) and DMEM cell culture media with 10% FBS (gray bars). (B) Schematic presentations of the interaction between DNA–GNP and serum proteins: positively charged serum proteins (or protein domains) may electrostatically adsorb to the strongly negatively charged DNA–GNP, leading to a significantly increased Dh. (C) A dense PEG shield on the PEGylated DNA–GNP can prevent the adsorption of serum proteins, leading to effectively no change of Dh.
The un-PEGylated GNP–M1/MC2 displays a Dh of 50 ± 4 nm in MES buffer, while those with various PEG-modifications, i.e. GNP–M1/MC2(EG6), GNP–M1/MC2(PEG17), and GNP–M1/MC2-(EG12)3, all show a larger Dh of 55 ± 6, 61 ± 8, and 70 ± 5 nm, respectively (Figure 2A). Therefore, the size of the GNP–DNA carrier gradually increases with the increasing number of total PEG units grafted to each MC2 strand. This result agrees well with our design that the MC2 strands hybridize to the GNP–M1 to form the GNP–M1/MC2 carrier, leaving the terminal PEG grafts extending outward. As a result, the higher the number of the PEG units grafted on each MC2 strand the bigger the volume it will occupy and hence the bigger the overall carrier Dh.
In serum-containing media, the Dh of the un-PEGylated GNP–M1/MC2 is increased significantly (by ∼30 nm) to ∼80 nm, indicating significant adsorption of serum proteins onto the carrier. This is most likely due to electrostatic adsorption of some positively charged proteins (or domains) onto such a strongly negatively charged nanocarrier (Figure 2B). This result agrees well with those of unmodified DNA–GNPs reported in earlier literature.29,44 In contrast, the Dh of the PEGylated GNP–M1/MC2s (except for GNP–M1/MC2(EG6) which shows a small increase of ∼4 nm) in the cell culture media shows effectively no changes over those in the MES buffer, indicating no nonspecific adsorption of serum proteins onto the PEGylated nanocarriers. This result confirms the success of our PEGylation strategy for the DNA–GNP system. PEGylation is a well-established strategy for resisting nonspecific adsorption of biomolecules on surfaces. It has been widely used to improve the pharmacokinetic properties and to reduce nonspecific uptake for therapeutic biomolecules.45,46 In those cases, a few strands of relatively long PEGs (with molecular weight of ∼5–40 kDa, containing ∼110–900 PEG units each) are conjugated to each protein to complete the PEGylation. Here we find that ∼110 strands of short PEGs (each containing 17 PEG units) are sufficient to completely inhibit the nonspecific adsorption of proteins on such a large (Dh ∼ 50 nm) and strongly negatively charged DNA–GNP conjugate presumably because many such short PEGs create a uniform, flexible, neutral, hydrophilic, and relatively dense shield on the particle outer surface that can sterically limit the access to the underneath DNAs by serum proteins to initiate electrostatic adsorption.47−49 As a result, the sizes of the PEGylated DNA–GNP carriers, particularly those with a moderate length or branched multichain PEGs, show no measurable changes after exposure to the serum containing culture media. This result also agrees well with the earlier reports that longer PEG chains and higher PEG density can provide greater shielding efficiency.50−53
PEGylation Improves Carrier Resistance to DNase I Digestion
In addition to resisting nonspecific adsorption, an effective drug nanocarrier should have sufficient stability in vivo. This has been a significant challenge for any DNA-based drug carriers because of exposure to numerous nucleases under the in vivo environment that can degrade them rapidly. It has been reported that a dense DNA packing on the DNA–GNP can increase the resistance of DNA to nuclease degradation by ∼3 fold, primarily through inhibition of nuclease activity by the high local salt (counterion) concentration surrounding the strongly negatively charged DNA–GNP.31 However, the 3-fold improvement may still not be enough to satisfy the more challenging in vivo conditions.
To investigate whether our PEGylation strategy can improve the carrier resistance to nuclease degradation, the dsDNA–GNPs are treated with a DNA digestive enzyme, DNase I (Figure 3A). This process is monitored by following a literature protocol31 but using a different signal readout strategy. Here a DNA intercalating dye, YO-PRO-1, is used instead of a covalently attached fluorophore at the end of the complementary strand.31 Compared to the literature approach, this strategy has several advantages: First, YO-PRO-1 binds strongly to dsDNA by intercalation which is very similar to that of anticancer drug (e.g., DOX) loading. Therefore, the stability of dsDNA–GNP–YO-PRO-1 against nuclease degradation should mimic more closely that of the dsDNA–GNP–DOX system. Second, unlike the covalent labeling strategy where each DNA strand contains just one fluorophore, multiple YO-PRO-1 molecules can bind to each dsDNA strand, allowing for a stronger fluorescence readout signal. Third, unlike DOX which intercalates preferentially to the GC base pairs,54 YO-PRO-1 intercalation does not have base pair preference and takes place throughout the whole dsDNA structure.55 Therefore, the YO-PRO-1 fluorescence intensity change should present a better reflection of the whole dsDNA degradation process than relying on terminal labeling or DOX intercalation. Finally, free YO-PRO-1 is effectively nonfluorescent. Its fluorescence intensity is enhanced by >1000 fold after dsDNA binding. This property allows for unambiguous differentiation of the DNA-bound and free YO-PRO-1 states after DNase I digestion.
Figure 3.

Schematic presentations of the YO-PRO-1 loaded (A) dsDNA and (B) PEGylated dsDNA–GNP systems under treatment of DNase I. The dsDNA only system is quickly degraded by DNase I, but the PEG-shield on the dsDNA–GNP can provide protection against DNase I digestion. (C) Normalized time-dependent fluorescence changes for the YO-PRO-1 loaded M1/MC2 and GNP–M1/MC2 (with or without PEGylation) conjugates after treatment with DNase I. (D) Comparison of initial rate of degradation velocities (%/min) over the first 30 min derived from C.
A series of samples containing the M1/MC2 duplex only, and GNP–M1/MC2s (with or without PEG modification, with ∼85 M1 strands per GNP) with identical effective final M1/MC2 strand concentrations (80 nM) and DNA strand loading per GNP (85) are mixed with YO-PRO-1 (400 nM, M1/MC2:YO-PRO-1 molar ratio = 1:5) for 10 min before DNase I (2 U/L) is introduced. The resulting time-dependent fluorescence intensity change of YO-PRO-1 (λEX/λEM: 491/509 nm) for each sample is monitored and shown in Figure 3C. The fluorescence decreases are all normalized by that of the M1/MC2 duplex only (80 nM) with YO-PRO-1 (400 nM). The fluorescence intensity changes within the first 30 min for all samples are approximately linear; hence, the slopes of the resulting linear fits are used to quantify their relative enzymatic digestion rates (Figure 3D). As shown in Figure 3C, free M1/MC2 duplex is rapidly digested by DNase I. The whole digestion process is complete in ∼50 min with an initial rate of 3.03%/min. In contrast, degradation of the GNP–M1/MC2 is much slower, with an initial rate of 1.13%/min, ∼1/3 that of the free duplex DNA alone. This result is in excellent agreement with an earlier report that the DNA stability against nuclease degradation can be improved by ∼3 fold upon GNP conjugation.31 The improved resistance is assigned to a high local Na+ concentration at the DNA–GNP surface (to balance its strong negative surface charge) that can inhibit the DNase I activity.31
All of the PEGylated GNP–M1/MC2 carriers exhibit a slower degradation rate than the un-PEGylated GNP–M1/MC2. For single-PEG chain-modified systems, GNP–M1/MC2(EG6) and GNP–M1/MC2(PEG17), they both show very similar initial degradation rates of ∼0.70%/min, which is ∼21% that of the M1/MC2 duplex alone. Significantly, the three-PEG-chain-modified GNP–M1/MC2(EG12)3 exhibits the slowest degradation rate, 0.32%/min, which is less than half that of the single-PEG-chain systems and only ∼1/10 that of the free M1/MC2 duplex alone. This indicates that modification of GNP–M1/MC2 with a branched three-chain PEG greatly enhances its resistance to DNase I-mediated enzyme degradation.
The enhanced resistance of the PEGylated DNA–GNPs to DNase I degradation is likely to originate from a combined effect of steric hindrance and high local Na+ concentration. A dense PEG “shield” on the dsDNA–GNP outer surface (Figure 3B) can restrict the enzyme access to the underneath DNA structure, just like their ability to resist nonspecific adsorption of serum proteins observed above.38,46 These highly flexible, hydrophilic PEG chains produce a vast number of conformations constantly switching from one to another, acting as a “PEG shield” that can significantly reduce the possibility of digestive enzymes to reach the underneath objects. Meanwhile, the dense negative charge of the DNAs underneath the “PEG shield” still induces a high local Na+ concentration that can inhibit the activity of any enzymes managed to penetrate the “PEG shield”. Therefore, all three PEGylated DNA–GNPs exhibit slower enzymatic degradation rates than the un-PEGylated GNP–M1/MC2. The GNP–M1/MC2(EG12)3, which has a surface PEG density three times as high as the single-chain PEGs, can produce a much denser and hence more effective steric shield to prevent the access of DNase I to the DNA structures, leading to the slowest enzymatic degradation rate.50,56−58
A further insight into the resistance to DNase I degradation is obtained by examining the effects of the DNA (hence PEG as each MC2 strand is PEGylated) packing density on the GNP surface. Figure 4A shows the initial degradation rates of the un-PEGylated GNP–M1/MC2s with M1 strand loadings of 60, 85, and 110 per GNP, respectively (the M1:MC2 molar ratio is always maintained at 1:1). It clearly shows that the higher the DNA strand loading per GNP, the slower the degradation rate. For example, the initial degradation rate for the conjugate with 110 M1 strands per GNP (1.02%/min) is 46% slower than that with 60 strands (1.89%/min) and ∼11% slower than that with 85 strands (1.13%/min). This is consistent with the mechanism that the higher the DNA (negative charge) density, the higher the local Na+ ion concentration and hence the more effective inhibition of DNase I activity. A similar trend is also observed for the three-PEG-chain-modified GNP–M1/MC2(EG12)3 (Figure 4B). The initial rate of degradation is decreased from 0.32 to 0.25%/min as the DNA strand loading is increased from 85 to 110, a reduction of 22%, which is about twice that observed for the non-PEGylated system (∼11%). This result indicates that the stability of the PEGylated DNA–GNP against DNase I digestion can be further enhanced by increasing the GNP surface DNA loading. The combined effect of high DNA density (hence high local Na+ concentration for inhibiting DNase activity) and PEGylation (steric restriction of DNase access to underneath DNA structure) makes it more resistant to DNase degradation. This result thus provides useful guidance toward the design of highly stable DNA–GNP-based drug nanocarriers.
Figure 4.
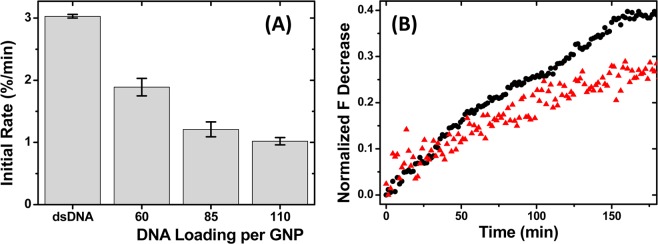
(A) Comparison of initial degradation rates for M1/MC2 duplex and un-PEGylated GNP–M1/MC2s at different DNA loadings per GNP. (B) Time-dependent fluorescence intensity changes of the GNP–M1/MC2(EG12)3 at M1 strand loadings of 85 (black dots) and 110 (red triangles) per GNP.
GNP–M1/MC2(EG12)3 for Intracellular Delivery of DNA Binding Reagents
The excellent resistance of the GNP–M1/MC2(EG12)3 against serum protein adsorption and DNase I degradation makes it highly attractive for drug delivery. We have previously shown that the GNP–M1/MC2 can be used for efficient delivery and pH-responsive release of DOX inside cancer cells, leading to high cytotoxicity.12 Here we report that the GNP–M1/MC2(EG12)3 can deliver not only DOX (a widely used clinical anticancer drug for treating bladder, breast, stomach, lung, ovaries, thyroid, soft tissue sarcoma, multiple myeloma, some leukemias, and Hodgkin’s lymphoma, Figure 5B) but also propidium iodide (PI), a cell membrane-impermeable fluorescent dye, to live human cervical cancer cells (HeLa cells). PI is widely used to stain dead cells but not live cells. As shown in Figure 5C, live HeLa cells are clearly stained by PI after exposure to PI mixed with the DNA–GNP nanocarrier. The DNA–GNPs have been previously reported to be internalized by cells mainly via the scavenger receptor-mediated endocytosis route.30 As a result, they should be mainly located in intracellular endosomes or lysosomes. Transmission electron microscopy (TEM) analysis of HeLa cells after incubation with the GNP–M1/MC2(EG12)3 for 3 h reveals that this is indeed the case. The GNPs are found to be exclusively located in endo/lysosomal-like intracellular compartments (Figure 5A), suggesting that modification of the GNP–M1/MC2 with the three-chain-PEG does not alter its cell uptake pathway. Therefore, its intracellular delivery mechanism is likely to be as follows: after cell uptake, the gradual acidification of the local environment following the natural endosomal maturation process (the local pH in late endosome or lysosome can be as low as 4.3)59 will trigger the formation of intramolecular i-motifs, leading to release of the intercalated PI molecules into the cytoplasm. The released PI molecules can then diffuse into the nucleus, staining live HeLa cells with a strong red fluorescence as shown in Figure 5C.
Figure 5.
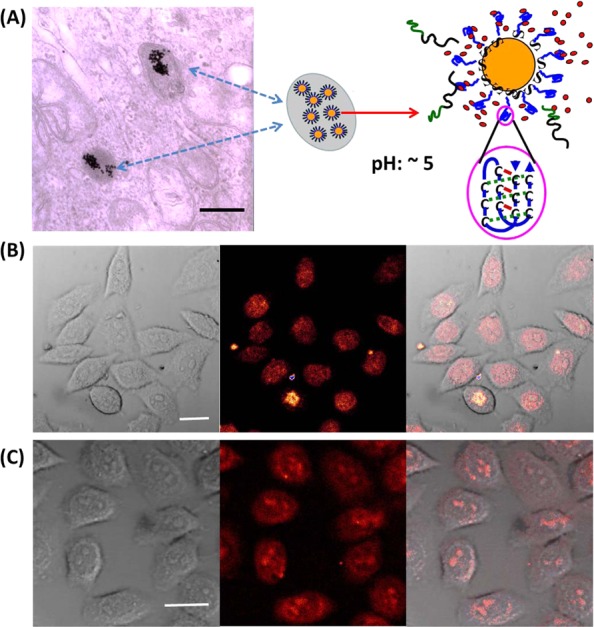
(A) A representative TEM image of HeLa cells after incubation with the GNP–M1/MC2(EG12)3 for 3 h at 37 °C, scale bar = 1 μm. (B) Confocal phase contrast (left), fluorescence (middle), and merged optical/fluorescence (right) images of HeLa cells after incubation with GNP–M1/MC2(EG12)3–DOX for 1.5 h at 37 °C, scale bar = 25 μm. (C) Confocal phase contrast (left), fluorescence (middle), and merged optical/fluorescence (right) images of HeLa cells after incubation with GNP–M1/MC2(EG12)3–PI for 3 h at 37 °C, scale bar = 25 μm.
Besides the ability of delivering PI molecules to live cells, the GNP–M1/MC2(EG12)3 also shows significantly higher stability in vitro than the un-PEGylated GNP–M1/MC2. For example, it shows no observable aggregation or change of physical appearance for at least 24 h even after exposure to excess free PI or DOX molecules in solution, whereas the un-PEGylated GNP–M1/MC2 is found to have aggregated and precipitated out of the solution under such conditions. The greatly improved stability of the GNP–M1/MC2(EG12)3 is most likely due to the dense branched EG chains on its outer surface that can provide a sufficient hydrophilic physical barrier to prevent DNA–GNP aggregation resulting from the PI/DOX intercalation-induced DNA charge neutralization (both PI and DOX molecules are positively charged). In contrast, the un-PEGylated GNP–M1/MC2 is mainly stabilized by electrostatic repulsion among such negatively charged nanoparticles. It aggregates readily and precipitates out of solution once its negative charges are neutralized.
To demonstrate the general use of the GNP–M1/MC2(EG12)3 for intracellular delivery of other types of DNA binding agents, we have further employed it to deliver a fluorescent diruthenium(II) complex, [(bpy)2Ru(tpphz)Ru(bpy)2]4+, denoted as BPY (Figure 6A). Unlike DOX and PI molecules which bind to DNA mainly through intercalation, BPY is a DNA groove binder.60 BPY has been shown to be impermeable to live cell membranes and therefore cannot enter cells on its own.60 This property is further confirmed from our results shown in Figure 6B: 3 h incubation of free BPY with HeLa cells produces negligible BPY fluorescence inside the cells, suggesting no significant cell uptake. In contrast, incubation of HeLa cells with the BPY mixed with the GNP–M1/MC2(EG12)3 for 3 h yields strong BPY fluorescence inside HeLa cells, suggesting that the GNP–M1/MC2(EG12)3 can effectively carry the BPY molecules and successfully deliver them into live HeLa cells. Together, these results demonstrate that the GNP–M1/MC2(EG12)3 reported herein has great potential for intracellular delivery of a wide range of DNA-intercalating agents. Its excellent stability and resistance against nonspecific adsorption and enzymatic degradation, together with high cell uptake, should make it an effective nanocarrier for intracellular delivery of any DNA-binding/intercalating reagents. Given a large number of drug molecules and metal complexes are known to be DNA-binders,61 the robust, versatile PEGylated DNA–GNP nanocarrier reported herein should have broad applications in bioimaging, drug delivery, and therapeutics, possibly even at the in vivo level.
Figure 6.

Delivery of a cell-membrane-impermeable diruthenium complex to live cancer cells by using the GNP–M1/MC2(EG12)3. (A) Chemical structure of the diruthenium(II) complex, BPY. (B) Confocal phase contrast (left), fluorescence (middle), and merged optical/fluorescence images (right) of HeLa cells after treatment with the BPY for 3 h at 37 °C. (C) Confocal phase contrast (left), fluorescence (middle), and merged optical/fluorescence (right) images of HeLa cells after incubation with GNP–M1/MC2(EG12)3–BPY for 3 h at 37 °C.
Conclusions
In summary, we have developed an effective PEGylation approach for polyvalent DNA–GNPs by terminal PEGylation of the complementary DNA strand. Hybridization of the PEGylated MC2s to the GNP–M1 conjugates produces a dense PEG “shield” on the carrier surface that can efficiently mask the strong negative charges, providing high resistance to nonspecific adsorption of serum proteins and greatly improved stability against enzymatic degradation. Particularly, the three-chain PEG-modified DNA–GNP nanocarrier is completely resistant to nonspecific adsorption of serum proteins and displaying >10-fold higher stability against DNase I-based enzymatic digestion over the corresponding dsDNA alone. Its stability may be further improved by increasing the PEG length, the number of PEG branches, and/or the GNP surface DNA density. Importantly, the PEGylated DNA–GNP still retains high cell uptake property. It can be used as a general, efficient intracellular delivery nanocarrier for a wide range of DNA-binding/intercalating reagents, including those which are cell-membrane impermeable on their own. Such stable and highly resistant DNA–GNP nanocarriers should have broad applications in bioimaging, drug delivery, and therapeutics.
Experimental Section
Materials
Hydrogen tetrachloroaurate (III) hydrate, 99.9% (metals basis), and 2-(N-morpholino)ethanesulfonic acid monohydrate (MES, 98%) were purchased from Alfa Aesar (UK). Tris-sodium citrate (99%), HCl (36%), HNO3 (70%), NaOH, NaCl (99.99%), and doxorubicin hydrochloride were purchased from Fisher Scientific UK limited (Milton Keynes, UK). DMEM (Dulbecco’s Modified Eagle’s Medium), PBS (phosphate buffered saline), MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), FBS (fetal bovine serum), and penicillin–streptomycin (10 000 units/mL penicillin, 10 mg/mL streptomycin), and anhydrous DMSO (≥99.7%) were all purchased from Sigma-Aldrich UK limited (Dorset, UK). High purity deionized water (resistance >18.2 MΩ·cm), purified by an ELGA Purelab classic UVF system, was used for all experiments and for making buffers. All buffers were filtered through a Whatman syringe filter (0.20 μm pore size, Whatman Plc.) before use. HPLC-purified DNA oligos, MC2, MC2-SH, and MC2(EG6) were purchased commercially from IBA GmbH (Göttingen, Germany). MC2(PEG17) was prepared in house, and its preparation and characterization details have been described in our recent paper.12 (Methyl-EG12)3-EG4-maleimide (TMM) was purchased from Thermo Scientific (UK). YO-PRO-1 was purchased from Life Technologies (UK). DNase I (1 U/μL) was purchased from Fisher Bio Reagents (Milton Keynes, UK). All chemicals and reagents were used as received unless otherwise stated.
Preparation of Gold Nanoparticle
HAuCl4 (80 mg) was dissolved in 200 mL of ultrapure water. The solution was then transferred to a freshly cleaned 250 mL three-necked flask and heated to reflux in an oil bath under magnetic stirring. When the solution began to reflux, an aqueous solution of trisodium citrate (228 mg in 20 mL water) was quickly added and the resulting solution was continuously refluxed. The color of the solution changed from yellow to deep red in ∼1 min. After refluxing for another 50 min, a stable deep red solution was obtained. The heating bath was then removed, and the solution was allowed to cool to room temperature naturally. The prepared GNP solution was transferred to a clean glass container and stored at room temperature. This produced a ∼14 nm GNP stock (as confirmed by TEM imaging see, Figure S1 in the SI) with a concentration of ca. 15 nM.
Preparation of MC2(EG12)3
A 100 nmol amount of MC2-SH was dissolved in 1 mL of freshly filtered (Whatman syringe filter with 0.22 μm pore size) MES buffer (50 mM MES, 0.15 M NaCl, pH 7.4) to make a 100 μM stock. TMM was dissolved in anhydrous DMSO to make a TMM stock solution of 40 mM. A 0.5 mL amount of the MC2-SH stock solution (50 nmol) was then mixed with 50 μL of TMM stock (the molar ratio of MC2-SH:TMM = 1:40) to ensure high DNA conversion. The resulting solution was allowed to stand overnight at room temperature to form MC2(EG12)3 via Michael addition between the DNA thiol group and the maleimide group in TMP (see Figure 1B).
Both RP-HPLC analysis and purification of MC2(EG12)3 were performed on a Gynkotek HPLC Instrument at room temperature using a Phenomenex C18 column (4.6 × 250 mm, 5 μm) with mobile phase consisting of TEAA buffer (A) and acetonitrile (B). UV absorbance was monitored by a Gynkotek (UVD 340S) detector at 260 nm. The solvent gradient used for analysis and purification of the MC2(EG12)3 was 10–70% (B) over 30 min. The resulting HPLC eluting profiles for MC2-SH and MC2(EG12)3 were shown in SI, Figures S2 and S3, respectively. The fractions containing the purified MC2(EG12)3 were combined, lyophilized, and stored at −20 °C until use. Its identity was confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) (see SI, Figure S4).
Preparation of PEGylated DNA–GNPs
The DNA–GNPs were prepared by following our previously established procedures. Briefly, a batch of three 2.2 mL GNP stock solutions (15 nM) obtained above were mixed with 33, 66, and 100 μL of DNA M1 aqueous stock solution (100 μM) overnight (GNP:M1 molar ratios = 1:100; 1:200; 1:300, respectively). The resulting solutions were then salt-aged (0.30 M NaCl) overnight. The samples were then centrifuged at 14800 rpm for 60 min to remove any unconjugated free DNAs that remained in the supernatant, yielding the GNP–M1 as an oily pellet that could be rapidly redispersed in water. The amounts of unbound free DNAs in the clear supernatants were determined as 13.2, 38, and 62.7 pmol by monitoring the UV absorption at 260 nm using an extinction coefficient of εM1 = 2.65 × 105 cm–1 M–1. The amounts of DNA conjugated onto the GNP were thus determined as 19.8, 28, and 37.3, nmol, respectively. Given 0.33 pmol of GNP was used for each sample, the M1 strand loading per GNP was thus determined as 60, 85, and 110, respectively, for the above samples.12 Afterward, the complementary MC2 strands (MC2, MC2(EG6), MC2(PEG17), or MC2(EG12)3) were added to the GNP–M1 (under a fixed M1:MC2 molar ratio of 1:1) and were allowed to hybridize in an MES buffer for 1 h to make GNP–M1/MC2, GNP–M1/MC2(EG6), GNP–M1/MC2(PEG17), and GNP–M1/MC2(EG12)3 nanocarriers.
Dynamic Light Scattering (DLS) Measurement
The hydrodynamic diameter (Dh) of the DNA–GNP (with M1 strand loading of 110 per GNP) was measured in both MES buffer (pH 7.4) and in complete DMEM media with 10% FBS. Briefly, 30 μL of the dsDNA–GNP stock solution (0.46 μM GNP) was mixed with 1.2 mL of MES buffer or complete DMEM and then filtered through a Whatman syringe filter (0.22 μm pore size). After 3 h, their Dh was measured on a Brookhaven Instruments Corp. BI-200SM laser light scattering goniometer with a BI-APD detector, using a He–Ne laser at 633 nm (scattering angle: 90°).12
DNase I Digestion Experiments
The dsDNA–GNP samples were mixed with YO-PRO-1 and then diluted to 200 μL with the enzyme working buffer (10 mM Tris-HCl, 2.5 mM MgCl2, and 0.5 mM CaCl2, pH 7.5) to give a final concentration of 80 nM for the dsDNA and 400 nM for YO-PRO-1. After 10 min equilibration at 37 °C, the DNase I was added to yield a final DNase I concentration of 2 U/L. The resulting fluorescence intensity change for each sample was measured on a fluorescence plate reader every 90 s for 3 h (λEX = 491 nm; λEM = 509 nm) and normalized against that of YO-PRO-1 + dsDNA sample.
GNP–M1/MC2(EG12)3 for PI Delivery
All confocal fluorescence imaging were carried out on a Leica TCS SP5 confocal laser scanning microscope with a fixed excitation wavelength of (λEX) of 488 nm. The GNP–M1 conjugate was mixed with MC2(EG12)3 (M1:MC2(EG12)3 molar ratio = 1:1) in an MES buffer (pH 7.4) and hybridized for 3 h to make a GNP–M1/MC2(EG12)3 carrier. The PI stock solution (1 mg/mL in water) was then added to form the GNP–M1/MC2(EG12)3–PI system (M1:PI molar ratio = 1:6). An amount of 105 HeLa cells per well was seeded in a 24-well plate, incubated overnight, and then treated with the GNP–M1/MC2(EG12)3–PI (containing 10 μM PI) for 3 h. The spent medium was removed, and the cells were washed with PBS three times before being imaged on a confocal laser scanning microscope, using 488 nm excitation and fluorescence detection over 600–630 nm.
Delivery of DOX
The DOX stock solution (500 μM) was mixed with GNP–M1/MC2-(EG12)3 to form the GNP–M1/MC2(EG12)3–DOX system (M1:DOX molar ratio = 1:3). An amount of 105 HeLa cells per well was seeded in a 24-well plate, incubated overnight, and then treated with the GNP–M1/MC2-(EG12)3–DOX (containing 5 μM DOX) for 1.5 h. The spent medium was then removed, and the cells were washed with PBS three times. They were then imaged on a confocal laser scanning microscope using 488 nm excitation and fluorescence detection over 580–600 nm.
Delivery of Diruthenium(II) Complex, BPY
BPY was dissolved in water and mixed with GNP–M1/MC2-(EG12)3 to prepare GNP–M1/MC2-(EG12)3–BPY (the molar ratio of M1 to BPY is 1:9). The HeLa cells treated with GNP–M1/MC2-(EG12)3–BPY (containing 30 μM BPY) for 3 h. The spent medium was then removed, and the cells were washed with PBS three times as above. The cells were then imaged by confocal laser scanning microscopy using 488 nm excitation and fluorescence detection over 630–670 nm.
Transmission Electron Microscopy
An amount of 5 × 105 HeLa cells per well was seeded in six-well plates and incubated overnight at 37 °C. The cells were treated with the GNP–M1/MC2(EG)3 nanocarrier in media for 3 h at 37 °C. After washing with PBS, the cells were detached and centrifuged. The cell pellets were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer for 2.5 h, dehydrated using an ascending alcohol series (20, 40, 60, 80, and 100% twice) for 20 min for each change, and embedded in Araldite resin at 65 °C overnight. A 70 nm section was placed on a TEM grid and stained with saturated uranyl acetate and 0.2% Reynolds lead citrate before TEM imaging.12
Acknowledgments
We thank the University of Leeds for funding this project and for providing a fully funded international research scholarship (FFIRS) to L.S. We also thank Mr. Martin Fuller for help in TEM imaging. Y.G. thanks the Wellcome Trust for funding a career-reentry fellowship (grant no. 097354/Z/11/Z). S.G. thanks the EU FP7 for providing an Marie Curie international incoming fellowship (grant no. PIIF-GA-2012-331281). R.C. and D.R. thank Imperial College London for funding support. D.R. is supported by an Industrial CASE Ph.D. Studentship funded jointly by the Biotechnology and Biological Sciences Research Council (BBSRC, UK) and MedImmune (UK).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.5b05228.
TEM image of the GNP. HPLC profiles showing the purification and characterization of the three chain PEG-modified DNA, MC2(EG12)3, and its MS characterization (MALDI-TOF) (PDF)
Author Contributions
D.Z. designed and jointly supervised the research with R.C. and S.G.; L.S. performed all of the research with help from Y.G. and D.R. except the synthesis of MC2(PEG17) (performed by C.C., Z.Y., and D.L.); DNA MALDI-TOF analysis (performed by M.Y.) and the synthesis of the diruthenium complex (performed by C.G., S.S., and J.A.T.); L.S. and D.Z. analyzed data; L.S. and D.Z. wrote the paper. All authors have read, commented on, and approved the final version of the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Mirkin C. A.; Letsinger R. L.; Mucic R. C.; Storhoff J. J. A DNA-Based Method for Rationally Assembling Nanoparticles into Macroscopic Materials. Nature 1996, 382, 607–609 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- Elghanian R.; Storhoff J. J.; Mucic R. C.; Letsinger R. L.; Mirkin C. A. Selective Colorimetric Detection of Polynucleotides Based on the Distance-Dependent Optical Properties of Gold Nanoparticles. Science 1997, 277, 1078–1081 10.1126/science.277.5329.1078. [DOI] [PubMed] [Google Scholar]
- Rosi N. L.; Mirkin C. A. Nanostructures in Biodiagnostics. Chem. Rev. 2005, 105, 1547–1562 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- Cao Y. W. C.; Jin R. C.; Mirkin C. A. Nanoparticles with Raman Spectroscopic Fingerprints for DNA and RNA Detection. Science 2002, 297, 1536–1540 10.1126/science.297.5586.1536. [DOI] [PubMed] [Google Scholar]
- Nam J. M.; Thaxton C. S.; Mirkin C. A. Nanoparticle-Based Bio-Bar Codes for the Ultrasensitive Detection of Proteins. Science 2003, 301, 1884–1886 10.1126/science.1088755. [DOI] [PubMed] [Google Scholar]
- Sharma J.; Chhabra R.; Yan H.; Liu Y. pH-driven Conformational Switch of ″i-Motif’’ DNA for the Reversible Assembly of Gold Nanoparticles. Chem. Commun. 2007, 477–479 10.1039/B612707J. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Cao L.; Ouyang J.; Wang M.; Wang K.; Xia X. H. Reversible Plasmonic Probe Sensitive for pH in Micro/Nanospaces Based on i-Motif-Modulated Morpholino-Gold Nanoparticle Assembly. Anal. Chem. 2013, 85, 1053–1057 10.1021/ac302915a. [DOI] [PubMed] [Google Scholar]
- Wang W. X.; Liu H. J.; Liu D. S.; Xu Y. R.; Yang Y.; Zhou D. J. Use of the Interparticle i-Motif for the Controlled Assembly of Gold Nanoparticles. Langmuir 2007, 23, 11956–11959 10.1021/la702273a. [DOI] [PubMed] [Google Scholar]
- Alexander C. M.; Maye M. M.; Dabrowiak J. C. DNA-Capped Nanoparticles Designed for Doxorubicin Drug Delivery. Chem. Commun. 2011, 47, 3418–3420 10.1039/c0cc04916f. [DOI] [PubMed] [Google Scholar]
- Alexander C. M.; Dabrowiak J. C.; Maye M. M. Investigation of the Drug Binding Properties and Cytotoxicity of DNA-Capped Nanoparticles Designed as Delivery Vehicles for the Anticancer Agents Doxorubicin and Actinomycin D. Bioconjugate Chem. 2012, 23, 2061–2070 10.1021/bc3002634. [DOI] [PubMed] [Google Scholar]
- Xiao Z. Y.; Ji C. W.; Shi J. J.; Pridgen E. M.; Frieder J.; Wu J.; Farokhzad O. C. DNA Self-Assembly of Targeted Near-Infrared-Responsive Gold Nanoparticles for Cancer Thermo-Chemotherapy. Angew. Chem., Int. Ed. 2012, 51, 11853–11857 10.1002/anie.201204018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L.; Ho V. H. B.; Chen C.; Yang Z. Q.; Liu D. S.; Chen R. J.; Zhou D. J. Efficient, pH-Triggered Drug Delivery Using a pH-Responsive DNA-Conjugated Gold Nanoparticle. Adv. Healthcare Mater. 2013, 2, 275–280 10.1002/adhm.201200112. [DOI] [PubMed] [Google Scholar]
- Qiao G. M.; Zhuo L. H.; Gao Y.; Yu L. J.; Li N.; Tang B. A Tumor mRNA-Dependent Gold Nanoparticle-Molecular Beacon Carrier for Controlled Drug Release and Intracellular Imaging. Chem. Commun. 2011, 47, 7458–7460 10.1039/c1cc11490e. [DOI] [PubMed] [Google Scholar]
- Rosi N. L.; Giljohann D. A.; Thaxton C. S.; Lytton-Jean A. K. R.; Han M. S.; Mirkin C. A. Oligonucleotide-modified Gold Nanoparticles for Intracellular Gene Regulation. Science 2006, 312, 1027–1030 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- Kim E. Y.; Schulz R.; Swantek P.; Kunstman K.; Malim M. H.; Wolinsky S. M. Gold Nanoparticle-mediated Gene Delivery Induces Widespread Changes in the Expression of Innate Immunity Genes. Gene Ther. 2012, 19, 347–353 10.1038/gt.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Hao L. L.; Hurst S. J.; Mirkin C. A. Antibody-Linked Spherical Nucleic Acids for Cellular Targeting. J. Am. Chem. Soc. 2012, 134, 16488–16491 10.1021/ja306854d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissuwan D.; Niidome T.; Cortie M. B. The Forthcoming Applications of Gold Nanoparticles in Drug and Gene Delivery Systems. J. Controlled Release 2011, 149, 65–71 10.1016/j.jconrel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Giljohann D. A.; Seferos D. S.; Prigodich A. E.; Patel P. C.; Mirkin C. A. Gene Regulation with Polyvalent siRNA–Nanoparticle Conjugates. J. Am. Chem. Soc. 2009, 131, 2072–2073 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen S. A.; Day E. S.; Ko C. H.; Hurley L. A.; Luciano J. P.; Kouri F. M.; Merkel T. J.; Luthi A. J.; Patel P. C.; Cutler J. I.; Daniel W. L.; Scott A. W.; Rotz M. W.; Meade T. J.; Giljohann D. A.; Mirkin C. A.; Stegh A. H. Spherical Nucleic Acid Nanoparticle Conjugates as an RNAi-Based Therapy for Glioblastoma. Sci. Transl. Med. 2013, 5, 209ra152. 10.1126/scitranslmed.3006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar S.; Daniel W. L.; Giljohann D. A.; Mirkin C. A.; Lippard S. J. Polyvalent Oligonucleotide Gold Nanoparticle Conjugates as Delivery Vehicles for Platinum(IV) Warheads. J. Am. Chem. Soc. 2009, 131, 14652–14653 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. Q.; Xu X. Y.; Lam R.; Giljohann D.; Ho D.; Mirkin C. A. Strategy for Increasing Drug Solubility and Efficacy through Covalent Attachment to Polyvalent DNA– Nanoparticle Conjugates. ACS Nano 2011, 5, 6962–6970 10.1021/nn201446c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamner K. L.; Alexander C. M.; Coopersmith K.; Reishofer D.; Provenza C.; Maye M. M. Using Temperature-Sensitive Smart Polymers To Regulate DNA-Mediated Nanoassembly and Encoded Nanocarrier Drug Release. ACS Nano 2013, 7, 7011–7020 10.1021/nn402214e. [DOI] [PubMed] [Google Scholar]
- Alexander C. M.; Hamner K. L.; Maye M. M.; Dabrowiak J. C. Multifunctional DNA-Gold Nanoparticles for Targeted Doxorubicin Delivery. Bioconjugate Chem. 2014, 25, 1261–1271 10.1021/bc500136r. [DOI] [PubMed] [Google Scholar]
- Kim D.; Jeong Y. Y.; Jon S. A Drug-Loaded Aptamer–Gold Nanoparticle Bioconjugate for Combined CT Imaging and Therapy of Prostate Cancer. ACS Nano 2010, 4, 3689–3696 10.1021/nn901877h. [DOI] [PubMed] [Google Scholar]
- Liu D.; Balasubramanian S. A Proton-Fuelled DNA Nanomachine. Angew. Chem., Int. Ed. 2003, 42, 5734–6 10.1002/anie.200352402. [DOI] [PubMed] [Google Scholar]
- Liu D. S.; Bruckbauer A.; Abell C.; Balasubramanian S.; Kang D. J.; Klenerman D.; Zhou D. J. A Reversible pH-Driven DNA Nanoswitch Array. J. Am. Chem. Soc. 2006, 128, 2067–2071 10.1021/ja0568300. [DOI] [PubMed] [Google Scholar]
- Dong Y. C.; Yang Z. Q.; Liu D. S. DNA Nanotechnology Based on i-Motif Structures. Acc. Chem. Res. 2014, 47, 1853–1860 10.1021/ar500073a. [DOI] [PubMed] [Google Scholar]
- Peer D.; Karp J. M.; Hong S.; FaroKhzad O. C.; Margalit R.; Langer R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat. Nanotechnol. 2007, 2, 751–760 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- Giljohann D. A.; Seferos D. S.; Patel P. C.; Millstone J. E.; Rosi N. L.; Mirkin C. A. Oligonucleotide Loading Determines Cellular Uptake of DNA-Modified Gold Nanoparticles. Nano Lett. 2007, 7, 3818–3821 10.1021/nl072471q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P. C.; Giljohann D. A.; Daniel W. L.; Zheng D.; Prigodich A. E.; Mirkin C. A. Scavenger Receptors Mediate Cellular Uptake of Polyvalent Oligonucleotide-Functionalized Gold Nanoparticles. Bioconjugate Chem. 2010, 21, 2250–2256 10.1021/bc1002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seferos D. S.; Prigodich A. E.; Giljohann D. A.; Patel P. C.; Mirkin C. A. Polyvalent DNA Nanoparticle Conjugates Stabilize Nucleic Acids. Nano Lett. 2009, 9, 308–311 10.1021/nl802958f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H. S.; Liu W.; Misra P.; Tanaka E.; Zimmer J. P.; Itty Ipe B.; Bawendi M. G.; Frangioni J. V. Renal Clearance of Quantum Dots. Nat. Biotechnol. 2007, 25, 1165–1170 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexis F.; Pridgen E.; Molnar L. K.; Farokhzad O. C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharmaceutics 2008, 5, 505–515 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. E.; Chen Z. G.; Shin D. M. Nanoparticle Therapeutics: An Emerging Treatment Modality for Cancer. Nat. Rev. Drug Discovery 2008, 7, 771–782 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- Cho K.; Wang X.; Nie S.; Chen Z. G.; Shin D. M. Therapeutic Nanoparticles for Drug Delivery in Cancer. Clin. Cancer Res. 2008, 14, 1310–1316 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- Dobrovolskaia M. A.; Aggarwal P.; Hall J. B.; McNeil S. E. Preclinical Studies to Understand Nanoparticle Interaction with the Immune System and Its Potential Effects on Nanoparticle Biodistribution. Mol. Pharmaceutics 2008, 5, 487–495 10.1021/mp800032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmali P. P.; Simberg D. Interactions of Nanoparticles with Plasma Proteins: Implication on Clearance and Toxicity of Drug Delivery Systems. Expert Opin. Drug Delivery 2011, 8, 343–357 10.1517/17425247.2011.554818. [DOI] [PubMed] [Google Scholar]
- Knop K.; Hoogenboom R.; Fischer D.; Schubert U. S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem., Int. Ed. 2010, 49, 6288–6308 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- Ikeda Y.; Kawasaki H.; Ichinohe S.; Nagasaki Y. Facile Solid-Phase Synthesis of a Highly Stable Poly(ethylene glycol)–Oligonucleotide Conjugate. J. Mater. Chem. B 2013, 1, 529–535 10.1039/C2TB00031H. [DOI] [PubMed] [Google Scholar]
- Prime K. L.; Whitesides G. M. Adsorption of Proteins onto Surfaces Containing End-Attached Oligo(Ethylene Oxide): A Model System Using Self-Assembled Monolayers. J. Am. Chem. Soc. 1993, 115, 10714–10721 10.1021/ja00076a032. [DOI] [Google Scholar]
- Zhou D.; Bruckbauer A.; Ying L. M.; Abell C.; Klenerman D. Building Three-Dimensional Surface Biological Assemblies on the Nanometer Scale. Nano Lett. 2003, 3, 1517–1520 10.1021/nl0345926. [DOI] [Google Scholar]
- Mout R.; Moyano D. F.; Rana S.; Rotello V. M. Surface Functionalization of Nanoparticles for Nanomedicine. Chem. Soc. Rev. 2012, 41, 2539–2544 10.1039/c2cs15294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D. J.; Ying L. M.; Hong X.; Hall E. A.; Abell C.; Klenerman D. A Compact Functional Quantum Dot–DNA Conjugate: Preparation, Hybridization, and Specific Label-Free DNA Detection. Langmuir 2008, 24, 1659–1664 10.1021/la703583u. [DOI] [PubMed] [Google Scholar]
- Massich M. D.; Giljohann D. A.; Schmucker A. L.; Patel P. C.; Mirkin C. A. Cellular Response of Polyvalent Oligonucleotide–Gold Nanoparticle Conjugates. ACS Nano 2010, 4, 5641–5646 10.1021/nn102228s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessner A.; Paulke B. R.; Muller R. H.; Goppert T. M. Protein Rejecting Properties of PEG-Grafted Nanoparticles: Influence of PEG-Chain Length and Surface Density Evaluated by Two-Dimensional Electrophoresis and Bicinchoninic Acid (BCA)-Protein Assay. Pharmazie 2006, 61, 293–297. [PubMed] [Google Scholar]
- Torchilin V. P.; Omelyanenko V. G.; Papisov M. I.; Bogdanov A. A.; Trubetskoy V. S.; Herron J. N.; Gentry C. A. Poly(ethylene glycol) on the Liposome Surface - on the Mechanism of Polymer-Coated Liposome Longevity. Biochim. Biophys. Acta, Biomembr. 1994, 1195, 11–20 10.1016/0005-2736(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Immordino M. L.; Dosio F.; Cattel L. Stealth Liposomes: Review of the Basic Science, Rationale, and Clinical Applications, Existing and Potential. Int. J. Nanomed. 2006, 1, 297–315. [PMC free article] [PubMed] [Google Scholar]
- Erbacher P.; Bettinger T.; Belguise-Valladier P.; Zou S.; Coll J. L.; Behr J. P.; Remy J. S. Transfection and Physical Properties of Various Saccharide, Poly(ethylene glycol), and Antibody-Derivatized Polyethylenimines (PEI). J. Gene Med. 1999, 1, 210–222. [DOI] [PubMed] [Google Scholar]
- Caliceti P.; Veronese F. M. Pharmacokinetic and Biodistribution Properties of Poly(ethylene glycol)-Protein Conjugates. Adv. Drug Delivery Rev. 2003, 55, 1261–1277 10.1016/S0169-409X(03)00108-X. [DOI] [PubMed] [Google Scholar]
- Pasut G.; Guiotto A.; Veronese F. Protein, Peptide and Non-peptide Drug PEGylation for Therapeutic Application. Expert Opin. Ther. Pat. 2004, 14, 859–894 10.1517/13543776.14.6.859. [DOI] [Google Scholar]
- Perry J. L.; Reuter K. G.; Kai M. P.; Herlihy K. P.; Jones S. W.; Luft J. C.; Napier M.; Bear J. E.; Desimone J. M. PEGylated PRINT Nanoparticles: The Impact of PEG Density on Protein Binding, Macrophage Association, Biodistribution, and Pharmacokinetics. Nano Lett. 2012, 12, 5304–5310 10.1021/nl302638g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prencipe G.; Tabakman S. M.; Welsher K.; Liu Z.; Goodwin A. P.; Zhang L.; Henry J.; Dai H. J. PEG Branched Polymer for Functionalization of Nanomaterials with Ultralong Blood Circulation. J. Am. Chem. Soc. 2009, 131, 4783–4787 10.1021/ja809086q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logie J.; Owen S. C.; McLaughlin C. K.; Shoichet M. S. PEG-Graft Density Controls Polymeric Nanoparticle Micelle Stability. Chem. Mater. 2014, 26, 2847–2855 10.1021/cm500448x. [DOI] [Google Scholar]
- Bagalkot V.; Farokhzad O. C.; Langer R.; Jon S. An Aptamer-Doxorubicin Physical Conjugate as a Novel Targeted Drug-Delivery Platform. Angew. Chem., Int. Ed. 2006, 45, 8149–8152 10.1002/anie.200602251. [DOI] [PubMed] [Google Scholar]
- Marie D.; Vaulot D.; Partensky F. Application of the Novel Nucleic Acid Dyes YOYO-1, YO-PRO-1, and PicoGreen for Flow Cytometric Analysis of Marine Prokaryotes. Appl. Environ. Microbiol. 1996, 62, 1649–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monfardini C.; Schiavon O.; Caliceti P.; Morpurgo M.; Harris J. M.; Veronese F. M. A Branched Monomethoxypoly(ethylene glycol) for Protein Modification. Bioconjugate Chem. 1995, 6, 62–69 10.1021/bc00031a006. [DOI] [PubMed] [Google Scholar]
- Fee C. J. Size Comparison between Proteins PEGylated with Branched and Linear Poly(ethylene glycol) Molecules. Biotechnol. Bioeng. 2007, 98, 725–731 10.1002/bit.21482. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Yang K.; Martinez A.; Basu A.; Chintala R.; Liu H. C.; Janjua A.; Wang M.; Filpula D. Linear and Branched Bicin Linkers for Releasable PEGylation of Macromolecules: Controlled Release in Vivo and in Vitro from Mono- and Multi-PEGylated Proteins. Bioconjugate Chem. 2006, 17, 341–351 10.1021/bc050270c. [DOI] [PubMed] [Google Scholar]
- Lee R. J.; Wang S.; Low P. S. Measurement of Endosome pH Following Folate Receptor-Mediated Endocytosis. Biochim. Biophys. Acta, Mol. Cell Res. 1996, 1312, 237–242 10.1016/0167-4889(96)00041-9. [DOI] [PubMed] [Google Scholar]
- Gill M. R.; Garcia-Lara J.; Foster S. J.; Smythe C.; Battaglia G.; Thomas J. A. A Ruthenium (II) Polypyridyl Complex for Direct Imaging of DNA Structure in Living Cells. Nat. Chem. 2009, 1, 662–667 10.1038/nchem.406. [DOI] [PubMed] [Google Scholar]
- Liu H.-K.; Sadler P. J. Metal Complexes as DNA Intercalators. Acc. Chem. Res. 2011, 44, 349–359 10.1021/ar100140e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.