
Table 1. Statistics of data collection and refinement (molecular replacement).
Values in parentheses are for the highest resolution shell.
| PDB code | 4xw3 |
| X-ray source | Beamline X10SA, PSI |
| Wavelength () | 1.070 |
| Space group | P212121 |
| Unit-cell parameters (, ) | a = 45.06, b = 76.14, c = 122.66, = = = 90 |
| Resolution range () | 502.0 (2.12.0) |
| Observed reflections | 367329 (48403) |
| Unique reflections | 28891 (3815) |
| Multiplicity | 12.7 (12.7) |
| I/(I) | 23.6 (6.5) |
| Completeness (%) | 98.6 (97.6) |
| R meas † (%) | 7.9 (58.6) |
| Wilson B factor (2) | 32.7 |
| Refinement statistics | |
| Resolution () | 47.782.0 |
| No. of reflections used in refinement | 27446 |
| No. of reflections used for calculation of R free | 1445 |
| R work/R free ‡ (%) | 20.0/23.7 |
| No. of non-H atoms | |
| Total | 3144 |
| Protein | 3002 |
| Water molecules | 142 |
| Average B factors (2) | |
| Overall | 28.3 |
| Protein (chain A/B) | 28.0/28.0 |
| Water molecules | 34.0 |
| R.m.s. deviations from ideal geometry | |
| Bond lengths () | 0.009 |
| Bond angles () | 1.139 |
| Ramachandran plot | |
| Most favoured regions (%) | 88.1 |
| Additional allowed regions (%) | 11.9 |
| Generously allowed regions (%) | 0.0 |
| Disallowed regions (%) | 0.0 |
†
R
meas = 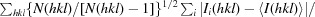
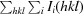 , where I(hkl) is the mean intensity of symmetry-equivalent reflections and N(hkl) is the redundancy.
, where I(hkl) is the mean intensity of symmetry-equivalent reflections and N(hkl) is the redundancy.
‡
R
work = 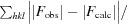
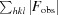 (working set, no cutoff applied); R
free is the R value calculated for 5% of the data set that was not included in refinement.
(working set, no cutoff applied); R
free is the R value calculated for 5% of the data set that was not included in refinement.