

Abstract Abstract
Little is known about the right ventricular (RV) proteome in human heart failure (HF), including possible differences compared to the left ventricular (LV) proteome. We used 2-dimensional differential in-gel electrophoresis (pH: 4–7, 10–150 kDa), followed by liquid chromatography tandem mass spectrometry, to compare the RV and LV proteomes in 12 explanted human hearts. We used Western blotting and multiple-reaction monitoring for protein verification and RNA sequencing for messenger RNA and protein expression correlation. In all 12 hearts, the right ventricles (RVs) demonstrated differential expression of 11 proteins relative to the left ventricles (LVs), including lesser expression of CRYM, TPM1, CLU, TXNL1, and COQ9 and greater expression of TNNI3, SAAI, ERP29, ACTN2, HSPB2, and NDUFS3. Principal-components analysis did not suggest RV-versus-LV proteome partitioning. In the nonischemic RVs (n = 6), 7 proteins were differentially expressed relative to the ischemic RVs (n = 6), including increased expression of CRYM, B7Z964, desmin, ANXA5, and MIME and decreased expression of SERPINA1 and ANT3. Principal-components analysis demonstrated partitioning of the nonischemic and ischemic RV proteomes, and gene ontology analysis identified differences in hemostasis and atherosclerosis-associated networks. There were no proteomic differences between RVs with echocardiographic dysfunction (n = 8) and those with normal function (n = 4). Messenger RNA and protein expression did not correlate consistently, suggesting a major role for RV posttranscriptional protein expression regulation. Differences in contractile, cytoskeletal, metabolic, signaling, and survival pathways exist between the RV and the LV in HF and may be related to the underlying HF etiology and differential posttranscriptional regulation.
Keywords: right ventricle, human heart failure, proteomics, protein expression
Right ventricular remodeling is a ubiquitous feature of common cardiovascular and pulmonary diseases, and right ventricular function is an independent predictor of survival in patients with left ventricular failure.1 In patients with primary pulmonary hypertension, right ventricular function is a better predictor of survival than pulmonary artery pressure or pulmonary vascular resistance.2 Thus, understanding the biology of right ventricular adaptive and maladaptive remodeling and transition to right ventricular failure is critical to improving outcomes in virtually all common cardiovascular and pulmonary diseases and conditions.3-5
Despite the clinical importance of the right ventricle (RV), surprisingly little is known about the molecular mechanisms and pathobiology of right ventricular adaptive or maladaptive remodeling and transition to failure in human heart failure (HF).6-8 The currently available data regarding right ventricular gene expression and proteomics have been derived from a variety of animal models of pulmonary hypertension.9-11 Although these models demonstrate distinct transcriptomic and proteomic signatures in progressive right ventricular remodeling and failure, such models (e.g., pulmonary artery banding, hypoxia, pulmonary artery embolization, and chemotherapy-associated pulmonary injury) incur a specific type of right ventricular injury and do not recapitulate the pathobiology of human right ventricular failure.
In this study, we investigated the right ventricular proteome signature in human subjects with end-stage left ventricular HF. First, we characterized and compared the right and left ventricular proteomes. Second, we compared ischemic and nonischemic right ventricular proteomes. Third, we performed RNA sequencing (RNAseq) to correlate messenger RNA (mRNA) and protein expression. We found distinctive proteomic differences between the RV and the left ventricle (LV) overall and between ischemic and nonischemic RVs in particular. Given the lack of consistent correlation between mRNA and protein expression, expression regulation for these differentially expressed proteins likely resides at the translational or posttranslational, rather than transcriptional, level. Our study has implications regarding the possible molecular mechanisms and pathways involved in human right ventricular remodeling during chronic HF.
Methods
Clinical characterization of study subjects
Before patients were enrolled in the Vanderbilt Human Heart Biorepository (VHHB), the study and protocol were approved by the Vanderbilt University Medical Center Institutional Review Board. Before transplantation, all subjects provided informed consent for enrollment in the VHHB. Twelve explanted hearts from subjects with ischemic (n = 6) or nonischemic (n = 6) HF were identified from the VHHB. Five unused human donor hearts were also obtained from the VHHB. Echocardiograms were performed in the usual care of patients listed and awaiting transplantation. Right ventricular size and function were quantified by visual inspection, tricuspid annular plane systolic excursion, the index of myocardial performance, and fractional area change on the echocardiogram performed most recently before heart explantation. Right ventricular echocardiographic function was classified as dysfunctional if mild or greater global hypokinesis was present. Pulmonary hypertension was defined as a mean pulmonary artery pressure greater than 25 mmHg by the right heart catheterization performed most recently before heart explantation. None of the subjects in this study underwent implantation of a left ventricular assist device before explantation.
Tissue preparation
At the time of explantation, right and left ventricular free-wall tissues in regions free of macroscopic infarction or fibrosis were collected from approximately the same cross-sectional area of both ventricles, immediately frozen in liquid nitrogen, and stored at −80ºC until use. Protein samples from both RVs and LVs were prepared on the basis of methods previously described.12
Differential in-gel electrophoresis (DIGE)
DIGE was performed (pH: 4–7, 10–150 kDa) with 3 spectrally resolvable fluorescent Cydye labels (Cy2 for all 12 RVs, Cy3 for all 12 LVs, and Cy5 for equal aliquots of all 12 RVs and all 12 LVs to serve as an internal control) to detect global and directed changes in patterns of intact protein expression by multiplexing differentially labeled samples onto the same gel, thereby removing analytical (gel-to-gel) artifacts. DIGE is standardized by using an internal control standard Cy5 consisting of equal aliquots of every sample to allow for multiple and replicate samples to be quantitatively intercompared with statistical confidence. DeCyder-2D suite, version 6.5 with Extended Data Analysis, was used for analysis of DIGE results and computation of average abundance ratios. We then re-reviewed the DIGE gel and average abundance ratios together to finalize a smaller set of more robust candidate proteins of interest. In-gel trypsin digestion, following staining by Spyro Ruby, was used to ensure accurate robotic gel protein excision of the final candidate proteins of interest by a spot-handling workstation.
Mass spectrometry
After staining, in-gel digestion, and excision, candidate proteins of interest were identified by liquid chromatography tandem mass spectrometry (LC/MS/MS) with a ThermoScientific LTQ-Orbitrap mass spectrometer. After review of the mass spectrometry raw data output for the digested proteins of interest, statistically significant candidate identifications were generated with the Sequest algorithm searching against the UniProt_KB human database.
Protein verification
We employed two methods to verify selected proteins of interest identified by DIGE and tandem mass spectrometry (MS/MS): (1) MS/MS multiple-reaction monitoring (MRM) and (2) Western blotting. For MRM for single-sample relative quantification, selected proteins underwent targeted quantification on individual patient samples with triple quadripole mass spectrometry (ThermoScientific Vantage spectrometer, coupled with Waters nonAcquity UPLC) after 1-dimensional gel separation and in-gel trypsin digestion of the molecular weight region of interest. MRM is less sensitive than Western blotting but more specific. For Western blotting, an equal amount of protein (an aliquot of the same preparation used for DIGE) from each sample was subjected to electrophoresis gel separation and immune-blotted with polyclonal or monoclonal antibodies (NOVUS Pharmaceuticals) against myosin heavy chain 7 (H00004625-A01), myosin light chain 3 (NBP1-51569), troponin I type 3 (NB110-57628), tropomyosin 1 alpha (NBP1-96659), alpha actinin 2 (NBP1-40428), and desmin (NB110-1790). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal loading control.
Proteomic data analyses
For determining global changes in proteomes between groups (all RVs vs. all LVs or ischemic RVs vs. nonischemic RVs), we performed principal-components analysis (PCA). For determining differences between individual proteins identified by DIGE, MS/MS, and Western blot, we used ANOVA. A P value of <0.05 was considered statistically significant. Network/pathway associations were explored by Ingenuity analysis.
RNAseq
We used 90–120 mg of cardiac tissue per study subject for RNA extraction. Total RNA was isolated with an RNeasy Mini kit (Qiagen) according to the manufacturer’s instructions. After a quality check (Agilent Bioanalyzer for integrity, Qubit RNA fluorometry for concentration), 100 ng of total RNA for each library underwent enrichment for poly-A-containing mRNA with poly-T oligoattached magnetic beads. Cleaved 120–201-bp RNA fragments were copied into first-strand complementary DNA (cDNA) with SuperScript II reverse transcriptase and random primers, followed by second-strand cDNA synthesis using DNA polymerase I and RNase H. The cDNA fragments then underwent an end-repair process, the addition of a single “A” base, and ligation of the Illumina multiplexing adapters. The products were purified and enriched with polymerase chain reaction (PCR) to create the final cDNA sequencing library. The cDNA library underwent a quality-control check via an Agilent Bioanalyzer HS DNA assay to confirm the final library size and via an Agilent Mx3005P quantitative PCR machine using the KAPA Illumina library quantification kit to determine concentration. From a 2-nM stock, samples were pooled by molarity for multiplexing. From the pool, 12 pM was loaded into each well for the flow cell on the Illumina cBot for cluster generation. The flow cell was loaded onto the Illumina HiSeq 2500 utilizing v3 chemistry and HTA 1.8 and was sequenced at paired-end 50 bp with a target of 30 million pass-filter reads per library. The raw sequencing reads in BCL format were processed through CASAVA-1.8.2 for FASTQ conversion and demultiplexing. The real-time analysis chastity filter was used, and only the pass-filter reads were retained for further analysis.
Raw sequence-derived data underwent quality controls to identify potential outliers before any advanced analysis, with tools such as Fastx Toolkit and FastQC. RNA read alignment and mapping were performed by Bowtie/TopHat, and transcriptome reconstruction was performed by Cufflinks for both mRNA and long intervening noncoding RNA (lincRNA). For Cufflinks, a minimum RPKM (reads per kilobase of exon model per million mapped reads) value of 1 was required for further analysis. Read counts were generated and differential gene expression was performed with the negative binomial method DESeq. Read counts for selected genes of interest were also compared by Student’s t test with the assumption of unequal variance.
Results
Demographic and clinical characteristics of patients
There were no significant demographic or clinical differences between patients with ischemic HF and those with nonischemic HF, with the exception of age (Table 1). The echocardiograms were performed at 111 ± 121 days (range: 0–434 days) after transplant, with no difference in days between groups (P = 0.94, Student’s t test). In both the ischemic and nonischemic groups, 1 patient had pulmonary hypertension, and in both the ischemic and nonischemic HF groups, 4 patients had right ventricular echocardiographic dysfunction (Table 2). Given the low prevalence of pulmonary hypertension, we did not analyze proteomic or RNAseq data between pulmonary hypertensive and non–pulmonary hypertensive subjects.
Table 1.
Clinical characteristics of the 12 study subjects
| Clinical characteristics | Nonischemic | Ischemic |
|---|---|---|
| Patients | 6 | 6 |
| Mean age (range), years** | 42 (27–60) | 59 (50–65) |
| Female sex | 1 | 1 |
| RV dysfunction | 2 | 6 |
| Pulmonary hypertension | 1 | 1 |
| Diabetes | 2 | 5 |
| ACEI/ARB | 4 | 4 |
| Beta blocker | 6 | 5 |
| Aldactone | 5 | 5 |
| Sildenafil | 1 | 2 |
| RA, mean (range), mmHg | 10 (1–17) | 9 (2–23) |
| PA, mean (range), mmHg | 28 (18–37) | 31 (13–47) |
| PCW, mean (range), mmHg | 18 (7–26) | 19 (4–25) |
| PVR, mean (range), Wood units | 2.5 (1.2–3.1) | 3.6 (2.3–6.7) |
| CI, mean (range) | 2.2 (1.7–2.7) | 2.1 (1.7–2.9) |
| IABP | 0 | 3 |
Data are no. of patients, unless otherwise noted. ACEI: angiotensin-converting enzyme inhibitor; ARB: angiotensin receptor blocker; CI: cardiac index; IABP: intra-aortic balloon pump; PA: pulmonary artery pressure; PCW: pulmonary capillary wedge pressure; PVR: pulmonary vascular resistance; RA: right atrial pressure; RV: right ventricle.
P < 0.01.
Table 2.
Echocardiographic features
| Echo to Tx, days | RV basal, cm | RV long axis, cm | Visual assessment | TAPSE, mm | RIMP, ms | FAC, % | RAP, mmHg | |
|---|---|---|---|---|---|---|---|---|
| Normal range | <4.2 | <8.6 | “Normal” | >16 | <40 | >35 | <8 | |
| Nonischemic patients | ||||||||
| 1a | 71 | 4.1 | 8.1 | Normal | 17 | 31.8 | 23.4 | NWS |
| 2 | 12 | 5.2 | 9.5 | Moderate HK | 11.5 | 94.4 | 24.6 | 15 |
| 3a | 191 | 5.1 | 9.7 | Normal | 15.4 | 41.7 | 24.4 | 8 |
| 4 | 174 | 3.9 | 8 | Mild HK | 13.1 | 89.9 | 19.4 | 3 |
| 5 | 90 | 5 | 9.8 | Severe HK | 4.5 | 94.4 | 6.4 | 15 |
| 6 | 138 | NWS | NWS | Mild–moderate HK | NWS | 49.3 | NWS | 3 |
| Ischemic patients | ||||||||
| 7 | 414 | 5.6 | 8.1 | Moderate HK | 7.5 | 94.4 | 17.4 | 15 |
| 8a | 44 | 4.1 | 6.8 | Normal | 14.8 | 35.1 | 42.6 | 3 |
| 9 | 106 | 5.7 | 7.9 | Mild HK | 9.7 | 28.8 | 30.1 | 10 |
| 10 | 63 | 4.1 | 6.7 | Mild apical HK | 13.8 | 55.5 | 32.3 | 15 |
| 11a | 7 | 4.2 | 6.9 | Normal | 10.9 | 69.7 | 36.7 | 5 |
| 12 | 256 | 4.6 | 8.1 | Mild HK | 8.9 | 55.8 | 19.4 | 3 |
FAC: RV fractional area change; HK: hypokinesis; NWS: not well seen; RAP: right atrial pressure; RIMP: right ventricular performance index; RV: right ventricle; TAPSE: tricuspid annular plane systolic excursion; Tx: cardiac transplantation.
Patients with echocardiographic normal function.
Two-dimensional (2D) DIGE
Our study was designed to detect relative expression differences between the right ventricular and left ventricular proteomes. The 2D-DIGE gel yielded a large number of differentially expressed proteins on the basis of average abundance ratios, 35 of which were identified for further comparative analysis after detailed review of signal-to-noise ratios and local signal integrity (Fig. 1). After protein identification by MS/MS, we then compared relative abundance ratio protein expression between prespecified groups: (1) pooled RVs (n = 12) versus pooled LVs (n = 12), (2) ischemic RVs (n = 6) versus nonischemic LVs (n = 6), and (3) RVs with echocardiographic normal function (n = 4) versus RVs with echocardiographic dysfunction (n = 8).
Figure 1.
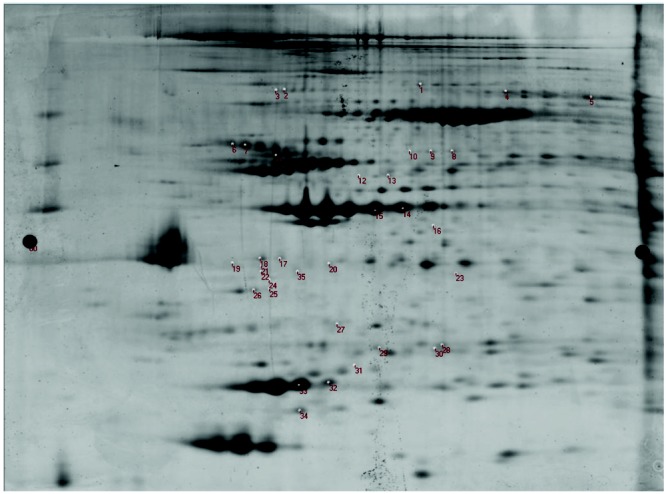
Two-dimensional (2D) differential in-gel electrophoresis. This figure shows the labeled 2D gel (protein isoelectric point plotted against protein molecular weight in kDa) of left and right ventricular proteins labeled by fluorescent dyes. The numbered positions on the gel represent the proteins differentially expressed in the right ventricle versus the left ventricle and selected for excision, digestion, and identification by tandem mass spectrometry analysis.
LV versus RV Proteomes
Pooled comparison
Compared to the 12 pooled LVs, the 12 pooled RVs demonstrated significant differential expression of 11 proteins, implicating possible differences in contractile/sarcomere, metabolic, and cell survival proteins and pathways (Table 3). The RVs demonstrated lesser expression of 5 proteins—CRYM, TPM1, CLU, TXNL1, and COQ9—and greater expression of 6 proteins—TNNI3, SAA1, ERP29, ACTN2, HSPB2, and NDUFS3. The protein fold change differences between RVs and LVs were, in general, modest, and PCA did not suggest significant partitioning of the overall right and left ventricular proteomes (Fig. 2). No proteins were exclusively expressed in either the RV or the LV alone. The top canonical pathways identified by exploratory gene ontology analysis in Ingenuity of these 11 differentially expressed proteins included LXR/RXR and FXR/RXR transcription factor activation, calcium signaling, calpain protease regulation, and adherens junction remodeling; top upstream regulators included GATA4, HAND2, APP, and TBX5; and top molecular and cellular functions included cell death and survival, cellular assembly and organization, and cell-to-cell signaling and interaction (Fig. 3).
Table 3.
Proteomic results for pooled left ventricles (n = 12) versus pooled right ventricles (n = 12)
| Unpaired | Paired | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cluster, protein | UniProt ID | Gene | MW, kDa | Unique peptides |
Spectral counts |
Peptide covered, % |
LV/RV AV ratio |
P, t test |
LV/RV AV ratio |
P, t test |
| Cluster 1 | 1.35 | 0.0005 | 1.31 | 0.0035 | ||||||
| Mu-crystallin homolog | Q14894 | CRYM | 34 | 1 | 1 | 3 | ||||
| Cluster 2 | 1.41 | 0.012 | 1.28 | 0.035 | ||||||
| Tropomyosin 1 alpha | B7Z596 | TPM1 | 33 | 13 | 14 | 41 | ||||
| Mu-crystallin homolog | Q14894 | CRYM | 34 | 9 | 12 | 23 | ||||
| Clusterin | P10909 | CLU | 52 | 5 | 7 | 13 | ||||
| Cluster 3 | 1.5 | 0.01 | 1.36 | 0.0057 | ||||||
| Tropomyosin 1 alpha | B7Z596 | TPM1 | 33 | 18 | 26 | 41 | ||||
| Mu-crystallin homolog | Q14894 | CRYM | 34 | 4 | 4 | 14 | ||||
| Cluster 4 | 1.74 | 0.003 | 1.49 | 0.0034 | ||||||
| Tropomyosin 1 alpha | B7Z596 | TPM1 | 33 | 19 | 25 | 42 | ||||
| Cluster 5 | 2.02 | 0.008 | 1.53 | 0.0066 | ||||||
| Thioredoxin-like protein 1 | O43396 | TXNL1 | 32 | 4 | 5 | 17 | ||||
| Cluster 6 | 1.35 | 0.002 | 1.33 | 0.00021 | ||||||
| Ubiquinone biosynthesis protein COQ9 (mitochondrial) | O75208 | COQ9 | 36 | 8 | 9 | 22 | ||||
| Cluster 7a | NS | NS | −1.29 | 0.0054 | ||||||
| Troponin I | P19429 | TNNI3 | 24 | 8 | 13 | 32 | ||||
| Serum amyloid P-component | P0DJI8 | SAA1 | 25 | 7 | 13 | 28 | ||||
| ER protein 29 | P30040 | ERP29 | 29 | 6 | 11 | 27 | ||||
| Cluster 8b | NS | NS | −1.4 | 0.0029 | ||||||
| Troponin I | P19429 | TNNI3 | 24 | 7 | 10 | 28 | ||||
| Serum amyloid P-component | P0DJI8 | SAA1 | 25 | 6 | 10 | 26 | ||||
| Cluster 9 | −2.07 | 0.0044 | −1.58 | 0.0039 | ||||||
| Actinin-2 fragment | P35609 | ACTN2 | 104 | 9 | 10 | 11 |
Unique peptides: no. of peptides unique to the parent protein in question identified by tandem mass spectrometry; spectral counts: no. of spectral counts identified by tandem mass spectrometry; peptide covered: percentage of the entire parent peptide identified by the digested peptide fragments identified by tandem mass spectrometry mapped back to the entire parent peptide. AV: average abundance; LV: left ventricle; MW: molecular weight; RV: right ventricle.
Weaker evidence for heat-shock protein beta-2 (HSPB2) and NADH dehydrogenase (ubiquinone) iron-sulfur protein 3, mitochondrial (NDUFS3).
Weaker evidence for heat-shock protein beta-2 (HSPB2).
Figure 2.
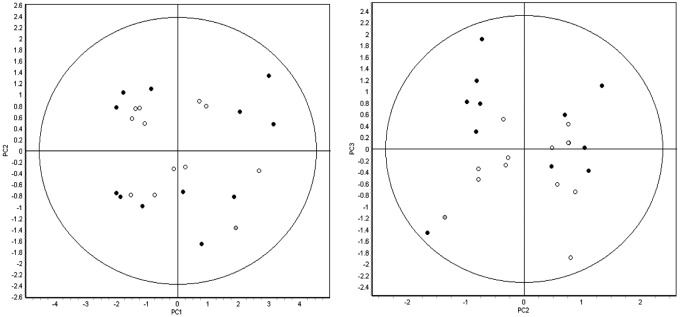
Principal-component analysis of the 24 pooled samples (12 left ventricular [LV], 12 right ventricular [RV]) using 407 features that were matched across all of the 12 gels. LV samples are represented by filled circles, and RV samples are represented by open circles. Both groups encompass explanted hearts from patients with or without ischemic and/or RV echocardiographic dysfunction. The first 3 principal components cumulatively represent more than 50% of the variation in the analysis (PC1: 37.4%, PC2: 10.3%, PC3: 9.8%) yet fail to segregate the samples into any discrete groups based on either biology of unanticipated technical variation. Thus, the magnitude and/or the number of statistically significant changes in protein expression between the right and left ventricles is expected to be low.
Figure 3.

Pooled right ventricle versus pooled left ventricle Ingenuity pathway prediction: the networked differentially expressed proteins in the pooled right versus the pooled left ventricles. The 11 differentially expressed proteins used as inputs into Ingenuity are displayed with respect to expression in the left versus the right ventricles, with relative upregulation of CRYM, TPM1, CLU, TXNL1, and COQ9 in the left ventricles (red) and relative downregulation of TNNI3, SAA1, ERP29, ACTN2, HSPB2, and NDUFS3 in the left ventricles (green). In addition to these 11 proteins, predicted changes included activation of TRIM63, IFNG, MB, SOAT1, MAPT, APP, and KCNA4 and inhibition of the 26s proteasome (see color code in inset).
Etiologic comparison
We analyzed the left and right ventricular proteomes of the 6 subjects with ischemic HF. Compared to the LVs, the RVs demonstrated lesser expression of 4 proteins: CRYM, TPM1, TXNL1, and COQ9 (Table S1). We also analyzed the left and right ventricular proteomes of the 6 subjects with nonischemic HF. Compared to the LVs, the RVs demonstrated lesser expression of 8 proteins: IMMT, CRYM, TPM1, COQ9, APT5H, MYL3, CATD, and PHB (Table S2).
Right ventricular echocardiographic function comparison
We analyzed the paired left and right ventricular proteomes of the 8 subjects with echocardiographic right ventricular dysfunction. Compared to the paired LVs, the RVs demonstrated lesser expression of 9 proteins: CRYM, TPM1, COQ9, TNNI3, SAAI, ERP29, HSPB2, NDUFS3, and ACTN2 (Table S3). Compared to the paired LVs, the 4 RVs with echocardiographic normal function demonstrated lesser expression of 11 proteins: TRFE, MYH7, RUVB2, FIBG, CRYM, TPM1, B7Z964, desmin, COQ9, TNNI3, and SAAI (Table S4). Among those 11 proteins, 5 proteins (CRYM, TPM1, COQ9, TNNI3, and SAAI) were less expressed in the RVs than in the LVs, regardless of right ventricular echocardiographic function.
RV proteomes alone
Etiologic comparison
We compared the proteomes of the 6 ischemic and the 6 nonischemic RVs (Table 4). The nonischemic RVs demonstrated increased expression of 5 proteins—CRYM, B7Z964, desmin, ANXA5, and MIME—and decreased expression of 2 proteins, SERPINA1 and ANT3. PCA demonstrated partitioning of the ischemic and nonischemic RV groups (Fig. 4). The top canonical pathways identified by exploratory Ingenuity analysis of these 7 differentially expressed proteins included the coagulation system, prothrombin activation, LXR/RXR activation, and atherosclerosis signaling,and the top network identified was cellular assembly or organization in cardiovascular disease with upstream regulators TUBB3, CST5, BMP1, and COL17A1 (Fig. 5). From RNAseq, read counts for CRYM and OGN were significantly decreased in unused donor RVs, compared to those in both nonischemic and ischemic RVs (Table 5). Although the RNAseq read counts for CRYM, WDR44, DES, ANXA5, OGN, SERPINA1, and SL25A6 were not significantly different between nonischemic and ischemic RVs by t test, the mean count directional difference between nonischemic and ischemic groups (i.e., either increased or decreased) was congruent with significant expression fold proteomic directional differences for 6 of these 7 proteins (Table 5). Of note, the read counts for DES were decreased in the nonischemic RVs, although desmin protein expression was increased in the nonischemic RVs. The lack of significant differences in read count ranges for the majority of these proteins and the inverse relationship of DES mRNA read counts and protein expression levels suggest that differential protein expression regulation likely resides at translational or posttranslational levels, rather than at the transcriptional level.
Table 4.
Nonischemic right ventricles (n = 6) vs. ischemic right ventricles (n = 6) proteomic results
| Cluster, protein | Gene | MW (kDa) | Unique peptides |
Spectral counts |
Peptide coverage, % |
AV ratio: non-ISCH RV/ISCH RV |
P, t test |
|---|---|---|---|---|---|---|---|
| Cluster 1 | −1.71 | 0.059 | |||||
| Alpha-1-antitrypsin (SERPINA1) | A1AT | 47 | 13 | 18 | 27 | ||
| Cluster 2 | −1.67 | 0.045 | |||||
| Alpha-1-antitrypsin (SERPINA1) | A1AT | 47 | 18 | 26 | 31 | ||
| Antithrombin-III (SERPINC1) | ANT3 | 53 | 8 | 10 | 13 | ||
| Cluster 3a | 1.25 | 0.054 | |||||
| WDR44b | B7Z964 | 37 | 23 | 34 | 49 | ||
| Desmin | DES | 54 | 8 | 9 | 19 | ||
| Cluster 4 | 1.61 | 0.026 | |||||
| Annexin A5 | ANXA | 36 | 10 | 13 | 27 | ||
| Mu-crystallin homolog | CRYM | 34 | 7 | 9 | 20 | ||
| Mimecan (osteoglycin) | OGN | 34 | 5 | 6 | 12 | ||
| Cluster 5 | 1.48 | 0.047 | |||||
| Annexin A5 | ANXA5 | 36 | 10 | 13 | 27 | ||
| Mu-crystallin homolog | CRYM | 34 | 7 | 9 | 20 | ||
| Mimecan (osteoglycin) | OGN | 34 | 5 | 6 | 12 |
Unique peptides: no. of peptides unique to the parent protein in question identified by tandem mass spectrometry; spectral counts: no. of spectral counts identified by tandem mass spectrometry; peptide covered: percentage of the entire parent peptide identified by the digested peptide fragments identified by tandem mass spectrometry mapped back to the entire parent peptide. AV: average abundance; ISCH: ischemic; MW: molecular weight; RV: right ventricle.
Also weaker evidence for heme oxygenase 2 (HMOX2), lactate dehydrogenase B (LDHB), and pyruvate dehydrogenase β (PDHB).
Highly similar to sarcolemma-associated protein (SLMAP).
Figure 4.
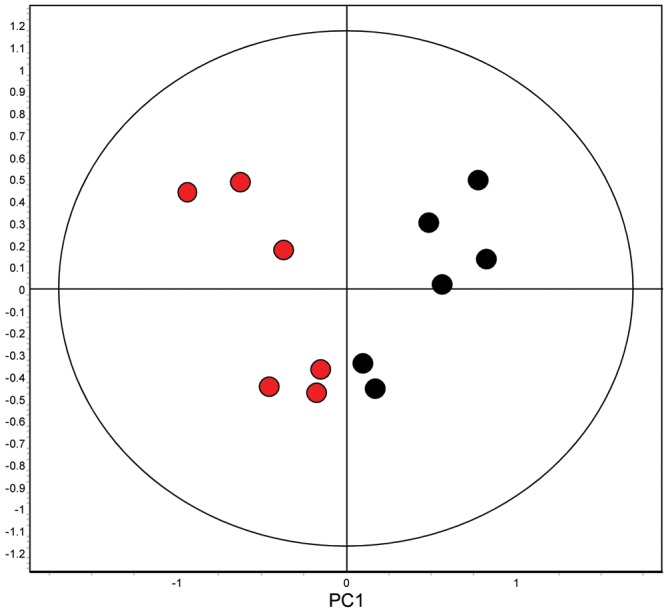
Principal-component analysis of the 12 ischemic (red) and nonischemic (black) right ventricle samples. Principal component 1 partitions the ischemic and nonischemic right ventricular subjects into different groups, supporting that the magnitude and/or number of statistically significant changes in protein expression is sufficient to discriminate the ischemic and nonischemic right ventricular groups.
Figure 5.

Nonischemic right ventricle versus ischemic right ventricle Ingenuity pathway prediction: networked differentially expressed proteins in the nonischemic and ischemic right ventricles. The 7 differentially expressed proteins used as inputs into Ingenuity are displayed with respect to expression in the nonischemic versus the ischemic right ventricles, with relative upregulation of CRYM, WDR44, DES, ANXA5, and OGN in the nonischemic right ventricles (red) and relative downregulation of SERPINA1 and SERPINC1 in the nonischemic right ventricles (green). In addition to these 7 proteins, predicted changes included activation of serine proteases, PRSS57, MHCII, SMPD2, Ccl6, elastase, CRHR1, TNF, F2, F8, and NFkB and inhibition of anti-inflammatory cytokines, OSMR, FCER2, p38-MAPK, and CX3CR1 (see color code in inset).
Table 5.
RNA sequencing read count results for selected proteins
| Read counts, mean ± SD | P, t test | |||||
|---|---|---|---|---|---|---|
| Gene | DON RV | N-ISCH RV | ISCH RV | Log fold difference N-ISCH RV vs. ISCH RV |
N-ISCH RV vs. ISCH RV |
DON RV vs. all RV |
| CRYMa | 944 ± 567 | 3,653 ± 1,549 | 3,226 ± 1,376 | 1.5 | 0.50 | 0.0000080 |
| WDR44 | 292 ± 102 | 256 ± 64 | 232 ± 82 | 1.2 | 0.45 | 0.37 |
| DES | 327,255 ± 142,355 | 311,880 ± 82,070 | 355,684 ± 132,284 | 1.2 | 0.36 | 0.93 |
| ANXA5 | 3,195 ± 342 | 2,216 ± 692 | 1,907 ± 442 | 1.5 | 0.23 | 0.00017 |
| OGNa | 161 ± 30 | 448 ± 215 | 316 ± 176 | 1.5 | 0.13 | 0.000059 |
| SERPINA1 | 26 ± 16 | 29 ± 13 | 36 ± 14 | −1.7 | 0.25 | 0.45 |
| SLC25A6 | 2,320 ± 430 | 2,747 ± 474 | 2,877 ± 889 | −1.7 | 0.67 | 0.07 |
| MYH7 | 393,338 ± 179,483 | 596,572 ± 122,839 | 679,245 ± 211,888 | … | 0.28 | 0.03 |
| MYL3 | 102,061 ± 19,558 | 96,138 ± 23,483 | 99,311 ± 20,123 | … | 0.74 | 0.67 |
| TNNI3 | 149,014 ± 73,644 | 273,192 ± 48,428 | 325,525 ± 172,839 | … | 0.35 | 0.0052 |
| TPM1 | 165,469 ± 65,292 | 210,182 ± 36,562 | 212,764 ± 63,368 | … | 0.91 | 0.20 |
| ACTN2 | 46,455 ± 14,611 | 57,939 ± 14,723 | 53,922 ± 12,533 | … | 0.50 | 0.24 |
Read counts: unadjusted read counts by RNA sequencing; DON RV: unused donor right ventricle; ISCH RV: ischemic right ventricle; N-ISCH RV: nonischemic right ventricle; T test, comparison of RNA sequencing read counts by a 2-way Student’s t test with the assumption of unequal variance and without adjustment for multiple comparisons.
CRYM and OGN were differentially expressed according to the DESeq method (adjusted P value < 0.05).
Preserved RV function versus RV dysfunction
We analyzed the right ventricular proteomes of the 4 hearts with echocardiographic preserved right ventricular function and the 8 hearts with echocardiographic right ventricular dysfunction. No significant protein expression differences were observed.
Summary
Table 6 tabulates the overall log fold differences found in all the analyses performed. Table 7 summarizes the overall results.
Table 6.
Overall DIGE and MS/MS log fold change proteomic expression summary results
| LV/RV ratios | ||||||
|---|---|---|---|---|---|---|
| Protein | Overall | ISCH | N-ISCH | N-RVF | RVF | N-ISCH RV/ISCH RV |
| Metabolic/signaling proteins | ||||||
| Mu-crystalin homolog | 1.4 | 1.7 | 1.3 | 1.4 | 1.3 | 1.5 |
| Thioredoxin-like protein 1 | 2.0 | 2.6 | ||||
| Ubiquinone COQ9 | 1.4 | 1.4 | 1.4 | 1.3 | ||
| ATP synthase subunit d | 1.3 | |||||
| IMMT (mitofilin) | 1.4 | |||||
| Prohibitin | 1.3 | |||||
| Transferrin | ||||||
| NDUFS3 | −1.3* | 1.2* | ||||
| RUVB2 | 1.4 | |||||
| Alpha-1 anti-trypsin | −1.7 | |||||
| Mimecan | 1.5 | |||||
| Heme oxygenase | 1.4 | |||||
| Lactate dehydrogenase | 1.4 | |||||
| Pyruvate dehydrogenase | 1.4 | |||||
| Contractile proteins | ||||||
| Tropomyosin 1 alpha | 1.4 | 2.0 | 1.5 | 2.0 | 1.4* | |
| Troponin I type 3 | −1.3* | 1.8* | 1.2* | |||
| Myosin H 7 | 1.4 | |||||
| Myosin L 3 | 1.3 | |||||
| Cytoskeletal proteins | ||||||
| Actinin-2 fragment | −1.6 | 2.7 | 1.9 | |||
| Desmin | 1.4 | 1.2 | ||||
| WDR44 | 1.4 | 1.2 | ||||
| Survival proteins | ||||||
| Annexin A5 | 1.5 | |||||
| Cathepsin | 1.3 | |||||
| Clusterin | 1.5 | |||||
| Chaperones/stress proteins | ||||||
| ER protein 29 | −1.3* | 1.2 | ||||
| Serum amyloid P (SAA1) | −1.3* | 1.2 | ||||
| HSPB2 | −1.3* | 1.8* | 1.2* | |||
| Miscellaneous/unknown proteins | ||||||
| Fibrinogen | 1.4 | |||||
| Anti-thrombin III | −1.7 | |||||
| RUVB2 (SLMAP) | 1.4 |
P < 0.05 unless otherwise noted. DIGE: differential in-gel electrophoresis; ISCH: ischemic; LV: left ventricle; N-ISCH: nonischemic; N-RVF: hearts without RVF; RV: right ventricle; RVF: right ventricular dysfunction; MS/MS: tandem mass spectrometry.
P = 0.05.
Table 7.
Summary: overall results of group proteomic analyses
| Table | Analyses | Findings |
|---|---|---|
| Table 6 | Various LV vs. RV | 30 proteins exhibited differential expression |
| Table 3 | Pooled all LVs (n = 12) vs. all RVs (n = 12) | 11 proteins were differentially expressed; expression changes were on the whole modest; no proteins were uniquely expressed in either LVs or RVs; PCA did not discriminate between LV and RV proteomes |
| Table S1 | Ischemic LVs (n = 6) vs. ischemic RVs (n = 6) | 4 proteins were differentially expressed; expression changes were similar to pooled all LV vs. RV changes |
| Table S2 | Nonischemic LVs (n = 6) vs. nonischemic RVs (n = 6) | 9 proteins were differentially expressed; these changes suggested a more “metabolic” and “survival” signature and revealed new additional proteins differentially expressed |
| Table S3 | LV (n = 4) vs. paired RV with echocardiographic normal function (n = 4) | 14 proteins were differentially expressed; expression changes were more “metabolic/cytoskeletal” in nature than overall changes |
| Table S4 | LV (n = 8) vs. paired RV with echocardiographic dysfunction (n = 8) | 9 proteins were differentially expressed; expression changes were similar to pooled all LV vs. RV changes |
| Table 4 | Ischemic RV (n = 6) vs. nonischemic RV (n = 6) | 7 proteins were differentially expressed; PCA did discriminate between ischemic and nonischemic RV proteomes |
| Table 4 | RV with echocardiographic normal function (n = 4) vs. RV with echocardiographic dysfunction (n = 8) | No differentially expressed proteins identified |
LV: left ventricle; PCA: principal-components analysis; RV: right ventricle.
Western blotting
We performed Western blotting on the contractile proteins identified after DIGE and MS/MS, using polyclonal antibodies. There were no significant quantitative differences between the unused donor LVs and the pooled HF LVs or between the normal RVs and the pooled HF RVs (Fig. 6A). There were no significant quantitative differences by Western blotting between the normal LVs and the ischemic LVs or between the normal LVs and the ischemic RVs (Fig. 6B). There were significant quantitative differences by Western blotting for MHC, MLC, TNI, and TRP between the normal RVs and the nonischemic RVs but no significant quantitative differences between the nonischemic RVs and the ischemic RVs (Fig. 6C). There were no significant differences between normal LVs, ischemic LVs, and nonischemic LVs (Fig. 6D). Of note, parallel increases or decreases in both RV and LV protein expression were demonstrated relative to controls for all right and left ventricular contractile proteins assessed.
Figure 6.

Western blotting results for contractile proteins: the protein/normalized glyceraldehyde 3-phosphate dehydrogenase (GAPDH) percentage change on the vertical axis (protein/GAPDH % change) plotted against various clinical subject comparison groups for the contractile proteins myosin heavy chain 7 (MHC), myosin light chain 3 (MLC), troponin I 3 (TNI), tropomyosin 1 alpha (TRP), alpha actinin 2 (ACT), and desmin (DSM). Statistically significant differences between groups are marked by asterisks above the error bars. “Norm” refers to unused donor samples. A, There were no significant quantitative differences for any of the contractile proteins between the unused donor left ventricles (LV) and the pooled heart failure left ventricles or between the unused donor right ventricles (RV) and the pooled heat failure right ventricles. B, There were no significant quantitative differences by Western blotting between the unused donor left ventricles and the ischemic left ventricles or between the unused donor right ventricles and the ischemic right ventricles. C, There were significant quantitative differences by Western blotting for MHC, MLC, TNI, and TRP between the unused donor right ventricles and the nonischemic right ventricles but no significant quantitative differences between the nonischemic right ventricles and the ischemic right ventricles. D, There were no significant differences between unused donor left ventricles and the ischemic or nonischemic left ventricles.
We also compared the Western blotting contractile protein findings to DIGE/MS/MS findings, cognizant of the potential quantitative limitations of Western blotting and the possible greater sensitivity of DIGE for isoforms or posttranslational modifications not detected by Western blotting (or vice versa). There was, in general, reasonable agreement between Western blotting and DIGE/MS/MS for relative quantification of most contractile proteins.
MRM
We performed MRM on a smaller number of proteins to confirm the presence of proteins identified by DIGE and MS/MS. MRM confirmed the presence of all proteins identified by tandem MS/MS. There was, in general, reasonable agreement between Western blotting and MRM for the detection and relative quantification of most contractile proteins.
Discussion
Proteomics of HF: prior human and animal studies
The human heart expresses more than 10,000 proteins at any given time, with a high dynamic abundance range (106 for cells), rapid turnover, and responsiveness to diverse physiologic and pathologic stressors. Current gel-based proteomic platforms are limited because they (1) resolve only up to 2,000 proteins, as a result of limitations imposed by protein comigration due to isoelectric focus and molecular weight, and (2) resolve a protein abundance range of 104. Meticulous cardiac protein sample preparation is critical to prevent protein modification (e.g., carbamylation) and/or degradation by proteases. The heart also contains cells other than cardiomyocytes (fibroblasts, smooth muscle cells, endothelial cells, blood cells), which can “contaminate” proteomic results. These pitfalls notwithstanding, proteomic platforms afford opportunities to study quantitative protein expression and posttranslational modification, protein localization and compartmentalization, organelle function, signal transduction, and membrane signaling and to identify novel tissue or plasma biomarkers.13-17
Studies employing a variety of proteomic platforms designed to annotate the normal human left ventricular proteome have reported the identification of 2,000–3,500 unique proteins, depending on proteomic platform, and low (22%–50%) overlap of protein identifications.18-21 In left ventricular biopsies from patients with inflammatory dilated cardiomyopathies compared to healthy human controls, label-free LC/MS/MS revealed 174 proteins with at least a 1.3-fold difference in expression, including proteins for metabolizing glucose; fatty acid and oxidative phosphorylation pathways; and mitochondrial, cytoskeletal, and extracellular matrix proteins.22 In human end-stage HF left ventricular myocardium, LC/MS/MS revealed that total carbonylated proteins, a marker of oxidative stress, were increased compared to those in human unused donor left ventricular myocardium.23 In a study using a 2D electrophoresis (2DE) LC/MS/MS platform, 34 proteins were differentially expressed in myocardium explanted from ischemic versus nonischemic failing human hearts, including proteins in glycolytic and oxidative phosphorylation pathways, stress-response proteins (e.g., heat-shock protein beta 1), and contractile proteins (e.g., myosin light chain 4 and 3, tropomyosin alpha 4).24 In a separate study of human end-stage failing and nonfailing myocardium using a 2DE MS/MS platform, 25 proteins in 3 broad categories exhibited at least a 1.5-fold change in expression: (1) metabolic proteins (e.g., NADH dehydrogenase and cytochrome d oxidase subunit), (2) cytoskeletal proteins (e.g., myosin light chain proteins, troponin I type 3, and transthyretin), and (3) stress-response proteins (αβ-crystallin, HSP27, and HSP20).25
Differential expression of proteins involved in metabolic and mitochondrial pathways has also been a consistent feature of animal models of HF proteomic studies. In a streptozotocin-treated diabetic-cardiomyopathy rat model, 12 of the 24 differentially expressed proteins identified by DIGE/MS/MS were mitochondrial oxidative stress–induced proteins indicative of a “type 1 diabetic cardiomyopathy.”12 After myocardial infarction in rabbits, ramipril increased antioxidative protein expression (e.g., glutathione peroxidase, superoxide dismutase, and heart-type fatty acid–binding protein) and decreased stress-response proteins (e.g., HSP27 and cyclophilin A).26 After aortic constriction in rats, comparative mitochondrial proteomics using a label-free LC/MS/MS platform found decreased mitochondrial fatty acid oxidation proteins, with variable effects on mitochondrial electron transport proteins and increased glucose and tricarboxylic acid cycle proteins.27 In a study of dogs that used a 2DE MS/MS proteomic platform, cardiac resynchronization (CRT) altered the expression of 31 mitochondrial proteins, consistent with a CRT-induced increase in Krebs cycle intermediates, oxidative phosphorylation, and parallel changes in mitochondrial chaperones and proteases.13 In a rabbit rapid-pacing HF model, lesser expression of respiratory-chain proteins and contractile proteins was found, relative to controls.28
This study: right and left ventricular comparisons
In this study, we found differences in contractile, cytoskeletal, metabolic, signaling, and survival pathway protein expression between the RVs and LVs in end-stage human HF. The expression fold differences were modest, and our PCA did not partition the right and left ventricular proteomes by principal components. We did not find proteins uniquely expressed in either ventricle. These results are congruent with prior right-versus-left ventricular proteome studies in normal and HF animal models. In normal mice, rabbits, and pigs, for example, differential right-versus-left ventricular expression of only 1%–2% of proteins has been reported, using a variety of proteomic platforms.29-31 In an ischemia/reperfusion (I/R) model in rabbits, 2DE followed by MS/MS found only 10 proteins expressed differentially between RVs and LVs under both aerobic and I/R conditions, including ATP synthase beta subunit, myosin light chain 2, myosin heavy chain fragments, peroxiredoxin 2, and several heat-shock proteins.32 Our findings in human HF are thus congruent with those of prior studies in animal models. Thus, global protein expression appears far more similar than dissimilar in normal and failing RVs and LVs, without striking or frequent fold changes in the expression of individual proteins or the unique expression of proteins in either ventricle alone.
The lack of striking global changes in protein expression between the RV and the LV in animal HF models and in end-stage human HF may not be surprising, for several reasons. First, several lines of experimental and clinical evidence support that the failing RV “converges” toward similar but nonidentical patterns of failing left ventricular transcription and translation, cardiodynamics, and phenotype.3,4 Second, the most common cause of right ventricular failure is left ventricular failure. Shared features of the end-stage human LV HF milieu (e.g., progressive right ventricular pressure and volume overload due to progressive LV dysfunction, pharmacologic therapy with high-dose inotropes and neurohormonal antagonists) may promote right ventricular transcriptional and translational convergence further toward the failing left ventricular phenotype. Third, the lack of striking global changes in protein expression between the RV and the LV does not exclude RV-specific pathobiology in end-stage human HF. Small but critical differences in regulatory enzyme isoform expression, phosphorylation states, enzymatic activity, nonphosphorylation protein posttranslational modification, substrate location, and/or availability and intracellular signals (e.g., small intracellular calcium fluxes) may all have significant, if not profound, effects on cardiomyocyte function. In particular, small changes in protein phosphorylation ratios or rates of hierarchical phosphorylation may have significant kinetic or regulatory effects in key enzymatic steps in multistep amplified biological pathways.33 Such changes would not be detected by the proteomic platform used in our study.
The etiology of HF does not appear to have widespread effects on right-versus-left ventricular protein expression. When the right and left ventricular proteomes in ischemic HF were compared, only 4 proteins were differentially expressed. These differentially expressed proteins “overlapped” with the proteins differentially expressed in the overall pooled right-versus-left ventricular analysis. In nonischemic HF, we found that 8 proteins were differentially expressed between the RV and the LV. These proteins provided a more “metabolic/survival” signature in nonischemic HF, with the inclusion of ATP synthase subunit d, mitofilin, prohibitin, cathepsin, and clusterin, all of which showed relatively increased expression in the LV compared to the RV. The right-versus-left ventricular expression differences for these proteins in the nonischemic subjects may in part have resulted from (1) the relative young age and absence of conventional atherosclerosis risk factors, such as diabetes mellitus, in the nonischemic subjects and (2) the lesser use of mechanical circulatory support in the nonischemic subjects at the time of explantation (Table 2).
We found 9 proteins differentially expressed between the RVs with echocardiographic dysfunction and the paired LVs and 14 proteins differentially expressed between the RVs with echocardiographic normal function and the paired LVs. The expression of several metabolic proteins, including heme oxygenase, lactate dehydrogenase, and pyruvate dehydrogenase, the expression of the cytoskeletal protein desmin, and the expression of the chaperone protein HSP27 were all decreased in the RVs with echocardiographic normal function, compared to those in the paired LVs (Table 6). Of these, only HSP27 was reduced in the RVs with echocardiographic dysfunction relative to the paired LVs. As left ventricular HF progresses, metabolic remodeling and mitochondrial remodeling are increasingly prominent, and increased desmin and HSP27 expression corresponds to increasing myocardial fibrosis. Intriguingly, the differential right-versus-left ventricular expression of these metabolic and fibrosis-associated proteins may belie a lesser degree of metabolic and mitochondrial remodeling and myocardial fibrosis signaled only in the RVs with echocardiographic normal function.
RV proteome
The etiology of HF does appear to have effects on right ventricular protein expression. The strongest signal for differential protein expression in this study was between ischemic and nonischemic RVs. We found differential expression of 7 proteins between the ischemic and nonischemic RVs, and PCA demonstrated partitioning of the ischemic and nonischemic right ventricular proteomes by principal components. Proteome partitioning between the ischemic and nonischemic RVs in human HF has not yet been reported. Differential expression of atherosclerosis-linked molecules and pathways appeared to account, at least in part, for this partitioning of protein expression in nonischemic and ischemic ventricles. The top canonical pathways identified by exploratory Ingenuity analysis of these 7 differentially expressed proteins included the coagulation system, prothrombin activation, LXR/RXR heterodimer activation, and atherosclerosis signaling, and the top network identified was cellular assembly or organization in cardiovascular disease with upstream regulators TUBB3, CST5, BMP1, and COL17A1. This partitioning may also have been due in part to clinical differences between ischemic and nonischemic subjects, as cited above, including (1) the relative young age and absence of conventional atherosclerosis risk factors, such as diabetes mellitus, in the nonischemic subjects and (2) the lesser use of mechanical circulatory support in the nonischemic subjects at the time of explantation (Table 2).
Interestingly, genomic analyses have revealed rather poor discrimination between ischemic and nonischemic LV cardiomyopathies by gene expression microarrays.34-36 In a recent meta-analysis of 28 microarray studies, coordinated and reciprocal regulation of major metabolic and signaling pathways was demonstrated.37 Major metabolic pathways were typically downregulated, and cell signaling pathways were upregulated. Although animal studies uniformly showed activation of a “fetal gene” program, human microarray studies displayed greater heterogeneity, with some studies even showing upregulation of metabolic and downregulation of signaling pathways in end-stage human HF. These results were attributed to both medical therapy and the heterogeneous transcriptional response observed in phenotypically similar HF.
We did not detect significant protein expression differences between the RVs with echocardiographic normal function and those with echocardiographic dysfunction. This may be due to the small number of subjects for comparison (4 normal vs. 8 dysfunctional) or to the variable time from the most recent echocardiogram to explanation (111 ± 121 days). In a rat pulmonary artery–banding RV hypertrophy model, a variety of proteomic changes have been reported in comparison to control RVs, including a general shift toward glycolytic pathway and away from oxidative phosphorylation, an increase in stress chaperones (e.g., HSP27 species) and antioxidant proteins (e.g., peroxiredoxin 2), and upregulation of desmin and αβ-crystallin.38,39 In pulmonary artery–banded neonatal piglets, 18 proteins were differentially expressed compared to control RVs, including 5 structural proteins (vinculin, tropomyosin b, tropomyosin1A, LIM domain protein CLP-36, and calsarcin-1), 6 metabolic proteins (enolase α, cytoplasmic malate dehydrogenase chain A, superoxide dismutase, ubiquinol cytochrome c reductase, cytochrome c1, and F1-ATOPase b-chain), 2 stress proteins (e.g., HSP70), and 5 miscellaneous proteins.40 Animal models may not represent human pathobiology, particularly given the chronicity of end-stage human failure.41
Protein verification: Western blotting and MRM
We found few discrepancies between the 2D DIGE-MS/MS results and the Western blotting results. These discrepancies are likely due to both (1) the polyclonal antibodies used in Western blotting and their inherent low sensitivity for detecting protein posttranslational modifications and (2) the high sensitivity of 2D DIGE-MS/MS for detecting posttranslational modifications.
The polyclonal antibodies we used for Western blotting would be expected to detect both nonmodified and modified protein species with insensitivity to specific protein posttranslational modifications, depending on the targeted protein epitope(s). DIGE-MS/MS is a highly sensitive method for detecting protein modifications, including posttranslational protein modifications. Since we selected the 2D DIGE spots of interest on the basis of relative differences in right-versus-left ventricular protein expression, we anticipated that some of the difference might due to posttranslational modifications that are less likely to be detected by Western blotting analysis, especially when polyclonal antibody was used.
The pathobiology of troponin I (TnI) and desmin provide possible insights into the challenges of proteomics in human HF. TnI illustrates the complexity of phosphorylation-related posttranslational modifications in human HF. Human TnI contains 209 amino acids and undergoes regulatory posttranslational phosphorylation at one of 14 potential sites, many of which have been recently discovered by MRM.33,42 In human failing myocardium, there is decreased phosphorylation at 2 known protein kinase A sites (S22 and S23) and 3 newly discovered N-terminal sites (S41, S43, and T142) and increased phosphorylation at 2 IT-arm domain sites (S76 and T77) and 3 C-terminal domain sites (S165, T180, and S198).33 HF therapies, such as CRT, alter this pattern of overall TnI phosphorylation.33 Given “functional flexibility” of multiple hierarchical phosphorylation regulatory mechanisms in a given critical protein such as TnI, changes in posttranslational modification phosphorylation, rather than changes in overall levels of protein expression, are likely much more physiologically relevant in the remodeled and/or failing human heart.43
Desmin illustrates the potential pitfalls of quantifying protein expression in human HF. Desmin is the main intermediate filament protein expressed in the heart, and it “links” (1) adjacent myofibrils to myofibrils at the Z-disc and (2) myofibrils to the sarcolemma at costameres, thereby providing a cytoskeletal network that maintains normal myofibril-to-myofibril and myofibril-to-sarcolemma spatial relationships, orientation, integrity, force transduction, and mechanochemical signaling.44,45 Desmin mutations may result in dilated,46,47 restrictive,48 or arrhythmogenic right ventricular cardiomyopathies, depending on the site and functional consequence of specific mutations.49 In animal HF models, desmin expression is increased.50,51 In end-stage human HF, however, studies have yielded conflicting findings, with either increased desmin by Western blotting19,52 or decreased desmin by PCR.53 Earlier gel-based proteomics approaches reported considerable variation in desmin expression between patients with apparently similar disease severity due to variation in mechanisms of remodeling and/or sample bias, since desmin tends to be expressed more abundantly in areas of fibrosis.19 Whether desmin is therefore increased or decreased in HF may depend on HF stage, etiology, additional genetic or environmental modifiers, inclusion of fibrotic myocardium in analysis, and type of assay employed.
Toward systems biology: the relationship of quantitative mRNA expression to quantitative protein expression in the RV
In this study, we found a generally inconstant relationship between the right ventricular RNAseq mRNA read counts and right ventricular log fold protein expression differences and thus frequently the lack of a predictable relationship between right ventricular mRNA and protein expression. From this inconstant relationship, an important caveat emerged, namely, that the prediction of biological pathways and processes from mRNA expression data alone (i.e., by microarray or RNAseq) may not be supported by parallel protein expression fold change detected in the proteome. This caveat has emerged from prior studies as well. The synthesis rate of a given protein is, in general, proportional to the concentration and translational efficiency of its mRNA.54 In general, eukaryotic mRNAs have long half-lives (>2 hours), so that fine-tuning protein levels is achieved by control of mRNA translational efficiency and protein degradation rates rather than by de novo mRNA transcription.54 Mammalian proteins have a mean protein half-life of between 0.5 and 35 hours, with many proteins exhibiting a bimodal distribution of half-lives around 0.5–2 hours and longer proteins in general exhibiting longer half-lives.55 The importance of mRNA translation, rather than mRNA transcription, in regulating protein levels was highlighted in a recent landmark study designed to quantify protein turnover and mRNA expression.56 The study found that only 40% of the variability in protein level was explainable by mRNA levels, leading the investigators to conclude that protein abundance was controlled mainly at the level of translation, not transcription.56 The proteome thus predominately reflects regulatory aspects of mRNA translation and proteostasis (e.g., integrated regulation of the proteome via protein chaperones, protein folding and protein degradation components, signaling pathways, and specialized compartmentalized protein autophaphy modules ) rather than mRNA transcription.55
Limitations
Our study was restricted to the relatively small number of explanted human hearts available at our center at the time this study was performed. The number of biological replicates may have limited the ability to identify principal components and reduced the power of the PCA method employed. End-stage human HF has a variable neurohormonal milieu, and subjects received different therapies, which would be expected to alter protein expression. The 2D DIGE platform used imposes limits: isoelectric focusing (pI: 4–7), molecular weight (10 kDA < Mr < 150 kDa), lower-abundance proteins (below 20 fmol), and hydrophobic integral membrane proteins (e.g., dystrophin and titin, both of which are resolvable but require 1.5%–2.0% gels). We used right and left ventricular free-wall tissue homogenates for protein sample preparation, which include micro–blood vessels, extracellular matrix, and myocyte protein components. We did not undertake complete verification for all proteins by either Western blotting or MRM but used MRM for`selected peptides and Western blotting for contractile proteins only. This observational data set lacked time-dependent standardization of RV failure definition, particularly with regard to the timing of echocardiography. The lack of hearts with pretransplant irreversible, moderate-to-severe pulmonary hypertension was due to selection bias operative in the selection of patients for cardiac transplantation. Irreversible moderate-to-severe pulmonary hypertension (pulmonary vascular resistance > 3 Wood units) is an absolute contraindication to cardiac transplantation at our center. Multiple studies have reported poor survival of cardiac transplant recipients with pretransplant pulmonary hypertension.57 Thus, we did not have the opportunity to study a sufficient number of explanted hearts with pretransplant irreversible, moderate-to-severe pulmonary hypertension.
Summary and conclusions
Differences in contractile, cytoskeletal, metabolic, signaling, and survival pathway proteins exist between RVs and LVs in end-stage human HF and may be related to the underlying etiology of HF. Although the right and left ventricular proteomes differ in end-stage human HF, we did not detect marked abundance differences in the types of proteins expressed or marked expression fold differences in the RVs versus the LVs in end-stage human HF. Thus, the right and left ventricular proteomes in end-stage human HF appear more similar than dissimilar overall, congruent with the results of animal studies to date. The proteomes of ischemic and nonischemic RVs differ significantly by a small number of proteins linked to atherosclerosis-related pathways. The proteomic differences observed in this study likely result from differences in rates of protein translation, modification, or degradation rather than from differences in rates of mRNA transcription, given the lack of consistent correlation of right ventricular protein expression to right ventricular mRNA expression. The roles of right ventricular (1) protein posttranslational modifications, (2) phosphoproteome, (3) protein expression localization, and (4) proteins not identifiable by this proteomic platform (e.g., low-abundance proteins such as kinases, basic proteins, and large proteins such as dystrophin and titin) all bear further study.
Acknowledgments
We thank the Vanderbilt Proteomics Core and Dr. David Friedman for his technical expertise and the Cardiology Core Laboratory and Ms. Kelsey Tomasek for her technical assistance.
The contents of this manuscript are solely the responsibility of the authors and do not represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Appendix.
Table S1.
Ischemic LV (n = 6) versus ischemic RV (n = 6) proteomic results
| Cluster, protein | Gene | MW, kDa | Unique peptides |
Spectral counts |
Peptide covered, % |
LV/RV AV ratio |
P, t test |
|---|---|---|---|---|---|---|---|
| Cluster 1 | 1.41 | 0.012 | |||||
| Mu-crystallin homolog | CRYM | 34 | 1 | 1 | 3 | ||
| Cluster 2 | 1.61 | 0.051 | |||||
| Tropomyosin 1 alpha | TPM1 | 33 | 18 | 26 | 41 | ||
| Mu-crystallin homolog | CRYM | 34 | 4 | 4 | 14 | ||
| Cluster 3 | 2.04 | 0.03 | |||||
| Tropomyosin 1 alpha | TPM1 | 33 | 19 | 25 | 42 | ||
| Cluster 4 | 2.61 | 0.021 | |||||
| Thioredoxin-like protein 1 | TXNL1 | 32 | 4 | 5 | 17 | ||
| Cluster 5 | 2.75 | 0.019 | |||||
| Actinin-2 fragment | ACTN2 | 104 | 9 | 10 | 11 |
Unique peptides: no. of peptides unique to the parent protein in question identified by tandem mass spectrometry; spectral counts: no. of spectral counts identified by tandem mass spectrometry; peptide covered: percentage of the entire parent peptide identified by the digested peptide fragments identified by tandem mass spectrometry mapped back to the entire parent peptide. AV: average abundance; LV: left ventricle; MW: molecular weight; RV: right ventricle.
Table S2.
Nonischemic LV (n = 6) versus nonischemic RV (n = 6) proteomic results
| Cluster, protein | Gene | MW, kDa | Unique peptides |
Spectral counts |
Peptide covered, % |
LV/RV AV ratio |
P, t test |
|---|---|---|---|---|---|---|---|
| Cluster 1 | 1.36 | 0.028 | |||||
| Mitochondrial inner membrane proteina | IMMT | 84 | 29 | 38 | 31 | ||
| Cluster 2 | 1.29 | 0.029 | |||||
| Mu-crystallin homolog | CRYM | 34 | 1 | 1 | 3 | ||
| Cluster 3 | 1.49 | 0.046 | |||||
| Tropomyosin 1 alpha | TPM1 | 33 | 13 | 14 | 41 | ||
| Mu-crystallin homolog | CRYM | 34 | 9 | 12 | 23 | ||
| Clusterin | CLUS | 52 | 5 | 7 | 13 | ||
| Cluster 4 | 1.43 | 0.026 | |||||
| Tropomyosin 1 alpha | TPM1 | 33 | 19 | 25 | 42 | ||
| Cluster 5 | 1.35 | 0.013 | |||||
| Ubiquinone biosynthesis protein COQ9 (mitochondrial) | COQ9 | 36 | 8 | 9 | 22 | ||
| Cluster 6b | 1.28 | 0.047 | |||||
| ATP synthase subunit d | ATP5H | 18 | 18 | 23 | 43 | ||
| Cluster 7 | 1.3 | 0.056 | |||||
| Cathepsin D | CATD | 45 | 8 | 12 | 14 | ||
| Prohibitin | PHB | 30 | 8 | 10 | 29 |
Unique peptides: no. of peptides unique to the parent protein in question identified by tandem mass spectrometry; spectral counts: no. of spectral counts identified by tandem mass spectrometry; peptide covered: percentage of the entire parent peptide identified by the digested peptide fragments identified by tandem mass spectrometry mapped back to the entire parent peptide. AV: average abundance; LV: left ventricle; MW: molecular weight; RV: right ventricle.
Mitofilin, p87/89, cell proliferation-inducing gene 4 protein.
Also some background from myosin light chain 3 (MYL3).
Table S3.
Paired LV (n = 8) versus RV dysfunction (n = 8) proteomic results
| Unpaired | Paired | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cluster, protein | Gene | MW, kDa | Unique peptides |
Spectral counts |
Peptide covered, % |
LV/RV AV ratio |
P, t test |
LV/RV AV ratio |
P, t test |
| Cluster 1 | 1.3 | 0.01 | 1.34 | 0.026 | |||||
| Mu-crystallin homolog | CRYM | 34 | 1 | 1 | 3 | ||||
| Cluster 2 | NS | NS | 1.37 | 0.04 | |||||
| Tropomyosin 1 alpha | TPM1 | 33 | 19 | 25 | 42 | ||||
| Cluster 3 | 1.31 | 0.035 | 1.35 | 0.012 | |||||
| Ubiquinone biosynthesis protein COQ9 (mitochondrial) | COQ9 | 36 | 8 | 9 | 22 | ||||
| Cluster 4a | NS | NS | 1.23 | 0.017 | |||||
| Troponin I | TNNI3 | 24 | 8 | 13 | 32 | ||||
| Serum amyloid P-component | SAMP | 25 | 7 | 13 | 28 | ||||
| ER protein 29 | ERP29 | 29 | 6 | 11 | 27 | ||||
| Cluster 5 | 1.81 | 0.022 | 1.88 | 0.023 | |||||
| Actinin-2 fragment | ACTN2 | 104 | 9 | 10 | 11 |
Unique peptides: no. of peptides unique to the parent protein in question identified by tandem mass spectrometry; spectral counts: no. of spectral counts identified by tandem mass spectrometry; peptide covered: percentage of the entire parent peptide identified by the digested peptide fragments identified by tandem mass spectrometry mapped back to the entire parent peptide. AV: average abundance; LV: left ventricle; MW: molecular weight; NS: not significant; RV: right ventricle.
Also weaker evidence for heat-shock protein 27 (HSP27) and NADH dehydrogenase (ubiquinone) iron-sulfur protein 3, mitochondrial (NDUFS3).
Table S4.
Paired LV (n = 4) versus RV no dysfunction (n = 4) proteomic results
| Unpaired | Paired | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cluster, protein | Gene | MW, kDa | Unique peptides |
Spectral counts |
Peptide covered, % |
LV/RV AV ratio |
P, t test |
LV/RV AV ratio |
P, t test |
| Cluster 1 | NS | NS | 1.37 | 0.011 | |||||
| Transferrin | TRFE | 77 | 17 | 27 | 21 | ||||
| Cluster 2a | 1.47 | 0.047 | 1.49 | 0.046 | |||||
| Myosin 7 | MYH7 | 223 | 15 | 19 | 7 | ||||
| RuvB-like 2 | RUVB2 | 51 | 14 | 19 | 28 | ||||
| Fibrin(ogen) gamma | FIBG | 52 | 10 | 13 | 18 | ||||
| Cluster 3 | 1.47 | 0.036 | NS | NS | |||||
| Mu-crystallin homolog | CRYM | 34 | 1 | 1 | 3 | ||||
| Cluster 4 | 1.55 | 0.042 | NS | NS | |||||
| Tropomyosin 1 alpha | TPM1 | 33 | 18 | 26 | 41 | ||||
| Mu-crystallin homolog | CRYM | 34 | 4 | 4 | 14 | ||||
| Cluster 5b | 1.36 | 0.048 | NS | NS | |||||
| WDR44 | B7Z964 | 37 | 23 | 34 | 49 | ||||
| Desmin | DES | 54 | 8 | 9 | 19 | ||||
| Cluster 6 | 2.68 | 0.014 | 2.61 | 0.024 | |||||
| Tropomyosin 1 alpha | TPM1 | 33 | 19 | 25 | 42 | ||||
| Cluster 7 | 1.42 | 0.018 | 1.41 | 0.0012 | |||||
| Ubiquinone biosynthesis protein COQ9 (mitochondrial) | COQ9 | 36 | 8 | 9 | 22 | ||||
| Cluster 8c | NS | NS | 1.77 | 0.0067 | |||||
| Troponin I | TNNI3 | 24 | 7 | 10 | 28 | ||||
| Serum amyloid P-component | SAMP | 25 | 6 | 10 | 26 |
Unique peptides: no. of peptides unique to the parent protein in question identified by tandem mass spectrometry; spectral counts: no. of spectral counts identified by tandem mass spectrometry; peptide covered: percentage of the entire parent peptide identified by the digested peptide fragments identified by tandem mass spectrometry mapped back to the entire parent peptide. AV: average abundance; LV: left ventricle; MW: molecular weight; NS: not significant; RV: right ventricle.
Also weaker evidence for desmin (DES).
Also weaker evidence for heme oxygenase 2 (HMOX2), lactate dehydrogenase B (LDHB), and pyruvate dehydrogenase β (PDHB).
Also heat-shock protein 27 (HSP27).
Source of Support: This work was supported in part by the Division of Cardiovascular Medicine, Vanderbilt University School of Medicine, and in part by the Clinical and Translational Science award UL1TR000445 from the National Center for Advancing Translational Sciences.
Conflict of Interest: None declared.
Supplements
AppendixPulmCirc-005-481.s001.pdf (434.9KB, pdf)
References
- 1.Ghio S, Gavazzi A, Campana C, Inserra C, Klersy C, Sebastiani R, Arbustini E, Recusani F, Tavazzi L. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol 2001;37(1):183–188. [DOI] [PubMed]
- 2.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009;119(16):2250–2294. [DOI] [PubMed]
- 3.Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation 2008;117(13):1717–1731. [DOI] [PubMed]
- 4.Haddad F, Hunt SA, Rosenthal DN, Murphy DJ. Right ventricular function in cardiovascular disease, part I: anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation 2008;117(11):1436–1448. [DOI] [PubMed]
- 5.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, et al. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 2006;114(17):1883–1891. [DOI] [PubMed]
- 6.Banerjee D, Haddad F, Zamanian RT, Nagendran J. Right ventricular failure: a novel era of targeted therapy. Curr Heart Fail Rep 2010;7(4):202–211. [DOI] [PubMed]
- 7.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009;135(3):794–804. [DOI] [PubMed]
- 8.Haddad F, Ashley E, Michelakis ED. New insights for the diagnosis and management of right ventricular failure, from molecular imaging to targeted right ventricular therapy. Curr Opin Cardiol 2010;25(2):131–140. [DOI] [PubMed]
- 9.Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, Gao Y, Dumur CI, Fawcett P, Voelkel NF, Natarajan R. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol 2011;45(6):1239–1247. [DOI] [PMC free article] [PubMed]
- 10.Gomez-Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Nataranjan R, Dos Remedios CG, Drake JI, et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail 2013;6(1):136–144. [DOI] [PMC free article] [PubMed]
- 11.Urashima T, Zhao M, Wagner R, Fajardo G, Farahani S, Quertermous T, Bernstein D. Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am J Physiol Heart Circ Physiol 2008;295(3):H1351–H1368. [DOI] [PMC free article] [PubMed]
- 12.Hamblin M, Friedman DB, Hill S, Caprioli RM, Smith HM, Hill MF. Alterations in the diabetic myocardial proteome coupled with increased myocardial oxidative stress underlies diabetic cardiomyopathy. J Mol Cell Cardiol 2007;42(4):884–895. [DOI] [PMC free article] [PubMed]
- 13.Agnetti G, Kaludercic N, Kane LA, Elliott ST, Guo Y, Chakir K, Samantapudi D, et al. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ Cardiovasc Genet 2010;3(1):78–87. [DOI] [PMC free article] [PubMed]
- 14.McGregor E, Dunn MJ. Proteomics of the heart: unraveling disease. Circ Res 2006;98(3):309–321. [DOI] [PubMed]
- 15.Sharma P, Cosme J, Gramolini AO. Recent advances in cardiovascular proteomics. J Proteomics 2012;81:3–14. [DOI] [PMC free article] [PubMed]
- 16.Walther TC, Mann M. Mass spectrometry-based proteomics in cell biology. J Cell Biol 2010;190(4):491–500. [DOI] [PMC free article] [PubMed]
- 17.Agnetti G, Husberg C, Van Eyk JE. Divide and conquer: the application of organelle proteomics to heart failure. Circ Res 2011;108(4):512–526. [DOI] [PMC free article] [PubMed]
- 18.Aye TT, Scholten A, Taouatas N, Varro A, van Veen TA, Vos MA, Heck AJ. Proteome-wide protein concentrations in the human heart. Mol Biosyst 2010;6(10):1917–1927. [DOI] [PubMed]
- 19.Corbett JM, Why HJ, Wheeler CH, Richardson PJ, Archard LC, Yacoub MH, Dunn MJ. Cardiac protein abnormalities in dilated cardiomyopathy detected by two-dimensional polyacrylamide gel electrophoresis. Electrophoresis 1998;19(11):2031–2042. [DOI] [PubMed]
- 20.Kline KG, Frewen B, Bristow MR, MacCoss MJ, Wu CC. High quality catalog of proteotypic peptides from human heart. J Proteome Res 2008;7(11):5055–5061. [DOI] [PMC free article] [PubMed]
- 21.Westbrook JA, Wheeler JX, Wait R, Welson SY, Dunn MJ. The human heart proteome: two-dimensional maps using narrow-range immobilised pH gradients. Electrophoresis 2006;27(8):1547–1555. [DOI] [PubMed]
- 22.Hammer E, Goritzka M, Ameling S, Darm K, Steil L, Klingel K, Trimpert C, et al. Characterization of the human myocardial proteome in inflammatory dilated cardiomyopathy by label-free quantitative shotgun proteomics of heart biopsies. J Proteome Res 2011;10(5):2161–2171. [DOI] [PubMed]
- 23.Brioschi M, Polvani G, Fratto P, Parolari A, Agostoni P, Tremoli E, Banfi C. Redox proteomics identification of oxidatively modified myocardial proteins in human heart failure: implications for protein function. PloS ONE 2012;7:e35841. doi:10.1371/journal.pone.0035841. [DOI] [PMC free article] [PubMed]
- 24.Roselló-Lletí E, Alonso J, Cortés R, Almenar L, Martínez-Dolz L, Sánchez-Lázaro I, Lago F, et al. Cardiac protein changes in ischaemic and dilated cardiomyopathy: a proteomic study of human left ventricular tissue. J Cell Mol Med 2012;16(10):2471–2486. [DOI] [PMC free article] [PubMed]
- 25.Li W, Rong R, Zhao S, Zhu X, Zhang K, Xiong X, Yu X, et al. Proteomic analysis of metabolic, cytoskeletal and stress response proteins in human heart failure. J Cell Mol Med 2012;16(1):59–71. [DOI] [PMC free article] [PubMed]
- 26.Chen CY, Lee BC, Hsu HC, Lin HJ, Chao CL, Lin YH, Ho YL, Chen MF. A proteomic study of the effects of ramipril on post-infarction left ventricular remodelling in the rabbit. Eur J Heart Fail 2008;10(8):740–748. [DOI] [PubMed]
- 27.Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, Nguyen TD, et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc Res 2010;85(2):376–384. [DOI] [PubMed]
- 28.Birner C, Dietl A, Deutzmann R, Schröder J, Schmid P, Jungbauer C, Resch M, et al. Proteomic profiling implies mitochondrial dysfunction in tachycardia-induced heart failure. J Card Fail 2012;18(8):660–673. [DOI] [PubMed]
- 29.Phillips D, Aponte AM, Covian R, Neufeld E, Yu ZX, Balaban RS. Homogenous protein programming in the mammalian left and right ventricle free walls. Physiol Genomics 2011;43(21):1198–1206. [DOI] [PMC free article] [PubMed]
- 30.Scholten A, Mohammed S, Low TY, Zanivan S, van Veen TA, Delanghe B, Heck AJ. In-depth quantitative cardiac proteomics combining electron transfer dissociation and the metalloendopeptidase Lys-N with the SILAC mouse. Mol Cell Proteomics 2011;10(10):O111.008474. doi:10.1074/mcp.O111.008474. [DOI] [PMC free article] [PubMed]
- 31.Comunian C, Rusconi F, De Palma A, Brunetti P, Catalucci D, Mauri PL. A comparative MudPIT analysis identifies different expression profiles in heart compartments. Proteomics 2011;11(11):2320–2328. [DOI] [PubMed]
- 32.Cadete VJ, Lin HB, Sawicka J, Wozniak M, Sawicki G. Proteomic analysis of right and left cardiac ventricles under aerobic conditions and after ischemia/reperfusion. Proteomics 2012;12(14):2366–2377. [DOI] [PubMed]
- 33.Zhang P, Kirk JA, Ji W, dos Remedios CG, Kass DA, Van Eyk JE, Murphy AM. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation 2012;126(15):1828–1837. [DOI] [PMC free article] [PubMed]
- 34.Kittleson MM, Ye SQ, Irizarry RA, Minhas KM, Edness G, Conte JV, Parmigiani G, et al. Identification of a gene expression profile that differentiates between ischemic and nonischemic cardiomyopathy. Circulation 2004;110(22):3444–3451. [DOI] [PubMed]
- 35.Kuner R, Barth AS, Ruschhaupt M, Buness A, Zwermann L, Kreuzer E, Steinbeck G, Poustka A, Sültmann H, Nabauer M. Genomic analysis reveals poor separation of human cardiomyopathies of ischemic and nonischemic etiologies. Physiol Genomics 2008;34(1):88–94. [DOI] [PubMed]
- 36.Sanoudou D, Vafiadaki E, Arvanitis DA, Kranias E, Kontrogianni-Konstantopoulos A. Array lessons from the heart: focus on the genome and transcriptome of cardiomyopathies. Physiol Genomics 2005;21(2):131–143. [DOI] [PubMed]
- 37.Barth AS, Kumordzie A, Frangakis C, Margulies KB, Cappola TP, Tomaselli GF. Reciprocal transcriptional regulation of metabolic and signaling pathways correlates with disease severity in heart failure. Circ Cardiovasc Genet 2011;4(5):475–483. [DOI] [PMC free article] [PubMed]
- 38.Faber MJ, Dalinghaus M, Lankhuizen IM, Bezstarosti K, Dekkers DH, Duncker DJ, Helbing WA, Lamers JM. Proteomic changes in the pressure overloaded right ventricle after 6 weeks in young rats: correlations with the degree of hypertrophy. Proteomics 2005;5(10):2519–2530. [DOI] [PubMed]
- 39.Faber MJ, Dalinghaus M, Lankhuizen IM, Bezstarosti K, Verhoeven AJ, Duncker DJ, Helbing WA, Lamers JM. Time dependent changes in cytoplasmic proteins of the right ventricle during prolonged pressure overload. J Mol Cell Cardiol 2007;43(2):197–209. [DOI] [PubMed]
- 40.Sheikh AM, Barrett C, Villamizar N, Alzate O, Valente AM, Herlong JR, Craig D, et al. Right ventricular hypertrophy with early dysfunction: a proteomics study in a neonatal model. J Thorac Cardiovasc Surg 2009;137(5):1146–1153. [DOI] [PubMed]
- 41.Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, et al. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res 2012;111(1):131–150. [DOI] [PubMed]
- 42.Solaro RJ, Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem 2011;286(12):9935–9940. [DOI] [PMC free article] [PubMed]
- 43.Donaldson C, Palmer BM, Zile M, Maughan DW, Ikonomidis JS, Granzier H, Meyer M, VanBuren P, LeWinter MM. Myosin cross-bridge dynamics in patients with hypertension and concentric left ventricular remodeling. Circ Heart Fail 2012;5(6):803–811. [DOI] [PMC free article] [PubMed]
- 44.Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 2009;119(7):1806–1813. [DOI] [PMC free article] [PubMed]
- 45.Goldfarb LG, Olivé M, Vicart P, Goebel HH. Intermediate filament diseases: desminopathy. In Laing NG, ed. The sarcomere and skeletal muscle disease. Adv Exp Med Biol 2008;642:131–164. [DOI] [PMC free article] [PubMed]
- 46.Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. New Engl J Med 2000;342(11):770–780. [DOI] [PubMed]
- 47.Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, Haubold K, et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007;115(10):1244–1251. [DOI] [PubMed]
- 48.Arbustini E, Pasotti M, Pilotto A, Pellegrini C, Grasso M, Previtali S, Repetto A, et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail 2006;8(5):477–483. [DOI] [PubMed]
- 49.Otten E, Asimaki A, Maass A, van Langen IM, van der Wal A, de Jonge N, van den Berg MP, et al. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm 2010;7(8):1058–1064. [DOI] [PubMed]
- 50.Agnetti G, Bezstarosti K, Dekkers DH, Verhoeven AJ, Giordano E, Guarnieri C, Caldarera CM, Van Eyk JE, Lamers JM. Proteomic profiling of endothelin-1-stimulated hypertrophic cardiomyocytes reveals the increase of four different desmin species and alpha-B-crystallin. Biochim Biophys Acta 2008;1784(7–8):1068–1076. [DOI] [PMC free article] [PubMed]
- 51.Sharov VG, Kostin S, Todor A, Schaper J, Sabbah HN. Expression of cytoskeletal, linkage and extracellular proteins in failing dog myocardium. Heart Fail Rev 2005;10(4):297–303. [DOI] [PubMed]
- 52.Heling A, Zimmermann R, Kostin S, Maeno Y, Hein S, Devaux B, Bauer E, et al. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ Res 2000;86(8):846–853. [DOI] [PubMed]
- 53.Di Somma S, Di Benedetto MP, Salvatore G, Agozzino L, Ferranti F, Esposito S, La Dogana P, et al. Desmin-free cardiomyocytes and myocardial dysfunction in end stage heart failure. Eur J Heart Fail 2004;6(4):389–398. [DOI] [PubMed]
- 54.Hershey JW, Sonenberg N, Mathews M. Principles of translational control: an overview. Cold Spring Harb Perspect Biol 2012;4(12):a011528. doi:10.1101/cshperspect.a011528. [DOI] [PMC free article] [PubMed]
- 55.Toyama B, Hetzer M. Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol 2013;14(1):55–61. [DOI] [PMC free article] [PubMed]
- 56.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature 2011;473(7347):337–342. [DOI] [PubMed]
- 57.Vakil K, Duval S, Sharma A, Adabag S, Abidi KS, Taimeh Z, Colvin-Adams M. Impact of pre-transplant pulmonary hypertension on survival after heart transplantation: a UNOS registry analysis. Int J Cardiol 2014;176(3):595–599. [DOI] [PubMed]