

INTRODUCTION
Three major clinical features of restless leg syndrome/Willis-Ekbom disease (RLS/WED) unique for common neurologic disorders enabled somewhat surprising pathophysiologic discoveries. RLS/WED has a well-defined phenotype, one accessible and well-defined environmental vector (iron), and dramatic response to increasing activity of one neurotransmitter system (dopamine). Studies based on these 3 pillars of RLS/WED found a complex underlying biology almost the opposite of expectations. The well-defined phenotype enabled important genetic discoveries for RLS. In sleep medicine, only RLS/WED and narcolepsy have well-defined genetic factors, and both have well defined phenotypes missing for the other sleep disorders. Study of the environmental vector of iron deficiency found an underlying iron pathophysiology. The dopamine treatment response drove studies of dopamine pathophysiology.
This article includes sections focused on pathophysiologic findings from each of these 3 areas: genetics, cortical-spinal excitability, and iron and dopamine. There are other less well-developed features of RLS/WED pathophysiology that could be considered, particularly cortical excitability, neuroanatomical considerations, and other neurotransmitter/neuromodulators. The summary in the last section includes a brief note of these.
GENETICS AND RESTLESS LEG SYNDROME/WILLIS-EKBOM DISEASE PATHOPHYSIOLOGY
The well-defined RLS/WED phenotype enabled successful genomewide association studies (GWAS) that have now identified RLS/WED risk alleles on 5 specific genomic regions for MEIS1, BTBD9, PTPRD, MAP2k/SKOR1, and TOX3/BC034767 and on an intergenic region on chromosome 2 (rs6747972).1–3 Most of these variants also seem to have some relation to the periodic leg movements (PLMS) motor sign of RLS/WED.4 An RLS/WED risk allele on BTBD9 is also strongly associated with both increased PLMS independent of RLS/WED and with decreased peripheral iron stores (decreased serum ferritin).5 Another BDBT9 allelic variant relates to RLS/WED diagnosis,3 increased PLMS,6 and greater decreases in peripheral iron stores with blood donations.7 Functional relation of BTBD9 has been further indicated by findings of increased peripheral iron for a BDBT9-mutant mouse8 and murine ventral midbrain iron content associated with a quantitative trait loci that includes BDBT9.9 Besides these findings relating in general BDBT9 to iron deficiency in RLS/WED, there has been no significantly substantial discovery of potential genetics pathways to RLS/WED pathophysiology. This commonly occurs for results from GWAS for common diseases, suggesting the need for different approaches.
IRON PATHOPHYSIOLOGY
RLS/WED has one major, well-defined primary environmental factor of iron deficiency. This deficiency was noted in the seminal RLS/WED studies of Ekbom10 and Nordlander.11 RLS/WED severity increases with decreased peripheral iron,12 and its prevalence is about 9 times greater in iron-deficient anemia than general populations.13 All conditions that compromise iron status have been associated with increased risk of RLS/WED (eg, pregnancy and end-stage renal disease). Moreover, in these cases, aggressive treatment of the iron deficiency reduces RLS/WED severity. But most RLS/WED patients have normal serum ferritin and little indication for abnormal peripheral iron stores. The pathophysiology appears to be less peripheral and more about central nervous system iron status. Reduced cerebrospinal fluid (CSF) ferritin was reported in 2 separate studies for RLS/WED patients who had normal peripheral iron measures.14, 15
The single best-documented biological abnormality for RLS/WED is brain iron deficiency. Initial reports showed decreased brain iron based on magnetic resonance imaging (MRI) of the substantia nigra and red nucleus as shown in Fig. 1.16 Brain iron deficiency for RLS/WED has now been confirmed in 6 studies using different methods in different laboratories.16-21 The brain areas most consistently showing the reduced iron include the substantia nigra and, to a lesser extent, the putamen and caudate. Recent studies with more sensitive measures have documented low iron in the thalamus.20 The iron deficiency seems to be more regional than global, and the affected regions include not only iron rich areas such as the substantia nigra but also iron-poor areas, particularly the thalamus. Some iron-rich areas have not consistently shown decreased iron for RLS/WED (eg, cerebellar dentate nucleus). Thus, the pathophysiology seems to involve a regional brain iron deficiency present in most RLS/WED patients despite normal iron status.
Fig. 1.
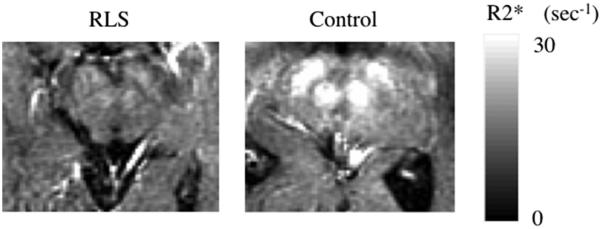
R2* images in a 70-year-old RLS patient and a 71-year-old control subject. Much lower R2* relaxation rates are apparent in the RLS case in both red nucleus and substantia nigra. (Adapted from Allen RP, Barker PB, Wehrl F, et al. MRI measurement of brain iron in patients with restless legs syndrome. Neurology 2001;56(2):263–5; with permission.)
Changes in iron regulatory proteins from RLS/WED autopsy studies present a surprisingly complicated interaction. H-ferritin but not L-ferritin is increased in the RLS/WED brains. The H-ferritin more than L-ferritin provides transport and storage of iron. In neuromelanin cells of the substantia nigra, transferrin receptor is decreased, contrary to expected response to reduced iron status. This finding may relate to a decreased activity of iron regulatory protein-1.22 The transport of iron into the brain shows a more complete picture. Autopsy evaluation of motor cortex microvessels found decreased iron regulatory protein-1 activity with associated decreased iron intake/storage proteins of transferrin receptor, transferrin, and H-ferritin but no change in the iron export protein, ferroportin. Thus, we have a consistent pattern of impaired iron transport into the brain and, more specifically, into neuromelanin cells of the substantia nigra.23 The failure of iron transport to the brain also occurs in the choroid plexus. The epithelial cells show decreased iron and H-ferritin consistent with iron deficiency, but they also show increased mitochondrial ferritin indicating increased mitochondrial iron uptake. The iron regulatory proteins in the choroid plexus are increased for the cellular iron intake (transferrin receptor, transferrin) but also iron export (ferroportin).23 Thus, these cells show a pattern consistent with a higher turnover of iron feeding an increased mitochondrial iron status. The increased mitochondrial ferritin was also found in the cells of the substantia nigra but not the putamen.24 In these cells there seemed to be more mitochondria and increased iron demand. Thus, the mitochondria seem to have acquired their iron surplus at the expense of cytosolic iron and overall cytosolic cellular iron deficiency.
Overall, the RLS/WED regional brain iron deficiency involves a failure to provide adequate iron transport across the blood-brain barrier compounded by a regional failure to import adequate iron into critical neuronal cells (eg, neuromelanin cells of the substantia nigra). A curious and possibly important finding of increased mitochondrial ferritin and iron turnover in the choroid plexus and possibly the substantia nigra may represent a fundamental feature of the iron pathophysiology deserving further consideration.
CONSEQUENCES OF IRON DEFICIENCY: HYPOXIC PATHWAY ACTIVATION AND MYELIN LOSS
The 2 major expected pathophysiologic consequences of brain iron deficiency have been documented: hypoxia and myelin loss. Oxygen transport depends on iron, and decreased iron should signal potential hypoxia. The hypoxic inducible factor 1-alpha has been found increased in the RLS/WED substantia nigra. The hypoxic inducible factor 2-alpha and the vascular endothelial growth factor were increased in the microvessels.25 These differences occurred despite the lack of any significant indication of actual hypoxia. Activation of these hypoxic pathways would be expected to lead to increased dopaminergic activity as described in later discussion.
Two studies found hypoxia in the leg muscles of RLS/WED patients that is not explained by levels of activity. These could represent a sequelae of a general iron regulation problem.26,27 Some recent studies have suggested an increased difference between morning and evening blood flow28 and relative hypoxia in leg muscles of RLS/WED patients.29 It may be that hypoxia or hypoxic pathway activation is one pathway to RLS/WED symptoms. This finding would explain the high prevalence of RLS/WED with chronic obstructive pulmonary disease.30–32
Myelin synthesis depends on iron, and brain iron deficiency in animals reduces myelin proteins, lipids, and cholesterol.33,34 The brain iron deficiency of RLS/WED would be expected to produce a mild but significant myelin deficit. This was confirmed by imaging showing significant decreases in white matter in the corpus callosum, anterior cingulum, and precentral gyrus. Postmortem analyses also found a 25% decrease in myelin proteins.35 This degree of myelin deficit could contribute to the RLS/WED symptoms, particularly in relation to sensorimotor integration that has time-dependent signaling.
DOPAMINE PATHOPHYSIOLOGY
The dramatic and immediate treatment benefits from levodopa led to a general view that RLS/WED has a significant brain dopamine deficiency. The search to document the dopamine abnormalities in RLS/WED turned out to be much more difficult than expected and produced surprising results. The initial CSF analyses showed no differences between RLS/WED and controls for the major proteins related to dopamine.36, 37 A repeat analysis of 3-orthymethyl dopamine (3-OMD) found significant increases in the CSF in 2 independent samples (Fig. 2).38 Moreover, the increases correlated with the dopamine metabolite, homovanillic acid (HVA). Given the metabolic pathways from tyrosine hydroxylase to dopamine (see Fig. 2), the increase in both 3-OMD and HVA is best explained as an increase in tyrosine hydroxylase activity leading to increased dopamine production.
Fig. 2.

RLS CSF samples show 3-OMD is increased and correlated with increased HVA. Top right panel shows increase in 2 separate sets of RLS patients for samples from evening and morning. Left panel shows metabolic pathways for levodopa to 3-OMD and HVA. Bottom right panel shows correlation of these 2 metabolites of levodopa from different metabolic pathways. The values correlate for both metabolites indicating likely increased levodopa rather than abnormalities in the 2 different metabolic pathways. These data are compatible with the autopsy data showing increased tyrosine hydroxylase in the substantia nigra. (Adapted from Allen RP, Connor JR, Hyland K, et al. Abnormally increased CSF 3-Ortho-methyldopa (3-OMD) in untreated restless legs syndrome (RLS) patients indicates more severe disease and possibly abnormally increased dopamine synthesis. Sleep Med 2009;10(1):124, 127; with permission.)
Brain imaging produced somewhat contradictory results. The early PET and single-photon emission computerized tomography studies found clear decreases in striatal D2 receptors,39,40 but this would be most consistent with a response to increased synaptic dopamine. A later study reported increased D2 receptor binding, indicating the opposite of a decreased synaptic dopamine, but that study had mostly mildly affected patients.41 The fluoro-l-dopa (fDOPA) studies, in contrast, have consistently shown decreased striatal fDOPA uptake. Given no cell loss in RLS/WED, the decreased fDOPA uptake would indicate a fast turnover of dopamine consistent with increased dopamine production. The final and probably most important imaging findings involve the dopamine transporter (DAT). Iron-deficient rodents show decreased D2 receptors similar to that seen in RLS/WED42; they also show a decreased DAT, mostly membrane-bound DAT.43 Initial single-photon emission computerized tomography studies of total DAT generally found no significant differences between RLS/WED and controls,39 but membrane-bound DAT was found decreased in 2 separate studies reported together.44 The decreased DAT was found with methylphenidate binding (Fig. 3). Thus, overall, the brain imaging studies also indicate increased striatal dopamine, not the expected decrease.45
Fig. 3.

Conceptualized basis for RLS augmentation. The dotted line indicates the critical postsynaptic dopamine signal level, decreasing below this produces RLS symptoms. The light white line is the RLS before treatment with symptoms at night. The dark white line represents initial treatment success with no symptoms. The red line represents the adjustment to the increased evening dopamine producing earlier and more intense RLS symptoms lasting longer during the night. Note that the morning and day stay protected from RLS symptoms. (Adapted from Earley CJ, Allen RP, Connor JR, et al. The dopaminergic neurons of the A11 system in RLS/WED autopsy brains appear normal. Sleep Med 2009;10:1155–7; with permission.)
DOPAMINE PATHOPHYSIOLOGY: BASIS FOR RESTLESS LEG SYNDROME/WILLIS-EKBOM DISEASE TREATMENT AUGMENTATION
The obvious problem arises: if in the RLS/WED brain dopamine is already abnormally increased, how does increasing this further by levodopa reduce the symptoms? Resolving this apparent contradiction requires appreciating the strong circadian aspect of both dopaminergic activity and RLS/WED symptoms. Increased dopaminergic stimulation will produce a postsynaptic down-regulation likely at both receptor and internal cellular function. The general pattern of decreased D2 receptors, especially for the more severe cases may represent part of this down-regulation of response. But dopamine has a clear circadian activity pattern decreasing in the evening and night and increasing in the morning. The RLS/WED postsynaptic adjustment to increased dopamine stimulation suffices for the daytime but seems to overcompensate when dopamine levels are lower during the evening and night. This produces a relative evening and nighttime dopamine deficit despite the overall dopamine increase. Thus, there is a circadian pattern of evening and night RLS/WED symptoms with, if anything, hyperalertness and arousal in the morning preventing the expected sleepiness for the short and disrupted RLS/WED sleep.
A small dose of dopamine in the evening and night serves to correct for this relative evening decrease in dopamine, but this is to some extent adding coals to the fire. The treatment with increased dopamine stimulation leads to increasing the down-regulation beyond that already occurring with the disease, thus making the underlying RLS/WED disease worse and augmenting the symptoms. The eventual adjustment to the treatment leads to a need for adding even more stimulation and higher doses to be effective. This initially looks like tolerance to the medication, but in this situation tolerance is essentially a worsening of the underlying pathology or augmentation. It is important to appreciate that tolerance and augmentation appear to be biologically the same for dopamine treatment of RLS. Tolerance is the first step.46 Eventually, over time, the adjustment of the system to even more increased dopamine produces a gradual worsening of the RLS. The dopamine agents continue to help sleep a little, leaving the patient struggling with increased daytime symptoms and dependent on the medications to avoid significant withdrawal symptoms. Fig. 3 demonstrates this problem. Thus, the pathophysiology of dopamine for RLS/WED indicates dopamine treatment needs to be done carefully at low doses. Because shorter-acting medications tend to require higher peek levels to support adequate duration, they tend to produce worse augmentation. Using longer-acting medications, particularly the continuous release transdermal rotigotine (Neupro, UCB pharma, Germany) reduces the risk and severity of RLS/WED augmentation. But even here the higher doses will produce significant augmentation (eg, rotigotine produces little augmentation over 5 years at the lower doses of 2 to 3 mg/24h but has significant augmentation at the 4-mg and higher doses).47 Overall, the RLS/WED pathophysiology of dopamine indicates that dopamine medications often have a limited lifespan for effective treatment, and other agents will be needed.
OTHER PATHOPHYSIOLOGIC FINDINGS IN RESTLESS LEG SYNDROME
Studies have found a range of other possible biological abnormalities in RLS/WED that have somewhat limited scientific support. Cortical excitability is one major exception. Transcranial magnetic stimulation (TMS) of the motor cortex for control of hand muscles has consistently shown decreased thresholds for response and reduced paired-pulse inhibition.48–51 The increased cortical excitability is partially reduced by dopamine treatment.52–54 The significance of these findings remains somewhat unclear. The increased cortical excitability has not been consistently related to any of the clinical features of RLS/WED.
Loss of cells has not generally been reported (eg, A11 dopamine cells55) except in one study that reported a decrease in thalamic cells staining for beta endorphin.56 This, finding, however, needs to be confirmed. Evaluations of amounts of gray matter have produced conflicting results.57–61 Overall, RLS/WED does not seem to be a neurodegenerative disorder, nor is there any convincing evidence of reduced number or death of cells. There are, however, consistent reports of regional white matter decrease in RLS/WED consistent with the expected effects of iron deficiency as noted before.35,62,63
An anatomic locus significant for RLS/WED symptoms has been suggested based on theoretic considerations and findings of dopamine, iron abnormalities, TMS, and MRI abnormalities. Much has been made of a “spinal theory” of RLS/WED involving abnormalities in the A11 descending dopamine system, but scant data have been provided supporting any abnormality in this system. The iron and brain imaging studies indicate changes in substantia nigra and striatum, particularly the putamen. The TMS findings indicate sensorimotor pathway abnormalities. Similarly, the MRI findings of white matter decreases seem to suggest abnormalities in cerebral sensorimotor pathways.63
SUMMARY
RLS/WED pathophysiology occurs in a wide range of locations and systems. It seems to have largely metabolic abnormalities mostly involving iron and the consequences of iron deficiency including increased dopamine. The iron-related abnormalities have even been found in the lymphocytes.64 But iron is probably not the whole picture. RLS/WED like other common diseases may have multiple pathways to disease, some less common than others (eg, hypoxia without iron deficiency producing tyrosine hydroxylase and dopamine increases).
The major pathophysiologic findings for RLS/WED have provided guidance for treatment advances for reducing the risk of dopamine augmentation and emphasizing the importance of developing better methods for iron treatment.
KEY POINTS.
Iron management is impaired in restless leg syndrome/Willis-Ekbom disease, leading to brain iron deficiency.
Brain iron deficiency acting partly through hypoxic pathway activation produces increased presynaptic and synaptic dopamine. This produces postsynaptic down-regulation that overcorrects for the normal evening and nocturnal decrease in dopamine-producing restless leg syndrome/Willis-Ekbom disease symptoms. Increasing dopamine activation in the evening and night corrects this problem reducing restless leg syndrome symptoms.
Eventual continued increased dopamine stimulation with long-term dopamine treatment leads to further postsynaptic desensitization and gradual worsening of restless leg syndrome/Willis-Ekbom disease, especially at the higher doses and possibly more for shorter-acting medications.
Brain iron deficiency also reduces myelin, possibly accounting for brain white matter decreases in restless leg syndrome/Willis-Ekbom disease.
Hypoxia (eg, with chronic obstructive pulmonary disease) activates hypoxic pathways leading to dopamine increases and restless leg syndrome/Willis-Ekbom disease independent of iron status.
Acknowledgments
Luipold pharmceuticals and has had research support from NIH (NINOS R01 NS075184, NIA P01 AG21190). Pharmacos mos and UCB Pharma.
Footnotes
Disclosures: Dr R.P. Allen in the last 2 years has served as a consultant for UCB pharma, Xenoport.
REFERENCES
- 1.Schormair B, Kemlink D, Roeske D, et al. PTPRD (protein tyrosine phosphatase receptor type delta) is associated with restless legs syndrome. Nat Genet. 2008;40(8):946–8. doi: 10.1038/ng.190. [DOI] [PubMed] [Google Scholar]
- 2.Winkelmann J, Czamara D, Schormair B, et al. Genome-wide association study identifies novel restless legs syndrome susceptibility loci on 2p14 and 16q12.1. PLoS Genet. 2011;7(7):e1002171. doi: 10.1371/journal.pgen.1002171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winkelmann J, Schormair B, Lichtner P, et al. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat Genet. 2007;39(9):1000–6. doi: 10.1038/ng2099. [DOI] [PubMed] [Google Scholar]
- 4.Moore H, 4th, Winkelmann J, Lin L, et al. Periodic leg movements during sleep are associated with polymorphisms in BTBD9, TOX3/BC034767, MEIS1, MAP2K5/SKOR1, and PTPRD. Sleep. 2014;37(9):1535–42. doi: 10.5665/sleep.4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stefansson H, Rye DB, Hicks A, et al. A genetic risk factor for periodic limb movements in sleep. N Engl J Med. 2007;357(7):639–47. doi: 10.1056/NEJMoa072743. [DOI] [PubMed] [Google Scholar]
- 6.Vilarino-Guell C, Soto AI, Young JE, et al. Susceptibility genes for restless legs syndrome are not associated with Parkinson disease. Neurology. 2008;71(3):222–3. doi: 10.1212/01.wnl.0000317101.67684.e3. [DOI] [PubMed] [Google Scholar]
- 7.Sorensen E, Grau K, Berg T, et al. A genetic risk factor for low serum ferritin levels in Danish blood donors. Transfusion. 2012;52(12):2585–9. doi: 10.1111/j.1537-2995.2012.03629.x. [DOI] [PubMed] [Google Scholar]
- 8.Deandrade MP, Johnson RL, Jr, Unger EL, et al. Motor restlessness, sleep disturbances, thermal sensory alterations and elevated serum iron levels in Btbd9 mutant mice. Hum Mol Genet. 2012;21(18):3984–92. doi: 10.1093/hmg/dds221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jellen LC, Unger EL, Lu L, et al. Systems genetic analysis of the effects of iron deficiency in mouse brain. Neurogenetics. 2012;13(2):147–57. doi: 10.1007/s10048-012-0321-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekbom KA. Restless legs. Ivar Haeggströms; Stockholm (Sweden): 1945. p. 123. [Google Scholar]
- 11.Nordlander NB. Therapy in restless legs. Acta Med Scand. 1953;145:453–7. [PubMed] [Google Scholar]
- 12.Sun ER, Chen CA, Ho G, et al. Iron and the restless legs syndrome. Sleep. 1998;21(4):371–7. [PubMed] [Google Scholar]
- 13.Allen RP, Auerbach S, Bahrain H, et al. The prevalence and impact of restless legs syndrome on patients with iron deficiency anemia. Am J Hematol. 2013;88(4):261–4. doi: 10.1002/ajh.23397. [DOI] [PubMed] [Google Scholar]
- 14.Earley CJ, Connor JR, Beard JL, et al. Abnormalities in CSF concentrations of ferritin and transferrin in restless legs syndrome. Neurology. 2000;54(8):1698–700. doi: 10.1212/wnl.54.8.1698. [DOI] [PubMed] [Google Scholar]
- 15.Mizuno S, Mihara T, Miyaoka T, et al. CSF iron, ferritin and transferrin levels in restless legs syndrome. J Sleep Res. 2005;14(1):43–7. doi: 10.1111/j.1365-2869.2004.00403.x. [DOI] [PubMed] [Google Scholar]
- 16.Allen RP, Barker PB, Wehrl F, et al. MRI measurement of brain iron in patients with restless legs syndrome. Neurology. 2001;56(2):263–5. doi: 10.1212/wnl.56.2.263. [DOI] [PubMed] [Google Scholar]
- 17.Earley CJ, Barker BP, Horska A, et al. MRI-determined regional brain iron concentrations in earlyand late-onset restless legs syndrome. Sleep Med. 2006;7(5):458–61. doi: 10.1016/j.sleep.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 18.Godau J, Klose U, Di Santo A, et al. Multiregional brain iron deficiency in restless legs syndrome. Mov Disord. 2008;23(8):1184–7. doi: 10.1002/mds.22070. [DOI] [PubMed] [Google Scholar]
- 19.Moon HJ, Chang Y, Lee YS, et al. T2 relaxometry using 3.0-tesla magnetic resonance imaging of the brain in early- and late-onset restless legs syndrome. J Clin Neurol. 2014;10(3):197–202. doi: 10.3988/jcn.2014.10.3.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rizzo G, Manners D, Testa C, et al. Low brain iron content in idiopathic restless legs syndrome patients detected by phase imaging. Mov Disord. 2013;28(13):1886–90. doi: 10.1002/mds.25576. [DOI] [PubMed] [Google Scholar]
- 21.Schmidauer C, Sojer M, Seppi K, et al. Transcranial ultrasound shows nigral hypoechogenicity in restless legs syndrome. Ann Neurol. 2005;58(4):630–4. doi: 10.1002/ana.20572. [DOI] [PubMed] [Google Scholar]
- 22.Connor JR, Wang XS, Patton SM, et al. Decreased transferrin receptor expression by neuromelanin cells in restless legs syndrome. Neurology. 2004;62(9):1563–7. doi: 10.1212/01.wnl.0000123251.60485.ac. [DOI] [PubMed] [Google Scholar]
- 23.Connor JR, Ponnuru P, Wang XS, et al. Profile of altered brain iron acquisition in restless legs syndrome. Brain. 2011;134:959–68. doi: 10.1093/brain/awr012. Pt 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder AM, Wang X, Patton SM, et al. Mitochondrial ferritin in the substantia nigra in restless legs syndrome. J Neuropathol Exp Neurol. 2009;68(11):1193–9. doi: 10.1097/NEN.0b013e3181bdc44f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patton SM, Ponnuru P, Snyder AM, et al. Hypoxiainducible factor pathway activation in restless legs syndrome patients. Eur J Neurol. 2011;18(11):1329–35. doi: 10.1111/j.1468-1331.2011.03397.x. [DOI] [PubMed] [Google Scholar]
- 26.Larsson BW, Kadi F, Ulfberg J, et al. Skeletal muscle morphology in patients with restless legs syndrome. Eur Neurol. 2007;58(3):133–7. doi: 10.1159/000104712. [DOI] [PubMed] [Google Scholar]
- 27.Wahlin-Larsson B, Ulfberg J, Aulin KP, et al. The expression of vascular endothelial growth factor in skeletal muscle of patients with sleep disorders. Muscle Nerve. 2009;40(4):556–61. doi: 10.1002/mus.21357. [DOI] [PubMed] [Google Scholar]
- 28.Oskarsson E, Wahlin-Larsson B, Ulfberg J. Reduced daytime intramuscular blood flow in patients with restless legs syndrome/Willis-Ekbom disease. Psychiatry Clin Neurosci. 2014;68(8):640–3. doi: 10.1111/pcn.12170. [DOI] [PubMed] [Google Scholar]
- 29.Salminen AV, Rimpila V, Polo O. Peripheral hypoxia in restless legs syndrome (Willis-Ekbom disease) Neurology. 2014;82(21):1856–61. doi: 10.1212/WNL.0000000000000454. [DOI] [PubMed] [Google Scholar]
- 30.Benediktsdottir B, Janson C, Lindberg E, et al. Prevalence of restless legs syndrome among adults in Iceland and Sweden: lung function, comorbidity, ferritin, biomarkers and quality of life. Sleep Med. 2010;11(10):1043–8. doi: 10.1016/j.sleep.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Kaplan Y, Inonu H, Yilmaz A, et al. Restless legs syndrome in patients with chronic obstructive pulmonary disease. Can J Neurol Sci. 2008;35(3):352–7. doi: 10.1017/s0317167100008957. [DOI] [PubMed] [Google Scholar]
- 32.Lo Coco D, Mattaliano A, Coco AL, et al. Increased frequency of restless legs syndrome in chronic obstructive pulmonary disease patients. Sleep Med. 2009;10:572–6. doi: 10.1016/j.sleep.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 33.Yu GS, Steinkirchner TM, Rao GA, et al. Effect of prenatal iron deficiency on myelination in rat pups. Am J Pathol. 1986;125(3):620–4. [PMC free article] [PubMed] [Google Scholar]
- 34.Ortiz E, Pasquini JM, Thompson K, et al. Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J Neurosci Res. 2004;77(5):681–9. doi: 10.1002/jnr.20207. [DOI] [PubMed] [Google Scholar]
- 35.Connor JR, Ponnuru P, Lee BY, et al. Postmortem and imaging based analyses reveal CNS decreased myelination in restless legs syndrome. Sleep Med. 2011;12(6):614–9. doi: 10.1016/j.sleep.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stiasny-Kolster K, Mignot E, Ling L, et al. The role of CNS dopaminergic, serotonergic and hypocretin (orexin) systems in restless legs syndrome. Sleep. 2003;26:A325–6. [Google Scholar]
- 37.Stiasny-Kolster K, Moller JC, Zschocke J, et al. Normal dopaminergic and serotonergic metabolites in cerebrospinal fluid and blood of restless legs syndrome patients. Mov Disord. 2004;19(2):192–6. doi: 10.1002/mds.10631. [DOI] [PubMed] [Google Scholar]
- 38.Allen RP, Connor JR, Hyland K, et al. Abnormally increased CSF 3-Ortho-methyldopa (3-OMD) in untreated restless legs syndrome (RLS) patients indicates more severe disease and possibly abnormally increased dopamine synthesis. Sleep Med. 2009;10(1):123–8. doi: 10.1016/j.sleep.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michaud M, Soucy JP, Chabli A, et al. SPECT imaging of striatal pre- and postsynaptic dopaminergic status in restless legs syndrome with periodic leg movements in sleep. J Neurol. 2002;249(2):164–70. doi: 10.1007/pl00007859. [DOI] [PubMed] [Google Scholar]
- 40.Turjanski N, Lees AJ, Brooks DJ. Striatal dopaminergic function in restless legs syndrome: 18Fdopa and 11C-raclopride PET studies. Neurology. 1999;52(5):932–7. doi: 10.1212/wnl.52.5.932. [DOI] [PubMed] [Google Scholar]
- 41.Cervenka S, Palhagen SE, Comley RA, et al. Support for dopaminergic hypoactivity in restless legs syndrome: a PET study on D2-receptor binding. Brain. 2006;129:2017–28. doi: 10.1093/brain/awl163. Pt 8. [DOI] [PubMed] [Google Scholar]
- 42.Erikson KM, Jones BC, Hess EJ, et al. Iron deficiency decreases dopamine D1 and D2 receptors in rat brain. Pharmacol Biochem Behav. 2001;69(3–4):409–18. doi: 10.1016/s0091-3057(01)00563-9. [DOI] [PubMed] [Google Scholar]
- 43.Erikson KM, Jones BC, Beard JL. Iron deficiency alters dopamine transporter functioning in rat striatum. J Nutr. 2000;130(11):2831–7. doi: 10.1093/jn/130.11.2831. [DOI] [PubMed] [Google Scholar]
- 44.Earley CJ, Kuwabara H, Wong DF, et al. The dopamine transporter is decreased in the striatum of subjects with restless legs syndrome. Sleep. 2011;34(3):341–7. doi: 10.1093/sleep/34.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Earley CJ, Kuwabara H, Wong DF, et al. Increased synaptic dopamine in the putamen in restless legs syndrome. Sleep. 2013;36(1):51–7. doi: 10.5665/sleep.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winkelman JW, Johnston L. Augmentation and tolerance with long-term pramipexole treatment of restless legs syndrome (RLS) Sleep Med. 2004;5(1):9–14. doi: 10.1016/j.sleep.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 47.Oertel W, Trenkwalder C, Benes H, et al. Long-term safety and efficacy of rotigotine transdermal patch for moderate-to-severe idiopathic restless legs syndrome: a 5-year open-label extension study. Lancet Neurol. 2011;10(8):710–20. doi: 10.1016/S1474-4422(11)70127-2. [DOI] [PubMed] [Google Scholar]
- 48.Gunduz A, Adatepe NU, Kiziltan ME, et al. Circadian changes in cortical excitability in restless legs syndrome. J Neurol Sci. 2012;316(1–2):122–5. doi: 10.1016/j.jns.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 49.Lanza G, Cantone M, Lanuzza B, et al. Distinctive patterns of cortical excitability to transcranial magnetic stimulation in obstructive sleep apnea syndrome, restless legs syndrome, insomnia, and sleep deprivation. Sleep Med Rev. 2015;19C:39–50. doi: 10.1016/j.smrv.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 50.Lanza G, Lanuzza B, Arico D, et al. Direct comparison of cortical excitability to transcranial magnetic stimulation in obstructive sleep apnea syndrome and restless legs syndrome. Sleep Med. 2015;16(1):138–42. doi: 10.1016/j.sleep.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 51.Scalise A, Cadore IP, Gigli GL. Motor cortex excitability in restless legs syndrome. Sleep Med. 2004;5(4):393–6. doi: 10.1016/j.sleep.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 52.Gorsler A, Liepert J. Influence of cabergoline on motor excitability in patients with restless legs syndrome. J Clin Neurophysiol. 2007;24(6):456–60. doi: 10.1097/WNP.0b013e31815a0038. [DOI] [PubMed] [Google Scholar]
- 53.Scalise A, Pittaro-Cadore I, Janes F, et al. Changes of cortical excitability after dopaminergic treatment in restless legs syndrome. Sleep Med. 2010;11(1):75–81. doi: 10.1016/j.sleep.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 54.Rizzo V, Arico I, Mastroeni C, et al. Dopamine agonists restore cortical plasticity in patients with idiopathic restless legs syndrome. Mov Disord. 2009;24(5):710–5. doi: 10.1002/mds.22436. [DOI] [PubMed] [Google Scholar]
- 55.Earley CJ, Allen RP, Connor JR, et al. The dopaminergic neurons of the A11 system in RLS/WED autopsy brains appear normal. Sleep Med. 2009;10:1155–7. doi: 10.1016/j.sleep.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walters AS, Ondo WG, Zhu W, et al. Does the endogenous opiate system play a role in the Restless Legs Syndrome?: a pilot post-mortem study. J Neurol Sci. 2009;279(1–2):62–5. doi: 10.1016/j.jns.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 57.Unrath A, Juengling FD, Schork M, et al. Cortical grey matter alterations in idiopathic restless legs syndrome: an optimized voxel-based morphometry study. Mov Disord. 2007;22(12):1751–6. doi: 10.1002/mds.21608. [DOI] [PubMed] [Google Scholar]
- 58.Chang Y, Chang HW, Song H, et al. Gray matter alteration in patients with restless legs syndrome: a voxel-based morphometry study. Clin Imaging. 2014;39(1):20–5. doi: 10.1016/j.clinimag.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 59.Celle S, Roche F, Peyron R, et al. Lack of specific gray matter alterations in restless legs syndrome in elderly subjects. J Neurol. 2010;257(3):344–8. doi: 10.1007/s00415-009-5320-2. [DOI] [PubMed] [Google Scholar]
- 60.Comley RA, Cervenka S, Palhagen SE, et al. A comparison of gray matter density in restless legs syndrome patients and matched controls using voxel-based morphometry. J Neuroimaging. 2012;22(1):28–32. doi: 10.1111/j.1552-6569.2010.00536.x. [DOI] [PubMed] [Google Scholar]
- 61.Hornyak M, Ahrendts JC, Spiegelhalder K, et al. Voxel-based morphometry in unmedicated patients with restless legs syndrome. Sleep Med. 2007;9(1):22–6. doi: 10.1016/j.sleep.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 62.Chang Y, Paik JS, Lee HJ, et al. Altered white matter integrity in primary restless legs syndrome patients: diffusion tensor imaging study. Neurol Res. 2014;36(8):769–74. doi: 10.1179/1743132814Y.0000000336. [DOI] [PubMed] [Google Scholar]
- 63.Unrath A, Muller HP, Ludolph AC, et al. Cerebral white matter alterations in idiopathic restless legs syndrome, as measured by diffusion tensor imaging. Mov Disord. 2008;23(9):1250–5. doi: 10.1002/mds.22074. [DOI] [PubMed] [Google Scholar]
- 64.Earley CJ, Ponnuru P, Wang X, et al. Altered iron metabolism in lymphocytes from subjects with restless legs syndrome. Sleep. 2008;31(6):847–52. doi: 10.1093/sleep/31.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]