

Abstract
Compelling evidence indicates that suppressor of cytokine signaling 3 (SOCS3) plays a pivotal regulatory role in inflammation. However, the function of SOCS3 in inflammatory responses mediated by Fcγ receptor (FcγR) remains largely unknown. In the current study, we found that SOCS3 expression was greatly enhanced in peritoneal macrophages treated with IgG immune complex (IgG IC). By over-expressing SOCS3 in macrophages, we observed that SOCS3 promoted IgG immune complex-induced production of inflammatory mediators, including IL-6, TNF-α, MIP-2, and MIP-1α. In contrast, SOCS3-defective peritoneal macrophages generated less inflammatory cytokines and chemokines when compared with their wild type counterparts during IgG IC-induced inflammatory responses. We further demonstrated that CCAAT/enhancer-binding protein (C/EBP) δ transcription factor was the major downstream target of SOCS3 in macrophages. These data suggested that SOCS3 was an inflammatory enhancer in IgG IC-treated macrophages by increasing C/EBPδ activity. To elucidate the role for myeloid-derived SOCS3 in IgG IC-induced inflammation in vivo, LysM-cre SOCS3fl/fl mice lacking SOCS3 in macrophages and neutrophils were generated. We found that SOCS3 deficiency greatly alleviated IgG IC-induced generation of pro-inflammatory mediators in lungs, consistent with the in vitro data. Our current findings may provide a new theoretical basis for designing drugs for treatment of IgG IC-associated diseases.
Keywords: SOCS3, IgG IC, peritoneal macrophage, C/EBPδ
Introduction
Suppressor of Cytokine Signaling 3 (SOCS3) belongs to SOCS family of proteins, and its expression can be induced by various inflammatory stimuli, such as bacterial elements, IgG immune complex, and cytokines [1-6]. SOCS3 was first found as a repressor of signal transducer and activator of transcription (STAT)/Janus kinase (JAK) signaling pathway by inhibiting JAK catalytic activity or inducing degradation of cytokine receptor/JAK [7]. Now, roles of SOCS3 in both innate and adaptive immune responses have been investigated. Depending on the distinct immune responses, SOCS3 may exert different effects on inflammation, referred to as pro- and anti-inflammatory functions. For instance, SOCS3 alleviates intestinal inflammation and rheumatoid arthritis by preventing STAT3 from hyperactivation [8, 9]. Besides STAT3, SOCS3 could also repress inflammatory responses through other mechanisms. Our recent studies have proved that SOCS3 decreases IL-6 expression in osteoblasts incubated with LPS by inhibiting C/EBPβ DNA binding activity [4]. In contrast to the above reports, SOCS3 could also act as an inflammatory promoter. SOCS3 exerts its pro-inflammatory function by the interaction of its Src Homology 2 (SH2) domain with gp130, which ensures the specific interference with gp130 signaling pathways that are efficient inhibitors of LPS signaling. Thus, IL-6 challenged peritoneal macrophages lacking SOCS3 produce less TNF-α in response to LPS, and mice are resistant to LPS-induced shock in the absence of SOCS3 in macrophages and neutrophils [10]. Taken together, these studies suggest that SOCS3 may be an attractive target for therapies of inflammatory diseases.
Macrophages play a central role in innate immune responses. In both IgG IC- and LPS-induced acute pulmonary inflammation, macrophage depletion significantly attenuates generation of pro-inflammatory mediators [11-15]. During inflammatory responses, high level of SOCS3 is expressed by myeloid-derived cells, including macrophages [16-18], and its influence on production of pro-inflammatory mediators has been elucidated in Toll-like receptor 4 (TLR4)-activated macrophages [19, 20]. However, its effect on inflammatory reactivities in macrophages upon Fcγ receptor (FcγR) cross-linking remains largely unknown. Here, by using over-expression and gene knockout technologies, we demonstrated that SOCS3 positively regulated IgG IC-induced inflammatory responses in peritoneal macrophages. In addition, we observed that myeloid-specific disruption of SOCS3 resulted in reduction of acute lung inflammation stimulated by IgG IC. Moreover, we found elevated transcriptional activity of the transcription factor-CCAAT/enhancer-binding protein (C/EBP) δ as an underlying mechanism by which SOCS3 enhanced IgG IC-induced expressions of pro-inflammatory mediators in macrophages.
Materials and methods
Animals and cell culture
The animal studies were conducted in accordance with guidelines approved by Southeast University and Harvard Medical School. LysM-cre mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA), and their Cre recombinase expressions are under the control of endogenous Lyz2 promoter/enhancer elements. When crossed with SOCS3fl/fl mice (The Jackson Laboratory, Bar Harbor, ME, USA), Cre-mediated recombination resulted in deletion of SOCS3 in myeloid cell lineage. All mice were used at the age of 8-12 weeks old. RAW264.7 cells were obtained from American Type Culture Collection (Manassas, VA, USA), and cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 2 mM L-glutamine, and 100 units/ml penicillin- streptomycin.
RNA extraction and real time PCR
Total RNAs were isolated from cells by using Trizol obtained from Invitrogen. Then RNAs were subjected to reverse transcription, and cDNAs were synthesized. PCR were conducted with primers for SOCS3: 5′ primer, 5′-CGG GCA GGG GAA GAG ACT GT-3′ and 3′ primer, 5′-GGA GCC AGC GTG GAT CTG C-3′. The PCR universal protocol was described as follows: activation at 95 °C for 5 seconds and, annealing/extension at 60 °C for 30 seconds. Accumulation of fluorescent products was measured in real time. The relative mRNA levels were normalized to levels of GAPDH mRNA in the same sample.
Stable Cell Construction
Control plasmids, or human SOCS3 over-expression plasmids were transduced into RAW264.7 cells by using Fugene®6 Transfection Reagent (Roche, Indianapolis, IN). 12 h after transfection, the cells were treated with 200 μg/ml G418 (MP Biomedicals, LLC, OH), and G418-resistant colonies were selected. SOCS3 expression was detected by Western blot.
Expression vectors and promoter reporters
The mouse C/EBPδ promoter-reporter, C/EBPβ and C/EBPδ expression plasmids, were kindly provided by Richard C. Schwartz (Michigan State University). The reporter plasmid (2 × C/EBP-Luc) containing two copies of a C/EBP binding site was a kind gift from Peter F. Johnson (NCI-Frederick). NF-κB promoter-reporter was purchased from Promega. Human SOCS3 over-expression vector was kindly provided by Dr. Akihiko Yoshimura (Keio University).
Luciferase assay
Plasmids were transduced into RAW264.7 cells by by using Fugene®6 Transfection Reagent as suggested by the supplier. 48 h after transfection, the cells were treated with or without 100 μg/ml IgG immune complex. 4 h later, luciferase activity was detected by using Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) as recommended by the manufacturer.
Preparation of immune complexes
Immune complexes could be formed by incubation of 100 μg BSA (Sigma-Aldrich) in 100 μl PBS with 25 μl 2.5 mg/ml anti-BSA IgG (MP Biomedicals, LLC, OH) at 37°C for 30 min.
Peritoneal macrophage isolation and culture
Mouse peritoneal macrophages were isolated as described previously [21]. Briefly, mice received peritoneal administration of 1 ml of 3% sterile thioglycollate. 5 days later, peritoneal macrophages were isolated by instillation and aspiration of 10 ml ice-cold PBS. Cell suspension was centrifuged at 1000 rpm at 4°C for 10 min. Then the macrophages were cultured in DMEM supplemented with 5% FBS.
IgG IC-induced acute lung inflammation
Mice were firstly anesthetized with intraperitoneal injection of 1.5% sodium pentobarbital. Then mice were treated intra-tracheally with rabbit anti-bovine serum albumin (BSA) IgG. Immediately after intra-tracheal injection of anti-BSA, 1 mg BSA was administrated intravenously. Negative control mice received intratracheal instillation of anti-BSA. 4 h later, bronchoalveolar lavage (BAL) fluids were harvested, and cell-free supernatants were subjected to ELISA.
Western Blot Analysis
RAW264.7 cells were lysed in cold RIPA buffer supplemented with protease inhibitors and PMSF following the manufacturer's guidelines. 30 μg proteins were subjected to electrophoresis in a 12% polyacrylamide gel and then transblotted onto a PVDF membrane followed by blocking with 5% nonfat milk solution. Rabbit anti-Myc antibody, rabbit anti-C/EBPδ antibody, rabbit anti-GAPDH antibody (Cell Signaling, Danvers, MA, USA), and rabbit anti-SOCS3 antibody (Proteintech, Wuhan, China) were applied, followed by a HRP-conjugated donkey anti-rabbit secondary IgG (GE Healthcare, Piscataway, NJ, USA). Immunoreactivity was visualized using enhanced chemiluminescence technique according to the manufacturer's direction (Thermo Fisher Scientific, Rockford, IL, USA).
ELISA
ELISA kits for mouse TNF-α, MIP-2, IL-6, MIP-1α, MCP-1 and MIP-1β were purchased from R&D Systems, and ELISA were performed following manufacture's protocols.
Gel shift assay
Gel shift assay were conducted as described previously [22, 23]. Cell nuclear proteins were prepared as follows. Cells were lysed in 2 mM MgCl2, 15 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol, 10 mM HEPES (pH 7.6), 0.5 mM PMSF, 0.1% (v/v) Nonidet P-40, and protease inhibitors on ice. Then nuclei were harvested, and proteins were extracted by using buffer C (420 mM NaCl, 0.2 mM EDTA, protease inhibitors, 25% (v/v) glycerol, 1 mM dithiothreitol, 0.5 mM PMSF, and 20 mM HEPES (pH 7.9). Protein concentrations were measured by Bio-Rad protein assay kit (Thermo Fisher Scientific, Rockford, IL). The gel shift probes for NF-κB were obtained from Promega, and were labeled with γ [32P] ATP (3,000 Ci/mmol at 10 mCi/ml, GE Healthcare, Piscataway, NJ). DNA binding reactions were conducted in a reaction system containing nuclear proteins, 5× binding buffer, 50 mM HEPES (pH 7.9), 5 mM EDTA, 5 mM dithiothreitol, 50 mM KCl, 0.1% Nonidet P-40, 1 μg of poly (dI-dC), 200 pg of probe, 0.06% bromphenol blue. Samples were electrophoresed through 5.5% polyacrylamide gels in 1× TBE at 190 V for approximately 3.5 h, dried under vacuum, and exposed to X-ray film.
Statistical Analysis
All values were expressed as the mean ± S. E. M. Significance was assigned where p < 0.05. Data sets were analyzed using Student's t test or one-way ANOVA, with individual group means being compared with the Student-Newman-Keuls multiple comparison test.
Results
SOCS3 enhanced IgG IC-induced inflammatory responses in macrophages
A variety of cells express SOCS3, including macrophages, osteoblasts, microglia, and neutrophils [4, 18, 24], and roles of SOCS3 in inflammation have been investigated. However, the effect of SOCS3 on inflammatory responses after FcγR cross-linking remains largely unknown. Therefore, we sought to determine the role of SOCS3 in IgG IC-stimulated inflammatory mediator's production in peritoneal macrophages that express abundant IgG IC receptors. We first dissected if SOCS3 expression in peritoneal macrophages could be induced by IgG IC at mRNA level. As shown in Fig. 1A, SOCS3 mRNA production was dramatically induced by IgG IC at all time points examined, though its abundance was slowly declined from 3 h to 24 h. Next, Western blot was conducted to examine IgG IC-triggered SOCS3 expression at protein level. We found that SOCS3 generation was dramatically elevated 3 h after IgG IC challenge, and the protein was only marginally detected at 24 h time point (Fig. 1B). We next constructed a stable cell line-RAW-SOCS3, which constitutively produce high level of SOCS3. As shown in Fig. 2A, RAW-SOCS3 cells expressed higher level of SOCS3 at protein level when compared with RAW-Neo cells, which was used as negative control. We challenged RAW-Neo cells and RAW-SOCS3 cells with 100 μg/ml IgG IC for different time periods. Supernatants were harvested and subjected to ELISA to measure generation of distinct inflammatory mediators. We found that, upon IgG IC stimulation, RAW-SOCS3 cells secreted more amounts of IL-6, TNF-α, MIP-2, and MIP-1α as compared with RAW-Neo cells at all time points tested (Fig. 2, B-E), indicating that SOCS3 augmented FcγR-mediated inflammation.
Figure 1.
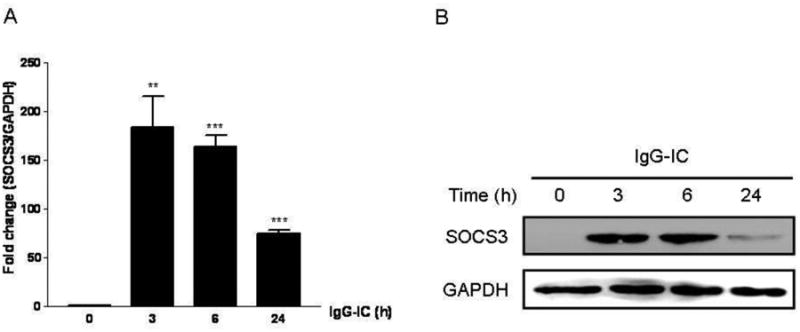
Effect of IgG IC on SOCS3 expression in RAW264.7 cells. RAW264.7 cells were stimulated by IgG IC for 0, 3 h, 6 h, and 24 h, respectively. A, total cellular RNAs were extracted, and real time-PCR was performed by using primers for SOCS3, and GAPDH, respectively. B, total cellular proteins were extracted, and Western blot was performed by using anti-SOCS3 antibody, and anti-GAPDH antibody, respectively. The level of GAPDH was shown at the bottom as a loading control. Data were expressed as means ± S. E. M. (n=3, biological replicates). *, and *** indicated statistically significant difference— p < 0.05, and p < 0.001, respectively.
Figure 2.
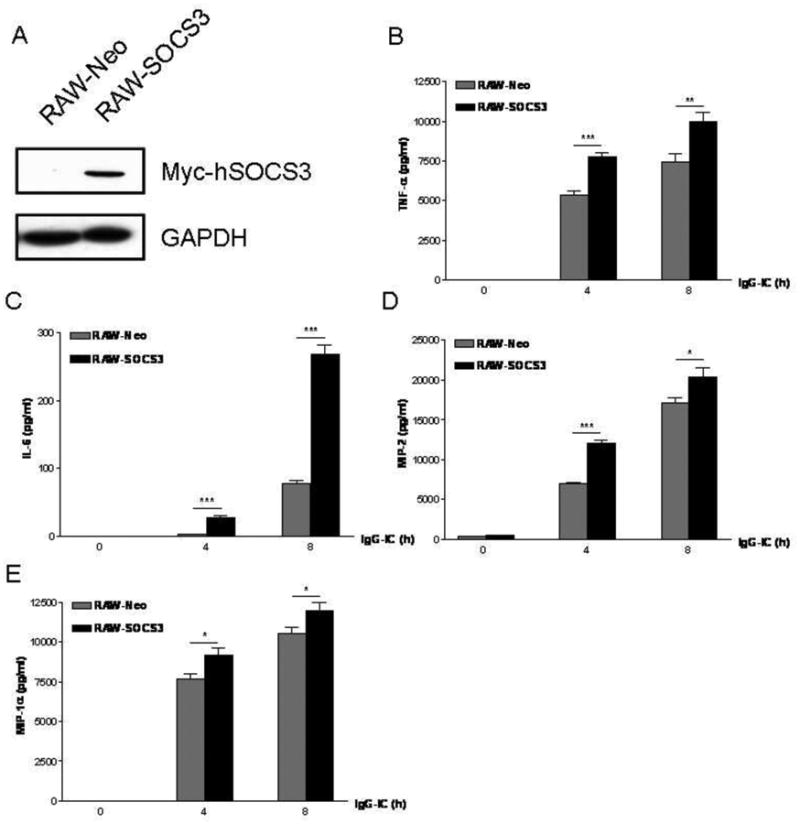
Over-expression of SOCS3 increased IgG IC-induced production of pro-inflammatory mediators in macrophages. A, two stable cell lines—RAW-Neo and RAW-SOCS3 were constructed. RAW-Neo cells were used as control. RAW-SOCS3 cells could constitutively express high level of SOCS3. Of note, the exogenous SOCS3 was linked to the tag—Myc at its C-terminal. Whole cell proteins were extracted from RAW-Neo and RAW-SOCS3, respectively, and Western blot was conducted by using rabbit anti-Myc antibody, and rabbit anti-GAPDH antibody, respectively. The level of GAPDH was shown at the bottom as a loading control. RAW-Neo and RAW-SOCS3 cells were treated with 100 μg/ml IgG IC for different time periods. Then supernatants were obtained and subjected to ELISA to detect expressions of IL-6 (B), TNF-α (C), MIP-2 (D), and MIP-1α (E). Data were expressed as means ± S. E. M. (n=8, biological replicates). *, and *** indicated statistically significant difference— p < 0.05, and p < 0.001, respectively.
To further verify if the regulatory role of SOCS3 in IgG IC-mediated inflammatory responses observed in macrophage-derived cell line—RAW264.7 cells was also applicable to primary cells, we generated LysM-Cre SOCS3fl/fl mice, which lack SOCS3 expression in macrophages and neutrophils as described previously [10]. We isolated SOCS3 sufficient and deficient peritoneal macrophages, and incubated them with IgG IC for different time points. We observed that SOCS3 deletion obviously reduced IgG IC-induced generation of IL-6, TNF-α, MIP-2, and MIP-1α in primary macrophages at all time periods examined (Fig. 3, A-D), which is consistent with the data obtained from RAW264.7 cells.
Figure 3.
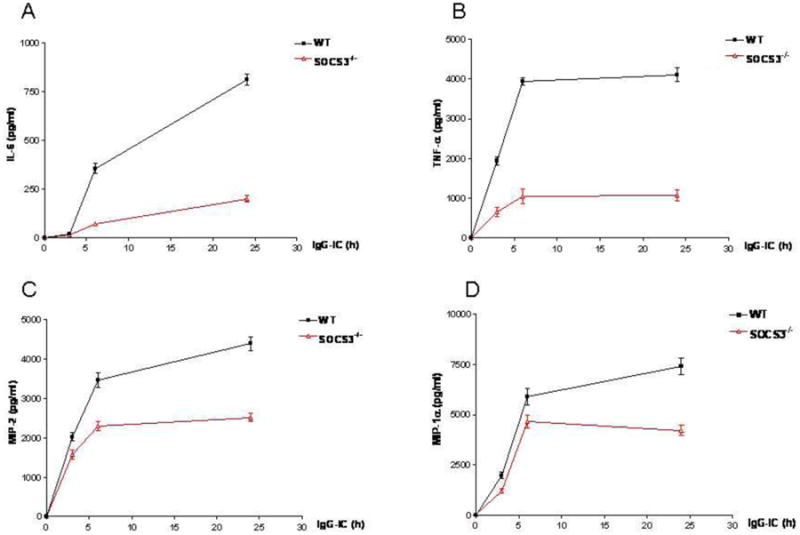
SOCS3 deletion led to reduction of IgG IC-induced inflammation in macrophages. Wild type and SOCS3 defective peritoneal macrophages were treated with 100 μg/ml IgG IC for different time periods. Then supernatants were obtained and subjected to ELISA to detect expressions of IL-6 (A), TNF-α (B), MIP-2 (C), and MIP-1α (D). Data were expressed as means ± S. E. M. (n=7, biological replicates).
SOCS3-enhanced IgG IC-mediated inflammatory responses was independent of NF-κB in macrophages
NF-κB, as a transcription factor, plays pivotal roles in expressions of many different types of pro-inflammatory mediators, and our previous studies have demonstrated that NF-κB is involved in IgG IC-stimulated inflammation in peritoneal macrophages [25]. Thus, to elucidate the underling mechanisms how IgG IC-induced inflammatory responses were regulated by SOCS3, we attempted to examine the influence of SOCS3 on IgG IC-induced NF-κB activation, which contributes to increased expressions of the pro-inflammatory mediators. Reporter assays demonstrated that IgG IC stimulation led to an over 6-fold increase of NF-κB transcriptional activity (Fig. 4A). Surprisingly, SOCS3 slightly decreased IgG IC-mediated NF-κB activation, though there was no significant difference (Fig. 4A). In addition, we performed gel shift assay, and found that SOCS3 over-expression had no influence on IgG IC-stimulated NF-κB DNA binding activity in macrophages (Fig 4B). Taken together, these data demonstrated that other transcription factor(s) but not NF-κB was involved in SOCS3-mediated increase of immune responses in IgG IC-challenged macrophages.
Figure 4.
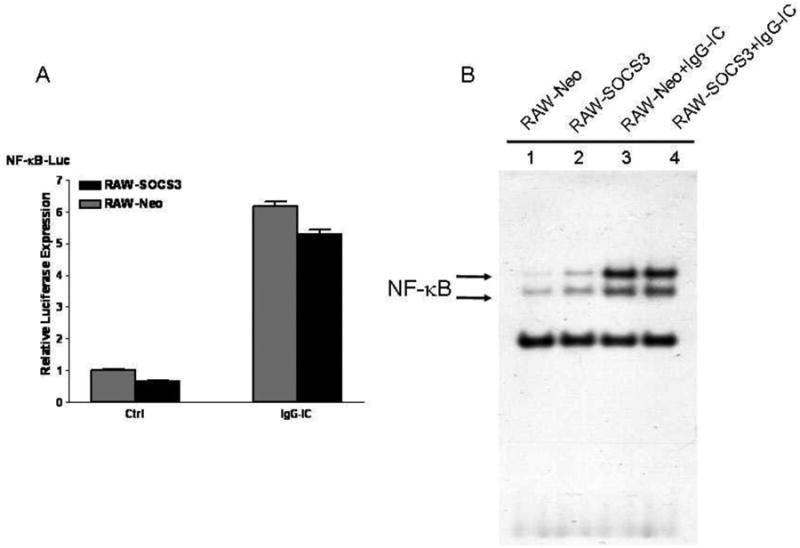
NF-κB signaling pathway was not required for SOCS3-enhanced generation of proinflammatory mediators in IgG IC-treated macrophages. A, indicated plasmids were transduced into RAW-Neo cells, and RAW-SOCS3 cells, respectively. 48 h later, the cells were treated with or without 100 μg/ml IgG IC for 4 h. Then cells were lysed and subjected to luciferase assays. Data were expressed as means ± S. E. M. (n=3, biological replicates). B, RAW-Neo and RAW-SOCS3 defective peritoneal macrophages were treated with or without 100 μg/ml IgG IC. 4 h later, nuclear proteins were extracted and subjected to gel shift assay.
Involvement of C/EBPδ in SOCS3 enhancement of pro-inflammatory mediator secretion in IgG IC-treated macrophages
It has been demonstrated the involvement of C/EBP family members in IgG IC-stimulated inflammatory responses in macrophages [25]. Thus, we sought to study the effect of SOCS3 on C/EBP transcriptional activity in IgG IC-treated macrophages. As shown in Fig. 5A, IgG IC stimulation triggered a 7.5-fold increase of C/EBP-Luc expression, and ectopic SOCS3 expression caused an increase of luciferase activity to 10-fold, suggesting that SOCS3 elevated IgG IC-triggered inflammatory responses via increasing C/EBP transcriptional activity. Our previous studies have shown that both C/EBPβ and C/EBPδ are indispensable for IgG IC-induced maximal immune reactivities in macrophages [25]. To further determine which C/EBP family member (s) might be regulated by SOCS3 in macrophages, we performed luciferase assays, and found that C/EBPδ-, but not C/EBPβ-driven gene transcription was increased by SOCS3 (Fig. 5B), indicating that SOCS3 enhances IgG IC-induced production of proinflammatory mediators through elevation of C/EBPδ activation. Because IgG IC stimulates C/EBPδ expression at both mRNA and protein levels [25], we next determined whether its production could be regulated by SOCS3. As shown in Fig. 5C, IgG IC-triggered C/EBPδ-Luc expression was significantly increased by SOCS3, indicating that C/EBPδ expression was positively regulated by SOCS3 at transcriptional level. We further examined whether this induction led to augmented accumulation of C/EBPδ at protein level. Western blot data suggested that SOCS3 over-expression led to an increase in the abundance of C/EBPδ protein stimulated by IgG IC (Fig. 5D). Together, our data indicated that the increased C/EBPδ but not NF-κB activation as a potential mechanism whereby SOCS3 augmented IgG IC-induced generation of pro-inflammatory mediators in macrophages.
Figure 5.
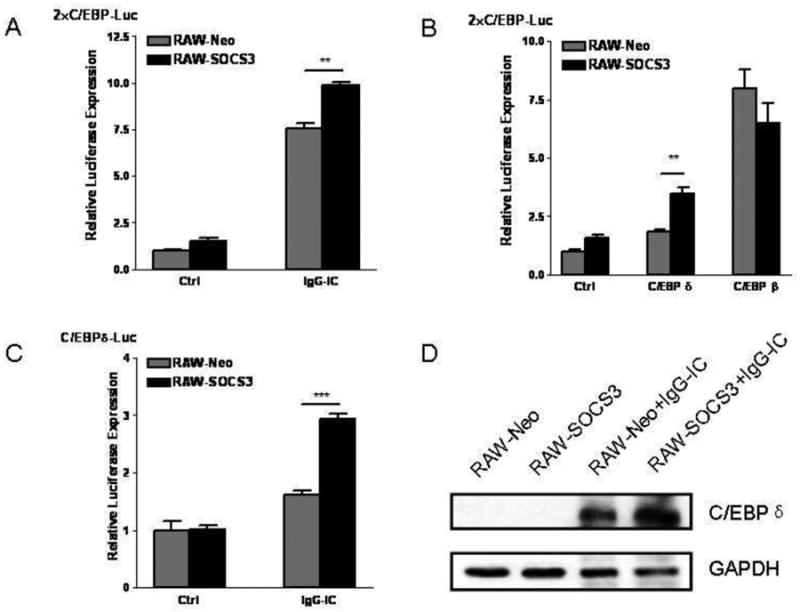
C/EPBδ was involved in SOCS3-enhanced inflammatory reactivities in IgG IC-treated macrophages. A, B, and C, indicated plasmids were transduced into RAW-Neo cells, and RAW-SOCS3 cells, respectively. 48 h later, the cells were treated with or without 100 μg/ml IgG IC for 4 h. Then cells were lysed and subjected to luciferase assays. Data were expressed as means ± S. E. M. (n=3, biological replicates). **, and *** indicated statistically significant difference— p < 0.01, and p < 0.001, respectively. D, RAW-Neo and RAW-SOCS3 cells were treated with or without 100 μg/ml IgG IC for 4 h. Then total proteins were extracted, and Western blot were performed by using rabbit anti-C/EBPδ antibody, and anti-GAPDH antibody, respectively. The level of GAPDH was shown at the bottom as a loading control.
Myeloid-specific disruption of SOCS3 attenuated IgG IC-induced production of pro-inflammatory mediators in lungs
Since macrophage plays a central role in IgG IC-induced acute lung inflammation, we generated LysM-Cre SOCS3fl/fl mice, which are deficient of SOCS3 expression in macrophages and neutrophils. Both wild type and LysM-Cre SOCS3fl/fl mice were treated intra-tracheally with or without IgG IC. Four hours later, BAL fluids were harvested, and subjected to ELISA. As shown in Fig. 6, there were no indicated pro-inflammatory mediators detected in BAL fluids from control-treated mice. However, IgG IC treatment dramatically induced MIP-1α, MIP-1β and MCP-1 expressions (Fig. 6, A-C). Of interest, in IgG IC-stimulated lungs, myeloid-specific deletion of SOCS3 resulted in obviously decreased generation of MIP-1α, MIP-1β and MCP-1 (by 81%, 55% and 49%, respectively) (Fig. 6, A-C), which is coincident with the in vitro phenomenon (Fig. 2, A-D).
Figure 6.
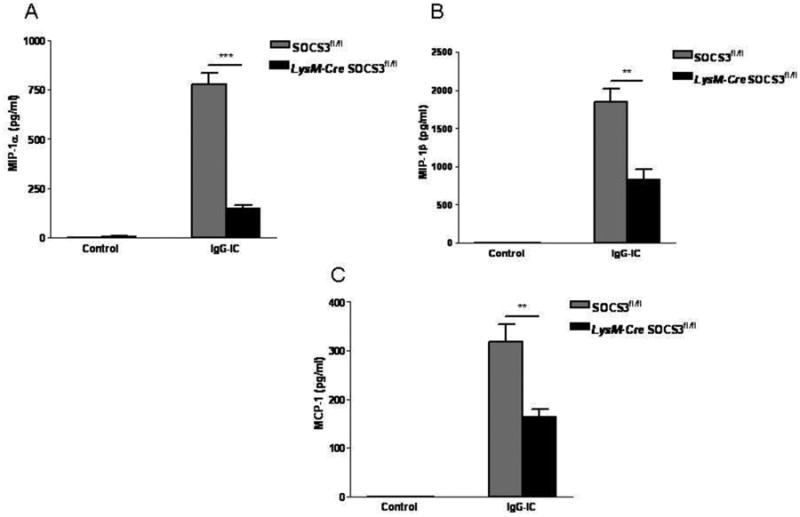
Myeloid-specific disruption of SOCS3 resulted in decreased pro-inflammatory mediator expressions in IgG IC-treated lungs. SOCS3fl/fl and LysM-Cre SOCS3fl/fl mice were treated intratracheally with IgG IC for 4 h, and cell-free supernatants were used to conduct ELISA to detect MIP-1α (A), and MIP-1β (B), and MCP-1 (C) expressions. Values represented means ± S. E. M. for ≥ 3 mice for each group. *, and ** suggested statistically significant difference— p < 0.05, and p < 0.01, respectively.
Discussion
Many inflammatory autoimmune diseases, such as immune thrombocytopenia, systemic lupus erythematosus, extrinsic allergic alveolitis, and rheumatoid arthritis, are caused by enhanced activation of immune cells in response to immune complex deposition [26-29]. Of note, macrophages are essential to both innate and adaptive immune systems, and play a central role in IC-associated diseases. For example, macrophage depletion significantly reduced IgG IC-stimulated alveolitis [11-14]. Thus, elucidation of the potential mechanisms underlying IC-induced inflammation in macrophages might contribute to design of a new generation of drugs required for treatment of IC-related illnesses.
Our previous studies verified that SOCS3 knockout significantly increases LPS-induced inflammatory responses [19], however, our current paper identified SOCS3 as a positive modulator of FcγR-induced inflammation in macrophages and lungs. The anti- and pro-inflammatory roles for SOCS3 in immune responses observed by us identified a novel paradigm for SOCS3 in inflammation. Intriguingly, not only SOCS3 but also a variety of other immune-associated proteins have dual functions in inflammatory reactivities. For instance, the inositol 3-phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is an enhancer of TLR4 signaling pathway, but a negative modulator of inflammation in macrophages after FcγR clustering [30]. Another example is Signal Transducer and Activator of Transcription 3 (STAT3). The anti-inflammatory function of IL-10 is obviously diminished in STAT3 deficient macrophages after TLR4 ligand treatment [31], indicating its negative role in inflammation, however, our previous studies found that down-regulation of STAT3 expression in macrophages attenuated inflammatory responses after FcγR cross-linking [32]. Using myeloid specific depletion of SOCS3 mice, we observed that SOCS3 deletion mice displayed decreased inflammation in IgG IC-treated lungs (Fig. 6), while increased inflammatory responses in lungs treated with LPS [19]. Collectively, these data indicated that macrophage-derived SOCS3 is a negative modulator of TLR4 activation-triggered inflammation, but a positive regulator of FcγR clustering-induced inflammatory reactivities. Additionally, the influence of SOCS3 on immune responses is still elusive, and is likely to be controversial.
The mechanisms whereby SOCS3 regulates inflammation have been widely investigated. SOCS3 represses cytokine receptor/Janus kinase/STAT signaling by direct inhibition of the catalytic activity of JAK1, JAK2, or TYK2, or by promoting unbiquitiantion of both JAK and cytokine receptors, and the following proteasomal degradation [33]. Additionally, another study reported that SOCS3 exerts its specific inhibitory function by simultaneously attaching to JAK and cytokine receptors [34]. Moreover, our recent studies revealed that SOCS3 inhibits TLR4-mediated pro-inflammatory mediator secretion via reduction of C/EBPβ DNA binding activity, and the following transcriptional activity [19]. In contrast, the underlying mechanisms how FcγR-mediated inflammatory reactivities are regulated by SOCS3 in macrophages remain unknown. In our current report, we found that SOCS3 amplifies expressions of pro-inflammatory mediators by increasing C/EBPδ transcriptional activity in macrophages incubated with IgG IC. Though both C/EBPβ and NF-κB are activated in IgG IC-treated macrophages, SOCS3 has no influence on their activities.
In this study, we demonstrated a unique role for SOCS3 in regulation of pro-inflammatory mediators' production in IgG IC-treated macrophage in vitro and in vivo. Ectopic expression of SOCS3 in macrophages heightened IgG IC-triggered generation of pro-inflammatory mediators, while macrophages deficient of SOCS3 showed attenuated responsiveness to IgG IC stimulation. Furthermore, in vivo experiments using an IgG IC-induced acute lung inflammation model verified that myeloid-specific disruption of SOCS3 promoted alleviation of inflammatory reactivities, which was due to decreased expressions of pro-inflammatory mediators. In addition, IgG IC-induced C/EBPδ activation was elevated by SOCS3 in macrophages. Collectively, these data proved that SOCS3 promoted IgG IC-triggered inflammatory responses in macrophages via the enhancement of C/EBPδ transcriptional activity. Thus, our current report found a novel signaling pathway—FcγR-SOCS3-C/EBPδ-proinflammatory mediators in IgG IC-treated macrophages, which is indispensible for IgG IC-induced maximal production of various pro-inflammatory mediators. Also, our current findings provide a new theoretical basis for design of drugs for treatment of IC-associated diseases.
Highlights.
IgG IC-stimulated inflammatory responses were enhanced by SOCS3 in macrophages.
C/EBPδ was required for SOCS3-mediated increase of inflammatory reaction in vitro.
Myeloid-derived SOCS3 was critical for full inflammatory mediator production in lung.
Acknowledgments
This project was funded by grants from the National Natural Science Foundation of Jiangsu Province Nos. BK20140622 (Chunguang Yan), the National Natural Science Foundation of China Nos. 31400751 (Chunguang Yan), and NIH Nos. 3R01HL092905-02S1 (Hongwei Gao).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bode JG, et al. LPS and TNFalpha induce SOCS3 mRNA and inhibit IL-6-induced activation of STAT3 in macrophages. FEBS Lett. 1999;463(3):365–70. doi: 10.1016/s0014-5793(99)01662-2. [DOI] [PubMed] [Google Scholar]
- 2.Cassatella MA, et al. Interleukin-10 (IL-10) selectively enhances CIS3/SOCS3 mRNA expression in human neutrophils: evidence for an IL-10-induced pathway that is independent of STAT protein activation. Blood. 1999;94(8):2880–9. [PubMed] [Google Scholar]
- 3.Stoiber D, et al. Lipopolysaccharide induces in macrophages the synthesis of the suppressor of cytokine signaling 3 and suppresses signal transduction in response to the activating factor IFN-gamma. J Immunol. 1999;163(5):2640–7. [PubMed] [Google Scholar]
- 4.Yan C, et al. Suppressor of cytokine signaling 3 inhibits LPS-induced IL-6 expression in osteoblasts by suppressing CCAAT/enhancer-binding protein {beta} activity. J Biol Chem. 285(48):37227–39. doi: 10.1074/jbc.M110.132084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin H, et al. IL-10 inhibits lipopolysaccharide-induced CD40 gene expression through induction of suppressor of cytokine signaling-3. J Immunol. 2006;177(11):7761–71. doi: 10.4049/jimmunol.177.11.7761. [DOI] [PubMed] [Google Scholar]
- 6.Gao H, et al. Adenoviral-mediated overexpression of SOCS3 enhances IgG immune complex-induced acute lung injury. J Immunol. 2006;177(1):612–20. doi: 10.4049/jimmunol.177.1.612. [DOI] [PubMed] [Google Scholar]
- 7.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7(6):454–65. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki A, et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med. 2001;193(4):471–81. doi: 10.1084/jem.193.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shouda T, et al. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest. 2001;108(12):1781–8. doi: 10.1172/JCI13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasukawa H, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4(6):551–6. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- 11.Lentsch AB, et al. NF-kappaB activation during IgG immune complex-induced lung injury: requirements for TNF-alpha and IL-1beta but not complement. Am J Pathol. 1998;152(5):1327–36. [PMC free article] [PubMed] [Google Scholar]
- 12.Gao H, Neff T, Ward PA. Regulation of lung inflammation in the model of IgG immune-complex injury. Annu Rev Pathol. 2006;1:215–42. doi: 10.1146/annurev.pathol.1.110304.100155. [DOI] [PubMed] [Google Scholar]
- 13.Lentsch AB, et al. Essential role of alveolar macrophages in intrapulmonary activation of NF-kappaB. Am J Respir Cell Mol Biol. 1999;20(4):692–8. doi: 10.1165/ajrcmb.20.4.3414. [DOI] [PubMed] [Google Scholar]
- 14.Yan C, et al. Critical role for CCAAT/enhancer-binding protein beta in immune complex-induced acute lung injury. J Immunol. 2012;189(3):1480–90. doi: 10.4049/jimmunol.1200877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koay MA, et al. Macrophages are necessary for maximal nuclear factor-kappa B activation in response to endotoxin. Am J Respir Cell Mol Biol. 2002;26(5):572–8. doi: 10.1165/ajrcmb.26.5.4748. [DOI] [PubMed] [Google Scholar]
- 16.Grutkoski PS, et al. Sepsis-induced SOCS-3 expression is immunologically restricted to phagocytes. J Leukoc Biol. 2003;74(5):916–22. doi: 10.1189/jlb.0303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin H, et al. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol. 2012;189(7):3439–48. doi: 10.4049/jimmunol.1201168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin H, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A. 2012;109(13):5004–9. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan C, et al. Myeloid depletion of SOCS3 enhances LPS-induced acute lung injury through CCAAT/enhancer binding protein delta pathway. FASEB J. 2013;27(8):2967–76. doi: 10.1096/fj.12-225797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hilberath JN, et al. Resolution of Toll-like receptor 4-mediated acute lung injury is linked to eicosanoids and suppressor of cytokine signaling 3. FASEB J. 2011;25(6):1827–35. doi: 10.1096/fj.10-169896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang S, et al. RasGRP3 limits Toll-like receptor-triggered inflammatory response in macrophages by activating Rap1 small GTPase. Nat Commun. 2014;5:4657. doi: 10.1038/ncomms5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao H, et al. C/EBP gamma has a stimulatory role on the IL-6 and IL-8 promoters. J Biol Chem. 2002;277(41):38827–37. doi: 10.1074/jbc.M206224200. [DOI] [PubMed] [Google Scholar]
- 23.Gao H, Schwartz RC. C/EBPzeta (CHOP/Gadd153) is a negative regulator of LPS-induced IL-6 expression in B cells. Mol Immunol. 2009;47(2-3):390–7. doi: 10.1016/j.molimm.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Qin H, et al. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179(9):5966–76. doi: 10.4049/jimmunol.179.9.5966. [DOI] [PubMed] [Google Scholar]
- 25.Yan C, et al. C5a-regulated CCAAT/enhancer-binding proteins beta and delta are essential in Fcgamma receptor-mediated inflammatory cytokine and chemokine production in macrophages. J Biol Chem. 2012;287(5):3217–30. doi: 10.1074/jbc.M111.280834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robles-Carrillo L, et al. Anti-CD40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. J Immunol. 2010;185(3):1577–83. doi: 10.4049/jimmunol.0903888. [DOI] [PubMed] [Google Scholar]
- 27.Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10(5):328–43. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuyler M, Gott K, French V. The role of MIP-1alpha in experimental hypersensitivity pneumonitis. Lung. 2004;182(3):135–49. doi: 10.1007/s00408-004-0311-7. [DOI] [PubMed] [Google Scholar]
- 29.Schaller M, Burton DR, Ditzel HJ. Autoantibodies to GPI in rheumatoid arthritis: linkage between an animal model and human disease. Nat Immunol. 2001;2(8):746–53. doi: 10.1038/90696. [DOI] [PubMed] [Google Scholar]
- 30.Cao X, et al. The inositol 3-phosphatase PTEN negatively regulates Fc gamma receptor signaling, but supports Toll-like receptor 4 signaling in murine peritoneal macrophages. J Immunol. 2004;172(8):4851–7. doi: 10.4049/jimmunol.172.8.4851. [DOI] [PubMed] [Google Scholar]
- 31.Bosmann M, et al. Interruption of macrophage-derived IL-27(p28) production by IL-10 during sepsis requires STAT3 but not SOCS3. J Immunol. 2014;193(11):5668–77. doi: 10.4049/jimmunol.1302280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang H, et al. An essential role for Stat3 in regulating IgG immune complex-induced pulmonary inflammation. FASEB J. 2011;25(12):4292–300. doi: 10.1096/fj.11-187955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Babon JJ, Nicola NA. The biology and mechanism of action of suppressor of cytokine signaling 3. Growth Factors. 2012;30(4):207–19. doi: 10.3109/08977194.2012.687375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babon JJ, et al. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. 2012;36(2):239–50. doi: 10.1016/j.immuni.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]