

Abstract
Docosahexaenoic acid (DHA) is generally reported to have anti-inflammatory properties, however, prior work has documented differential effects on individual pro-inflammatory cytokines: reduced IL-6, but not TNFα, mRNA expression in macrophages. To elucidate the mechanism, the roles of prostaglandin E2 (PGE2), cyclic AMP response element-binding protein (CREB), and NFκB were examined in RAW 264.7 macrophages. DHA did not influence CREB activity, but significantly reduced PGE2 production by 41% and NFκB activity by 32%. Exogenous PGE2 inhibited TNFα mRNA expression dose dependently. Unexpectedly, inhibiting PGE2 production with NS-398 also decreased TNFα mRNA expression, suggesting a concentration-dependent dual role of PGE2 in regulating TNFα expression. IL-6 expression was unaffected by endogenous or exogenous PGE2. Partial block of NFκB activation (SN50; 46%, or, BAY-11-7082; 41%) lowered IL-6 to a greater extent than TNFα mRNA expression. The differential effect of DHA on TNFα and IL-6 mRNA expression may be mediated via reduction in NFκB activity.
Keywords: TNFα, IL-6, TLR4, Macrophages, PGE2, CREB
1. Introduction
Docosahexaenoic acid (DHA) is a very long-chain omega-3 fatty acid found in high concentrations in marine animals and algae. In contrast to saturated fatty acids, DHA down-regulates toll-like receptor 4 (TLR4)-mediated production of pro-inflammatory cytokines. These effects are suggested to be primarily mediated by inhibition of nuclear factor κB (NFκB) activation as evidenced by decreased IκB phosphorylation and reduced nuclear levels of NFκB p65-p50 dimers [1]. However, DHA has been shown to reduce individual pro-inflammatory cytokines by varying degrees, and the anti-inflammatory mechanisms that underlie specific effects on individual pro-inflammatory cytokines remain unknown. We previously reported that DHA supplementation in cultured RAW 264.7 cells decreased interleukin 6 (IL-6) secretion to a greater extent than tumor necrosis factor α (TNFα) secretion [2]. TNFα and IL-6 influence the development of atherosclerotic plaque by promoting immune cell recruitment, macrophage foam cell formation, and destabilization of mature plaque [3–8]. Despite the importance of TNFα and IL-6 in atherosclerosis lesion progression, the differential effect of DHA on production of these cytokines in macrophages, as well as the regulatory mechanisms, has not been established. Although NFκB is a central regulator of TNFα and IL-6 production, other regulatory molecules that are up-regulated in TLR4-activated macrophages, including prostaglandin (PG) E2 and the transcription factor cAMP response element-binding protein (CREB), may have a gene-specific regulatory effect on the production of these cytokines.
PGE2 is perhaps the most prominent pro-inflammatory lipid mediator. PGE2 promotes inflammation and causes redness, swelling and pain in affected tissues [9]. Its synthesis has long been a pharmaceutical target for controlling inflammation. Among the diverse functions of PGE2 is the regulation of cytokine production in macrophages, which occurs in an autocrine-/paracrine-like manner [10,11]. Activation of TLR4 by lipopolysaccharide (LPS) increases PGE2 production in macrophages by inducing a series of steps including the release of arachidonic acid (AA) from membrane phospholipids, increasing the activity of cyclooxygenase 2 (COX2), the rate limiting enzyme in the conversion of AA into the intermediate product PGH2, and subsequent conversion to PGE2 by action of PGE synthase [12]. Through engagement of E prostanoid receptor 2 and/or 4 (EP2/EP4) expressed on the surface of macrophages, PGE2 decreases TNFα production and increases IL-6 production [13–17]. These effects are mediated through activation of the cAMP/protein kinase A (PKA) system [18,19].
Interestingly, studies in THP-1 and RAW 264.7 cells have suggested that triggering cAMP/PKA may be independently associated with inhibition of NFκB-mediated transcription of specific genes, including TNFα [20–22]. Transcription factor CREB, which can be phosphorylated and activated by PKA, may mediate the suppression and enhancement of TNFα and IL-6 mRNA expression, respectively, through cAMP/PKA activation [23]. Activated CREB inhibits transcription of select NFκB genes by binding to the cAMP-responsive element (CRE) in the promoter region and limiting the interaction between NFκB and the transcriptional co-activator, CREB-binding protein (CBP) [24,25]. However, CREB has been shown to enhance the transcription of some NFκB target genes including IL-6, which may occur through cooperative recruitment of CBP with NFκB, facilitated by the proximity of their binding sites [26]. CREB is phosphorylated by PKA. Hence, the effect of PGE2 on TNFα and IL-6 gene transcription may be mediated through the cAMP/PKA/CREB pathway [27–29].
The ability of DHA to reduce PGE2 production has been reported in a variety of cell types including LPS-stimulated RAW 264.7 cells [30–32]. Using this model, the aim of the present study was to determine the effect of DHA on PGE2 production and CREB and NFκB activities, and the role of PGE2 and NFκB in DHA-induced change in TNFα and IL-6 gene expression. We hypothesized that reduced PGE2 production by DHA may decrease the repressive effects of PGE2 on TNFα gene expression and thus diminish the inhibitory effect of DHA on TNFα but not IL-6 production. However, our results suggest that PGE2 is not a significant regulator of TNFα and IL-6 gene expression in this cell system. Instead, the effect of DHA could be mediated by a reduction in NFκB activation, which was found to have a greater influence on IL-6 gene expression compared to TNFα gene expression.
2. Materials and methods
2.1. Cell culture
RAW 264.7 cells, a murine macrophage-like cell line (ATCC, Manassas, VA), were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO), 100 U/mL penicillin and 100 μg/mL streptomycin (MP Biomedicals, LLC, Santa Anna, CA) at 37 °C in a 5% CO2 humidified incubator.
2.2. Fatty acid pretreatment and LPS stimulation
DHA sodium salts (Sigma-Aldrich, >95% purity) and MA sodium salts (Nu-Check Prep, Inc., Elysian, MN, >99% purity) were combined with fatty acid-free, low endotoxin bovine serum albumin (BSA; Sigma-Aldrich) at a 2:1 M ratio. Cells were pretreated with 100 μM DHA for 24 h. BSA without fatty acid was used as a control. Following the 24-h pretreatment, cells were stimulated with 100 ng/mL of ultra-pure LPS (Invivogen, San Diego, CA) from E. coli 0111:B4 strain for 3, 6, or 24 h in the presence of DHA, MA or BSA. Cell viability was determined by trypan blue exclusion. Cells were harvested and cellular protein concentration was measured by the bicinchoninic acid (BCA) method (Pierce Inc., Rockford, IL).
2.3. TNFα and IL-6 gene transcription
RNA was isolated from RAW 264.7 cells using an RNeasy mini kit (Qiagen, Valencia, CA). cDNA was synthesized from RNA using a Reverse Transcription System (Promega, Madison, WI) according to the manufacturer’s instructions. Real Time PCR was performed using SYBR green and Quantitect primer assays (Qiagen, Valencia, CA) for mouse TNFα (QT00104006), IL-6 (QT00098875), beta (β) actin (QT01136772) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (QT01658692) on a real-time PCR 7300 (Applied Biosystems, Foster City, CA). Relative quantification (ΔΔCt) was used to assess expression of target genes, using β-actin or GAPDH as an endogenous control.
2.4. Enzyme-linked immunosorbent assays (ELISA)
Commercially available ELISA kits were used to determine CREB phosphorylated at S133 in cell lysates (R&D Systems, Minneapolis, MN), and PGE2 concentration in the culture supernatants (Cayman Chemical Company, Ann Arbor, MI).
2.5. Exogenous PGE2 treatment
PGE2 (Cayman Chemicals, Ann Arbor, MI) dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO) was added to the culture media to achieve a final concentration of 2, 10, 50, 100 or 1000 nM. RAW 264.7 cells were pre-incubated in this PGE2 supplemented culture media for 45 min. Cells were then stimulated with ultra-pure LPS (100 ng/mL) for an additional 3 h. TNFα and IL-6 gene transcription were determined as described above.
2.6. Inhibition of NFκB and COX2
To inhibit nuclear translocation of the NFκB subunit p50, RAW 264.7 cells were pre-treated with a p50 inhibitor peptide, SN50, (Imgenex, San Diego, CA) dissolved in phosphate buffered saline (PBS) for 15 min. The concentration of SN50 in the culture media was 0, 40, 80, or 120 μM. Thereafter, cells were stimulated with ultra-pure LPS (100 ng/mL) for an additional 3 h.
RAW 264.7 cells were pretreated for 16 h with 10 μM of BAY-11-7082 (BAY) or 10 μM NS-398 (Cayman Chemicals, Ann Arbor, MI) dissolved in DMSO to inhibit NFκB and COX2, respectively, and then stimulated with ultra-pure LPS (100 ng/mL) for 3 or 6 h. The final concentration of DMSO in the medium of control groups matched that of treatment groups and did not exceed 0.05%. TNFα and IL-6 gene transcription and PGE2 secretion were determined as described above.
2.7. Western blotting for nuclear NFκB p50 and p65 proteins
RAW 264.7 cells were pretreated with BSA (fatty acid vehicle) for 24 h, and then treated with SN50 dissolved in PBS for 15 min at 37 °C at 10 and 100 μM. Immediately thereafter cells were stimulated with ultra-pure LPS (100 ng/mL) for 30 min. Nuclear protein was extracted using NE-PER® nuclear extraction reagents (Thermo Scientific, Rockford, IL). The extract (10 μg protein) was used to separate individual proteins through a 4–20% Criterion® Tris–HCl SDS–PAGE gradient gel (Bio-Rad, Hercules, CA) and transferred to a nitrocellulous membrane (Bio-Rad, Hercules, CA). After blocking, the membrane was incubated with primary antibodies for NFκB p50 (cat# ab32360, Abcam, Cambridge, MA), p65 (cat# 8242, Cell Signaling, Danvers, MA), and Histone 3 (H3, cat#9715, Cell Signaling, Danvers) or TATA binding protein (TBP; cat# ab818, Abcam, Cambridge, MA), a nuclear loading control, followed by peroxidase-conjugated detection antibody (cat# sc-2005 and sc-2030, Santa Cruz Biotechnology, Inc., Dallas, TX). Signals were visualized by chemiluminescence (Amersham Biosciences, Piscataway, NJ) and quantified using a GS-800 calibrated densitometer (Bio-Rad, Hercules, CA).
2.8. NFκB–DNA binding assay
The nuclear extracts prepared as described above were used to determine NFκB p50 binding to target DNA using a TransAM NFκB ELISA kit (Active Motif, Carlsbad, CA) according to the manufacturer’s protocol.
2.9. Statistical analysis
The significance of the differences in the mean values among three or more treatment groups from three independent experiments, each done in triplicate unless otherwise noted, was determined by one-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons. Two-way ANOVA followed by Sidak’s test for multiple comparisons was used when in addition to treatment, another factor such as time or the presence of ultra-pure LPS was considered. The repeated measures method was included in the analysis when the “concentration” or “fold-change” values were used in the analysis instead of “percent of control” value to account for the variation in control values among repeated experiments. Student’s t-test was used when one treatment was compared to a control. The statistical software GraphPad Prism version 6.03 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.comGraphPadPrism6 (La Jolla, CA), was used for statistical calculations. Significance was set at P<0.05.
3. Results
3.1. Effect of DHA on TNFα and IL-6 gene expression
We previously reported a significant and robust increase in TNFα and IL-6 secretion after incubating cells with 100 μM ultra-pure LPS (1–24 h), which was decreased when cells were pretreated with 100 μM EPA or DHA but not MA for 24 h, significantly enhancing the proportion of the respective fatty acids in cell membranes [2]. An assessment of cell viability indicated no significant effect of exposure of the cells to LPS or fatty acid bound to albumin at the concentrations used. Interestingly, EPA and DHA caused a greater reduction in LPS-induced IL-6 secretion compared with TNFα secretion [33]. The effect of EPA and DHA was observed during both the early (6 h) and late (24 h) phases of protein induction. In the current work, we investigated the differential effect of DHA on TNFα and IL-6 secretion. The effects of EPA were difficult to attribute to cellular membrane incorporation since a significant proportion was metabolized to DPA, therefore it was not further examined. We measured the mRNA levels of TNFα and IL-6 in unstimulated macrophages and in macrophages stimulated for 3, 6 or 24 h with ultra-pure LPS. The 3 and 6 h time points were chosen to capture the initial effects of DHA on gene induction, and to account for the difference in TNF and IL-6 mRNA induction patterns noticeable between 3 h and 6 h of stimulation found in preliminary work. After an initial induction, TNFα mRNA levels declined from 3 h to 6 h, while IL-6 mRNA levels continued to increase from 3 h to 6 h (data not shown). The 24 h time point was included to account for the delayed effect of DHA on TNFα secretion. Stimulation with ultra-pure LPS significantly upregulated the expression of both TNFα and IL-6 mRNA (data not shown). Pretreatment with DHA reduced baseline (unstimulated) IL-6 mRNA levels by 77% (significant only when MA is not included in the analysis, P<0.0001) and reduced LPS-induced IL-6 mRNA levels by 44% (P<0.001) in cells stimulated for 3 or 6 h compared to control cells (Fig. 1B). The effect of DHA compared to control was not significant after 24 h of LPS stimulation. In contrast, DHA pre-treatment did not significantly alter TNFα mRNA levels in unstimulated or stimulated cells (Fig. 1A). Treatment with MA significantly increased the expression of TNFα mRNA and IL-6 in non-stimulated cells and significantly increased the expression of IL-6 mRNA in cells stimulated for 3 and 24 h.
Fig. 1.
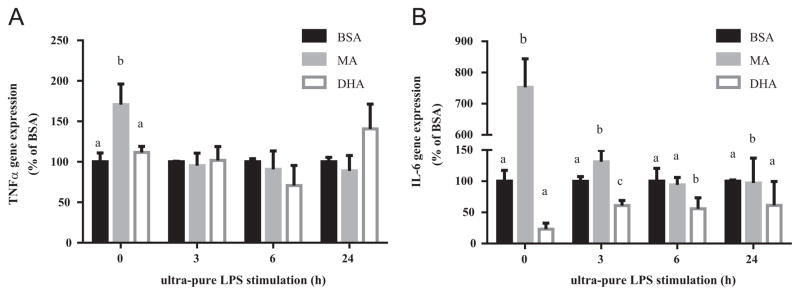
Effect of fatty acid on TNFα (A) and IL-6 (B) gene expression. RAW 264.7 cells were pretreated with DHA or MA (100 μM, 24 h) then stimulated with ultra-pure LPS (100 ng/mL) in the presence of treatment fatty acid for the times indicated. Bars without common letters within the same time group statistically differ at P<0.05 determined by one-way ANOVA, adjusted with Tukey’s post-hoc test for multiple comparisons. Values are mean±SD of three independent experiments.
3.2. Effect of DHA on PGE2 production and CREB activity
We next investigated whether DHA-induced changes in PGE2 production or CREB activity would account for the lack of effect of DHA on TNFα mRNA expression. In unstimulated RAW 264.7 cells PGE2 levels in the culture medium were below the detection limit. PGE2 concentrations reached approximately 3000 pg/mL after stimulation with ultra-pure LPS for 6 h and DHA pretreatment reduced PGE2 levels by 41% (P<0.05), while MA pretreatment had no effect (Fig. 2A). In response to ultra-pure LPS, P-CREB levels increased by approximately 3.5 fold after 30 min (Fig. 2B), which was consistent with previous reports [34,35]. Pretreatment of the cells with DHA compared with BSA or MA did not reduce basal or stimulated P-CREB levels (Fig. 2B). Based on these data we ruled out a possible role of CREB in mediating the effect of DHA on TNFα and IL-6 gene transcription.
Fig. 2.

Effect of fatty acid on PGE2 secretion and CREB activity in RAW 264.7 cells. (A) Cells were pretreated with MA or DHA (100 μM, 24 h) and then stimulated with ultra-pure LPS (100 ng/mL, 6 h). PGE2 concentration in culture supernatant was determined by ELISA. Values are mean±SD of three independent experiments. Bars without common letters statistically differ at P<0.05 determined by one-way ANOVA adjusted with Tukey’s post-hoc test for multiple comparisons. (B) Cells were pretreated with MA or DHA (100 μM, 24 h) and then stimulated with ultra-pure LPS (100 ng/mL, 30 min). P-CREB concentration in whole cell lysates was determined by ELISA. Values are mean±SD of four independent experiments. Bars without common letters within each group statistically differ at P<0.05 determined by two-way repeated measures ANOVA adjusted with Sidak’s post-hoc test for multiple comparisons.
3.3. Differential effect of PGE2 on TNFα and IL-6 gene transcription
Since DHA reduced PGE2 production in stimulated cells, we next determined whether PGE2 played a role in altering TNFα and IL-6 gene expression. Cells were pre-incubated with exogenous PGE2 over a wide concentration range: 0, 2, 10, 50, 100 and 1000 nM (10 nM=3525 pg/mL) and then stimulated with ultra-pure LPS. PGE2 suppressed TNFα mRNA expression at concentrations of 50 nM and higher (all P<0.05, Fig. 3A). The suppression was dose-dependent (P<0.01 for linear trend). PGE2 had no significant effect on IL-6 mRNA expression (Fig. 3B).
Fig. 3.

Effect of exogenous PGE2 on (A) TNFα and (B) IL-6 gene expression. RAW 264.7 cells were incubated with exogenous PGE2 at the concentrations indicated for 45 min, and then stimulated with ultra-pure LPS (100 ng/mL, 3 h). Bars without common letters statistically differ at P<0.05 determined by one-way ANOVA, adjusted with Tukey’s post-hoc test for multiple comparisons. Values are mean±SD of four independent experiments.
To confirm these findings we inhibited PGE2 production in RAW 264.7 cells using NS-398, a specific COX2 inhibitor. NS-398 reduced PGE2 secretion by 98% (Fig. 4A). TNFα and IL-6 gene expression was measured in the NS-398-treated cells 3 and 6 h post-stimulation, corresponding to the times when PGE2 concentration in culture supernatants was low (below detection) and high (>3000 pg/mL), respectively. Contrary to our hypothesis, TNFα mRNA expression decreased (21%, P<0.05) rather than increased in cells stimulated for 3 h (Fig. 4B). This effect was no longer present 6 h post-stimulation (Fig. 4B). NS-398 had no significant effect on IL-6 mRNA expression at either time point (Fig. 4C). Based on these data, endogenous PGE2 levels do not appear to inhibit TNFα gene expression in LPS-stimulated RAW 264.7 cells. Therefore, it is an unlikely mechanism for the differential effect of DHA on TNFα and IL-6 mRNA expression.
Fig. 4.

Effect of NS-398 on (A) PGE2 secretion, (B) TNFα, and (C) IL-6 gene expression. RAW 264.7 cells were pretreated with NS-398 (10 μM, 18 h) and then stimulated with ultra-pure LPS (100 ng/mL). (A) PGE2 in culture supernatant was determined by ELISA after 6 h of ultra-pure LPS stimulation. **P <0.01 vs. control determined by unpaired Student t test. (B) TNFα and (C) IL-6 gene expression were determined after 3 or 6 h of ultra-pure LPS stimulation. Values are mean±SD of three independent experiments. *P<0.05 determined by two-way repeated measures ANOVA adjusted by Sidak’s test for multiple comparisons. Values are mean±SD of three independent experiments.
3.4. Differential influence of NFκB on TNFα and IL-6 gene expression
Since changes in PGE2 and P-CREB levels did not influence the differential effect of DHA on TNFα and IL-6 mRNA levels, we next evaluated the influence of NFκB activity on TNFα and IL-6 gene expression. As expected, NFκB activation was induced after exposure to ultra-pure LPS as indicated by an increase in nuclear levels of p65 protein (Fig. 5A). DHA reduced NFκB-DNA binding activity by 32% compared to the control treated cells (P<0.05) (Fig. 5B).
Fig. 5.
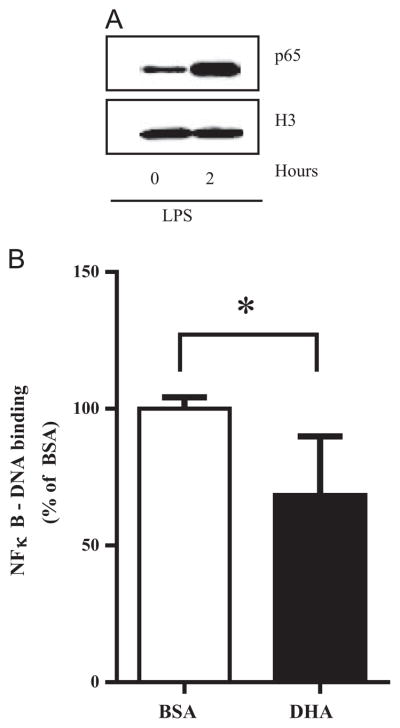
NFκB activity in RAW 264.7 cells. (A) Western blot of nuclear p65 protein expression before and after 2 h of ultra-pure LPS stimulation relative to histone 3 (H3) protein expression (nuclear protein loading control). One representative experiment is shown out of 3 independent experiments that had similar results. (B) Cells were pretreated with DHA (100 μM, 24 h), then stimulated with ultra-pure LPS (100 ng/mL, 30 min). NFκB-DNA binding in nuclear extracts was determined by ELISA. Values are mean±SD of five independent experiments. *P<0.05 determined by Student t test.
To assess the relationship between NFκB activity and TNFα or IL-6 gene expression, we blocked NFκB activation using two NFκB inhibitors. First, we pre-incubated cells with SN50, a p50-specific inhibitor that prevents the nuclear translocation of p50 subunit by acting as a p50 decoy. Pretreatment of ultra-pure LPS stimulated RAW 264.7 cells with 100 μM SN50 reduced nuclear p50 and p65 protein by 46% and 64%, respectively (Fig. 6A). However, while SN50 treatment decreased IL-6 mRNA expression in a dose-dependent manner, it had no significant effect on TNFα mRNA expression (Fig. 6B). These data suggest a greater dependence on NFκB activity by IL-6 than TNFα gene expression.
Fig. 6.

Effect of SN50 in RAW 264.7 cells. Cells were pretreated with SN50 (100 μM, 15 min) and then stimulated with ultra-pure LPS (100 ng/mL). (A) After 30 min of stimulation, nuclear protein expression of p50 and p65 were determined by western blot. TBP (TATA-binding protein) was used as a nuclear protein loading control. Values are mean of two independent samples of one experiment. (B) TNFα and (C) IL-6 mRNA expression after 3 h of stimulation was determined by RT-PCR. Values are mean±SD of three independent experiments. Bars without common letters statistically differ at P<0.05 determined by one-way ANOVA, adjusted with Tukey’s post-hoc test for multiple comparisons.
We further confirmed these effects using a second NFκB inhibitor, BAY-11-7082 (BAY). BAY inhibits the phosphorylation of IκB, resulting in decreased IκB degradation which in turn reduces the release of the NFκB p50-p65 heterodimer and its subsequent translocation into the nuclei [36]. Pretreatment of RAW 264.7 cells with 10 μM BAY reduced NFκB activity by 41% (Fig. 7A) in cells stimulated for 3 h with ultra-pure LPS. BAY was toxic to cells at 50 μM as assessed by the detachment of cells from the culture plate (data not shown). 10 μM BAY had no significant effect on PGE2 secretion (Fig. 7B). Pretreatment with BAY significantly reduced TNFα and IL-6 mRNA (62% vs. 32%, respectively) (Fig. 7C and D) measured after 3 h of stimulation. However, 6 h post-stimulation of RAW 264/7 cells with ultra-pure LPS, 10 μM BAY only reduced IL-6 (P<0.05), but not TNFα mRNA expression, similar to the effect of SN50.
Fig. 7.

Effect of BAY on inflammatory response of RAW 264.7 cells. Cells were pretreated with BAY (10 μM, 18 h) then stimulated with ultra-pure LPS (100 ng/mL). (A) NFκB-DNA binding determined by ELISA. Values are mean of triplicate samples of one experiment. (B) PGE2 in culture supernatant was determined after 6 h of ultra-pure LPS stimulation. (C) TNFα and (D) IL-6 gene expression were determined after 3 or 6 h of stimulation. Values are mean±SD of three independent experiments. *P≤0.05 determined by two-way repeated measures ANOVA adjusted with Sidak’s post-hoc test for multiple comparisons.
4. Discussion and conclusions
Consistent with our prior work documenting a greater reduction in IL-6 than TNFα secretion by DHA-treated RAW 264.7 cells stimulated with ultra-pure LPS [33], in the present experiment, we observed a significant reduction in mRNA expression of IL-6, but not TNFα, in both unstimulated and stimulated cells. These results are consistent with two prior studies using human THP-1 macrophages. The first reported a significant reduction in IL-6 but not TNFα mRNA expression after treatment with 100 μM DHA for 2 h followed by stimulation with LPS for 24 h [37]. The second documented that pre-treatment with DHA for a longer period, 48 h followed by 6 h of LPS stimulation, also reduced IL-6 but not TNFα mRNA expression [38]. Some studies have reported a down-regulated secretion or mRNA expression of both TNFα and IL-6 in THP-1 cells [39,40] or RAW 264.7 cells [41] using a wide range of treatment and stimulation conditions. The inconsistency between our findings and those reported previously may, at least in part, be related to the differences in the purity of LPS. Standard LPS (in contrast to ultra-pure) may contain lipoproteins capable of stimulating TLR2 signaling pathways at the high concentrations used in the aforementioned studies [42]. DHA has been shown to inhibit TLR2 activity and TNFα production induced by a TLR2 agonist [43]. The duration and dose of DHA and LPS treatments may have also affected the relative potency in inhibiting TNFα vs. IL-6 production.
Since the goal of the current study was to determine the effect of enhancing the proportion of DHA in cell membranes, we chose a relatively high concentration of DHA and LPS so as to maximize DHA incorporation into cell membranes and cytokine production while maintaining cell viability. However, since we did not examine the dose response of DHA and LPS treatments, we cannot eliminate the possibility that the observed effects of DHA are specific to the cell culture conditions used. Still, the large anti-inflammatory effect of DHA in unstimulated cells suggests that DHA is equally effective in reducing low-level, chronic inflammation. Nevertheless, there is limited data with which to assess the biological implications of our findings. A review of twenty-four studies published between 1991 and 2006 that examined the effect of EPA and DHA supplementation in healthy humans on the secretion of cytokines from LPS-stimulated isolated peripheral blood monocytes (PBMCs) concluded a minority of studies reported a reduction in TNFα and/or IL-6 [44]. Not addressed was whether the influence of EPA and DHA on TNFα and IL-6 differed. Regarding the majority of studies that reported negative findings, the review found no clear reason for the inconsistency in the data. More recent studies investigating the effect of EPA and DHA supplementation on the circulating levels of TNFα and IL-6 in plasma have likewise reported inconsistent findings, independent of subjects’ health statuses [45–49].
In terms of underlying mechanism(s) for our observations, we initially evaluated the influence of DHA on PGE2 production and CREB activity as they have each been shown to influence the transcription of NFκB target genes. Consistent with previous reports [30–32], DHA reduced PGE2 production in ultra-pure stimulated RAW 264.7 cells. However, we found that both pre-incubating cells with exogenous PGE2 or blocking production with a COX2 inhibitor, reduced TNFα mRNA expression. Thus, the possibility cannot be ruled out that the nature of PGE2’s effect on TNFα is concentration dependent. It has been previously demonstrated in primary mice macrophages that low PGE2 concentrations (0.1–10 ng/mL) stimulated, whereas high concentrations (>10 ng/mL) suppressed, TNFα release [50]. In the current study, the lowest concentration of exogenous PGE2 that significantly suppressed TNFα mRNA expression was 50 nM (17.6 ng/mL), which is several-fold greater than the average endogenous PGE2 concentration in the media (3.1 ng/mL) after 6 h of stimulation. Taken together, between 0 and 6 h after stimulation with ultra-pure LPS PGE2 production levels may have been insufficient to down-regulate TNFα in our cell system. A COX2 inhibitor, NS-398, was used to block de novo synthesis of PGE2. However, we cannot rule out the possibility that the effects observed may have been secondary to an effect of COX2 inhibition on the production of other prostaglandins and of lipoxygenase products such as leukotrienes [51].
In contrast to the large body of evidence supporting the role of CREB in regulating the transcription of NFκB-target genes including TNFα and IL-6, little is known about the effect of DHA on CREB activity [52]. In the only study identified to date, peritoneal macrophages isolated from DHA-fed mice had attenuated CREB activity and IL-6 expression in response to ex vivo treatment with deoxynivalenol (a fungus-derived mycotoxin found in wheat, barley, corn, rice and oats [53]); however, in vitro treatment of peritoneal macrophages with DHA did not affect deoxynivalenol-induced CREB activity [54]. Consistent with the latter findings, we observed no significant effect on ultra-pure LPS-induced P-CREB in RAW 264.7 cells pretreated with DHA compared to control treated cells. Of note, it has been reported that LPS-induced P-CREB in the absence of a cAMP inducer is transcriptionally inactive and is not necessary for LPS-induced TNFα production in RAW 264.7 cells [34]. Considering the available data we did not further investigate the role of CREB on TNFα and IL-6 expression.
The influence of NFκB was next assessed. DHA reduced NFκB activity by 32% in our cell system. This reduction was within the range previously reported [19,38,39,55]. Interestingly, we found that a greater inhibition of NFκB activity induced by SN50 or BAY also resulted in a significant reduction in IL-6 and a smaller or no reduction in TNFα, a pattern similar to the effect of DHA. BAY but not SN50 significantly reduced TNFα in cells stimulated for 3 h but not 6 h. The reason for this discrepancy may be related to different mechanisms of NFκB pathway inhibition and/or the level of the signaling pathway targeted by each inhibitor (BAY targets IκB phosphorylation while SN50 targets P50 nuclear transport). In addition to inhibiting NFκB activation, BAY has been reported to inhibit the activation of multiple kinases that activate nuclear transcription factors such as AP-1, which up-regulates TNFα and IL-6 transcription [56–59]. Similarly, SN50 has been reported to inhibit nuclear translocation of AP-1 and other transcription factors.
Our results support the hypothesis that IL-6 gene expression is more susceptible to reduced NFκB activity than TNFα gene expression. Differences in the transcriptional regulation of TNFα and IL-6 at the promoter region, which is also manifested by differences in induction timing, may underlie the difference in the level of dependence on NFκB activity. TNFα is induced early due to a “constitutively and immediately accessible” promoter region, while IL-6 induction occurs later as it depends on stimulus-induced chromatin remodeling [60,61], and the expression of early NFκB gene products that facilitate promoter activation [62–64]. These additional transcriptional requirements may make IL-6 gene transcription more susceptible to the inhibitory effects of DHA through reduced NFκB activity.
In summary, the results of this work demonstrate differential effects of DHA on TNFα and IL-6 gene expression in LPS-stimulated RAW 264.7 cells, an effect which may be mediated by a partial inhibition of the NFκB signaling pathway. These data expand observations from previous studies demonstrating that the anti-inflammatory effect of DHA is not a universal down-regulator of all pro-inflammatory cytokines, but of specific inflammatory cytokines and by differing degrees. The potential importance of our findings can be broadened to other areas which target inflammation or specific pro-inflammatory cytokines.
Acknowledgments
This study was supported by grants from the NIH NHLBI-T32-HL069772 (NLS) and the USDA agreement No. 58-1950-0-0014.
Any opinions, findings, conclusion, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the U.S. Department of Agriculture.
References
- 1.Calder PC. Long-chain fatty acids and inflammation. Proc Nutr Soc. 2012;71:284–289. doi: 10.1017/S0029665112000067. [DOI] [PubMed] [Google Scholar]
- 2.Honda KL, Lamon-Fava S, Matthan NR, Wu D, Lichtenstein AH. EPA and DHA exposure alters the inflammatory response but not the surface expression of toll-like receptor 4 in macrophages. Lipids. 2015;50:121–129. doi: 10.1007/s11745-014-3971-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calder PC, Ahluwalia N, Albers R, et al. A consideration of biomarkers to be used for evaluation of inflammation in human nutritional studies. Br J Nutr. 2013;109(Suppl 1):S1–S34. doi: 10.1017/S0007114512005119. [DOI] [PubMed] [Google Scholar]
- 4.Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipo-protein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:2137–2142. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 5.Sun Y, Yin M, Zhang L, Pan J. Characterization of the cytokine expression profiles of the aorta and liver of young tumor necrosis factor alpha mutant mice. Mol Cell Biochem. 2012;366:59–67. doi: 10.1007/s11010-012-1283-1. [DOI] [PubMed] [Google Scholar]
- 6.Xiao N, Yin M, Zhang L, et al. Tumor necrosis factor-alpha deficiency retards early fatty-streak lesion by influencing the expression of inflammatory factors in apoE-null mice. Mol Genet Metab. 2009;96:239–244. doi: 10.1016/j.ymgme.2008.11.166. [DOI] [PubMed] [Google Scholar]
- 7.McLaren JE, Michael DR, Ashlin TG, Ramji DP. Cytokines, macrophage lipid metabolism and foam cells: implications for cardiovascular disease therapy. Prog Lipid Res. 2011;50:331–347. doi: 10.1016/j.plipres.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Hashizume M, Mihara M. Atherogenic effects of TNF-alpha and IL-6 via up-regulation of scavenger receptors. Cytokine. 2012;58:424–430. doi: 10.1016/j.cyto.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 9.Legler DF, Bruckner M, Uetz-von Allmen E, Krause P. Prostaglandin E2 at new glance: novel insights in functional diversity offer therapeutic chances. Int J Biochem Cell Biol. 2010;42:198–201. doi: 10.1016/j.biocel.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Gomez I, Foudi N, Longrois D, Norel X. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot Essent Fat Acids. 2013;89:55–63. doi: 10.1016/j.plefa.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Medeiros A, Peres-Buzalaf C, Fortino Verdan F, Serezani CH. Prostaglandin E2 and the suppression of phagocyte innate immune responses in different organs. Mediat Inflamm. 2012;2012:327568. doi: 10.1155/2012/327568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akaogi J, Yamada H, Kuroda Y, Nacionales DC, Reeves WH, Satoh M. Prostaglandin E2 receptors EP2 and EP4 are up-regulated in peritoneal macrophages and joints of pristane-treated mice and modulate TNF-alpha and IL-6 production. J Leukoc Biol. 2004;76:227–236. doi: 10.1189/jlb.1203627. [DOI] [PubMed] [Google Scholar]
- 14.Treffkorn L, Scheibe R, Maruyama T, Dieter P. PGE2 exerts its effect on the LPS-induced release of TNF-alpha, ET-1, IL-1alpha, IL-6 and IL-10 via the EP2 and EP4 receptor in rat liver macrophages. Prostaglandins Other Lipid Mediat. 2004;74:113–123. doi: 10.1016/j.prostaglandins.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 15.Vassiliou E, Jing H, Ganea D. Prostaglandin E2 inhibits TNF production in murine bone marrow-derived dendritic cells. Cell Immunol. 2003;223:120–132. doi: 10.1016/s0008-8749(03)00158-8. [DOI] [PubMed] [Google Scholar]
- 16.Yamane H, Sugimoto Y, Tanaka S, Ichikawa A. Prostaglandin E(2) receptors, EP2 and EP4, differentially modulate TNF-alpha and IL-6 production induced by lipopolysaccharide in mouse peritoneal neutrophils. Biochem Biophys Res Commun. 2000;278:224–228. doi: 10.1006/bbrc.2000.3779. [DOI] [PubMed] [Google Scholar]
- 17.Williams JA, Pontzer CH, Shacter E. Regulation of macrophage interleukin-6 (IL-6) and IL-10 expression by prostaglandin E2: the role of p38 mitogen-activated protein kinase. J Interferon Cytokine Res. 2000;20:291–298. doi: 10.1089/107999000312423. [DOI] [PubMed] [Google Scholar]
- 18.Stafford JB, Marnett LJ. Prostaglandin E2 inhibits tumor necrosis factor-alpha RNA through PKA type I. Biochem Biophys Res Commun. 2008;366:104–109. doi: 10.1016/j.bbrc.2007.11.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wall EA, Zavzavadjian JR, Chang MS, et al. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci Signal. 2009;2:ra28. doi: 10.1126/scisignal.2000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ollivier V, Parry GC, Cobb RR, de Prost D, Mackman N. Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J Biol Chem. 1996;271:20828–20835. doi: 10.1074/jbc.271.34.20828. [DOI] [PubMed] [Google Scholar]
- 21.Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol. 2010;185:6413–6419. doi: 10.4049/jimmunol.1001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koga K, Takaesu G, Yoshida R, et al. Cyclic adenosine monophosphate suppresses the transcription of proinflammatory cytokines via the phosphorylated c-Fos protein. Immunity. 2009;30:372–383. doi: 10.1016/j.immuni.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 23.Gerlo S, Kooijman R, Beck IM, Kolmus K, Spooren A, Haegeman G. Cyclic AMP: a selective modulator of NF-kappaB action. Cell Mol Life Sci. 2011;68:3823–3841. doi: 10.1007/s00018-011-0757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parry GC, Mackman N. Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol. 1997;159:5450–5456. [PubMed] [Google Scholar]
- 25.Delgado M, Munoz-Elias EJ, Kan Y, et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit tumor necrosis factor alpha transcriptional activation by regulating nuclear factor-kB and cAMP response element-binding protein/c-Jun. J Biol Chem. 1998;273:31427–31436. doi: 10.1074/jbc.273.47.31427. [DOI] [PubMed] [Google Scholar]
- 26.Spooren A, Kooijman R, Lintermans B, et al. Cooperation of NFkappaB and CREB to induce synergistic IL-6 expression in astrocytes. Cell Signal. 2010;22:871–881. doi: 10.1016/j.cellsig.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 27.Fujino H, Salvi S, Regan JW. Differential regulation of phosphorylation of the cAMP response element-binding protein after activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. Mol Pharmacol. 2005;68:251–259. doi: 10.1124/mol.105.011833. [DOI] [PubMed] [Google Scholar]
- 28.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188:21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacKenzie KF, Clark K, Naqvi S, et al. PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J Immunol. 2013;190:565–577. doi: 10.4049/jimmunol.1202462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim YJ, Chung HY. Antioxidative and anti-inflammatory actions of docosahexaenoic acid and eicosapentaenoic acid in renal epithelial cells and macrophages. J Med Food. 2007;10:225–231. doi: 10.1089/jmf.2006.092. [DOI] [PubMed] [Google Scholar]
- 31.Saw CL, Huang Y, Kong AN. Synergistic anti-inflammatory effects of low doses of curcumin in combination with polyunsaturated fatty acids: docosahexaenoic acid or eicosapentaenoic acid. Biochem Pharmacol. 2010;79:421–430. doi: 10.1016/j.bcp.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 32.Norris PC, Dennis EA. Omega-3 fatty acids cause dramatic changes in TLR4 and purinergic eicosanoid signaling. Proc Natl Acad Sci USA. 2012;109:8517–8522. doi: 10.1073/pnas.1200189109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Honda KL, Lamon-Fava S, Matthan NR, Wu D, Lichtenstein AH. EPA and DHA exposure alters the inflammatory response but not the surface expression of toll-like receptor 4 in macrophages. Lipids. 2015;50:121–129. doi: 10.1007/s11745-014-3971-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avni D, Ernst O, Philosoph A, Zor T. Role of CREB in modulation of TNFalpha and IL-10 expression in LPS-stimulated RAW264.7 macrophages. Mol Immunol. 2010;47:1396–1403. doi: 10.1016/j.molimm.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 35.Eliopoulos AG, Dumitru CD, Wang CC, Cho J, Tsichlis PN. Induction of COX-2 by LPS in macrophages is regulated by Tpl2-dependent CREB activation signals. EMBO J. 2002;21:4831–4840. doi: 10.1093/emboj/cdf478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pierce JW, Schoenleber R, Jesmok G, et al. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 37.Wang S, Wu D, Lamon-Fava S, Matthan NR, Honda KL, Lichtenstein AH. In vitro fatty acid enrichment of macrophages alters inflammatory response and net cholesterol accumulation. Br J Nutr. 2009;102:497–501. doi: 10.1017/S0007114509231758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullen A, Loscher CE, Roche HM. Anti-inflammatory effects of EPA and DHA are dependent upon time and dose–response elements associated with LPS stimulation in THP-1-derived macrophages. J Nutr Biochem. 2010;21:444–450. doi: 10.1016/j.jnutbio.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 39.Weldon SM, Mullen AC, Loscher CE, Hurley LA, Roche HM. Docosahex-aenoic acid induces an anti-inflammatory profile in lipopolysaccharide-stimulated human THP-1 macrophages more effectively than eicosapentaenoic acid. J Nutr Biochem. 2007;18:250–258. doi: 10.1016/j.jnutbio.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Zhao G, Etherton TD, Martin KR, et al. Anti-inflammatory effects of polyunsaturated fatty acids in THP-1 cells. Biochem Biophys Res Commun. 2005;336:909–917. doi: 10.1016/j.bbrc.2005.08.204. [DOI] [PubMed] [Google Scholar]
- 41.Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 43.Lee JY, Plakidas A, Lee WH, et al. Differential modulation of toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res. 2003;44:479–486. doi: 10.1194/jlr.M200361-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Sijben JW, Calder PC. Differential immunomodulation with long-chain n-3 PUFA in health and chronic disease. Proc Nutr Soc. 2007;66:237–259. doi: 10.1017/S0029665107005472. [DOI] [PubMed] [Google Scholar]
- 45.Deike E, Bowden RG, Moreillon JJ, et al. The effects of fish oil supplementation on markers of inflammation in chronic kidney disease patients. J Ren Nutr. 2012;22:572–577. doi: 10.1053/j.jrn.2011.10.036. [DOI] [PubMed] [Google Scholar]
- 46.Derosa G, Cicero AF, Fogari E, et al. Effects of n-3 PUFAs on postprandial variation of metalloproteinases, and inflammatory and insulin resistance parameters in dyslipidemic patients: evaluation with euglycemic clamp and oral fat load. J Clin Lipidol. 2012;6:553–564. doi: 10.1016/j.jacl.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 47.Ferguson JF, Mulvey CK, Patel PN, et al. Omega-3 PUFA supplementation and the response to evoked endotoxemia in healthy volunteers. Mol Nutr Food Res. 2014;58:601–613. doi: 10.1002/mnfr.201300368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hassan KS, Hassan SK, Hijazi EG, Khazim KO. Effects of omega-3 on lipid profile and inflammation markers in peritoneal dialysis patients. Ren Fail. 2010;32:1031–1035. doi: 10.3109/0886022X.2010.510231. [DOI] [PubMed] [Google Scholar]
- 49.Koutsos A, Jackson KG, Lockyer S, Carvalho-Wells A, Minihane AM, Lovegrove JA. Greater impact of dietary fat manipulation than apolipoprotein E genotype on ex vivo cytokine production – insights from the SATgenepsilon study. Cytokine. 2014;66:156–159. doi: 10.1016/j.cyto.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Renz H, Gong JH, Schmidt A, Nain M, Gemsa D. Release of tumor necrosis factor-alpha from macrophages. Enhancement and suppression are dose-dependently regulated by prostaglandin E2 and cyclic nucleotides. J Immunol. 1988;141:2388–2393. [PubMed] [Google Scholar]
- 51.Martel-Pelletier J, Lajeunesse D, Reboul P, Pelletier JP. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann Rheum Dis. 2003;62:501–509. doi: 10.1136/ard.62.6.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 53.Pestka JJ, Smolinski AT. Deoxynivalenol: toxicology and potential effects on humans. J Toxicol Environ Health: B Crit Rev. 2005;8:39–69. doi: 10.1080/10937400590889458. [DOI] [PubMed] [Google Scholar]
- 54.Jia Q, Zhou HR, Shi Y, Pestka JJ. Docosahexaenoic acid consumption inhibits deoxynivalenol-induced CREB/ATF1 activation and IL-6 gene transcription in mouse macrophages. J Nutr. 2006;136:366–372. doi: 10.1093/jn/136.2.366. [DOI] [PubMed] [Google Scholar]
- 55.Komatsu W, Ishihara K, Murata M, Saito H, Shinohara K. Docosahexaenoic acid suppresses nitric oxide production and inducible nitric oxide synthase expression in interferon-gamma plus lipopolysaccharide-stimulated murine macrophages by inhibiting the oxidative stress. Free Radic Biol Med. 2003;34:1006–1016. doi: 10.1016/s0891-5849(03)00027-3. [DOI] [PubMed] [Google Scholar]
- 56.Lee J, Rhee MH, Kim E, Cho JY. BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediat Inflamm. 2007;53:111–117. doi: 10.1155/2012/416036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 58.Kawai T, Akira S. Toll-like receptor downstream signaling. Arthritis Res Ther. 2005;7:12–19. doi: 10.1186/ar1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boothby M. Specificity of sn50 for NF-kappa B? Nat Immunol. 2001;2:471–472. doi: 10.1038/88652. [DOI] [PubMed] [Google Scholar]
- 60.Ramirez-Carrozzi VR, Nazarian AA, Li CC, et al. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006;20:282–296. doi: 10.1101/gad.1383206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saccani S, Pantano S, Natoli G. Two waves of nuclear factor kappaB recruitment to target promoters. J Exp Med. 2001;193:1351–1359. doi: 10.1084/jem.193.12.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Motoyama M, Yamazaki S, Eto-Kimura A, Takeshige K, Muta T. Positive and negative regulation of nuclear factor-kappaB-mediated transcription by Ikap-paB-zeta, an inducible nuclear protein. J Biol Chem. 2005;280:7444–7451. doi: 10.1074/jbc.M412738200. [DOI] [PubMed] [Google Scholar]
- 63.Yamazaki S, Matsuo S, Muta T, Yamamoto M, Akira S, Takeshige K. Gene-specific requirement of a nuclear protein, IkappaB-zeta, for promoter association of inflammatory transcription regulators. J Biol Chem. 2008;283:32404–32411. doi: 10.1074/jbc.M802148200. [DOI] [PubMed] [Google Scholar]
- 64.Yamazaki S, Muta T, Takeshige K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J Biol Chem. 2001;276:27657–27662. doi: 10.1074/jbc.M103426200. [DOI] [PubMed] [Google Scholar]