
Fig. 1.
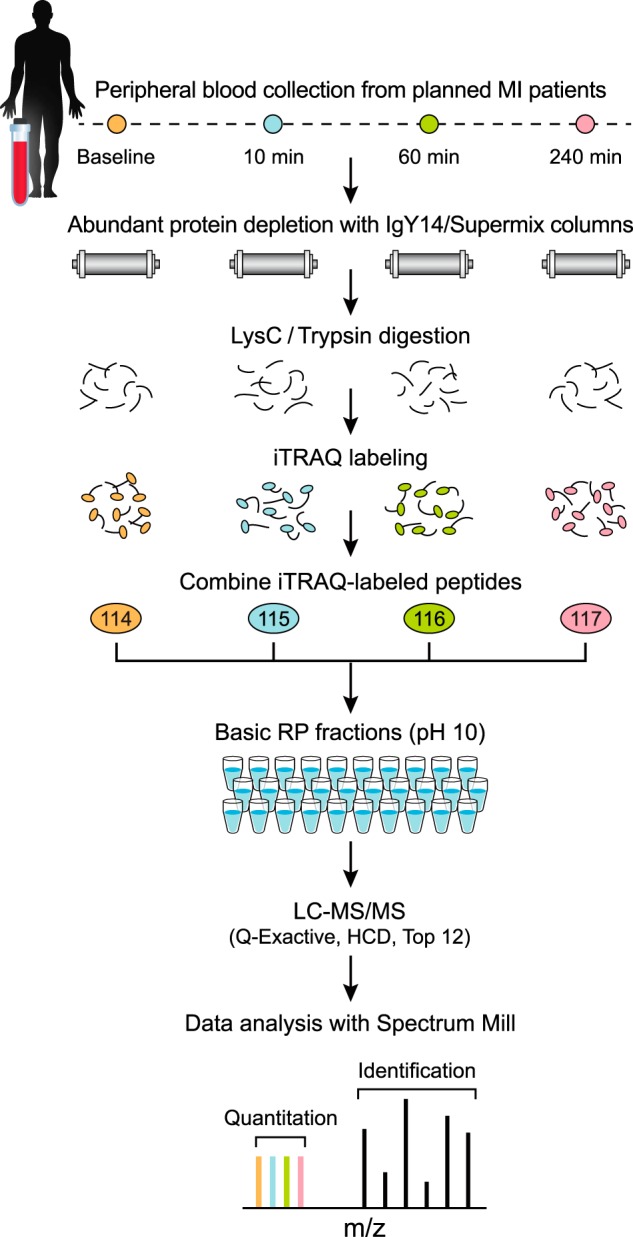
Diagram of improved workflow for discovery proteomics in plasma. Samples from four different time points of planned MI (PMI) patients were depleted from abundant proteins, reduced, alkylated and digested by LysC/Trypsin. Following desalting, samples were labeled by four-plex iTRAQ reagent, and mixed after evaluating label incorporation. Sample was then fractionated using reversed phase chromatography at high pH into 30 pooled, concatenated fractions. Fractions were analyzed by data dependent analysis on a Q Exactive mass spectrometer using 75 μm picofrit columns packed in-house with 1.9 μm beads to 20 cm length. See Methods for details.