

Background: BACE1 is the rate-limiting enzyme in the cleavage of amyloid precursor protein to Aβ peptides.
Results: Retinoic acid blocks the BACE1 gene up-regulation during inflammation via the inhibition of NFκB.
Conclusion: Retinoic acid suppresses aspects of neuroinflammation in rodent models of Alzheimer disease including elevated BACE1 transcription.
Significance: Retinoids may have an important role in limiting progression of Alzheimer disease.
Keywords: Alzheimer disease, beta-secretase 1 (BACE1), gene transcription, NFkappa B (NFKB), retinoic acid, retinoid signaling
Abstract
Insulin resistance and neuroinflammation have emerged as two likely key contributors in the pathogenesis of Alzheimer disease (AD), especially in those sporadic AD cases compromised by diabetes or cardiovascular disease. Amyloid-β (Aβ) deposition and its associated inflammatory response are hallmarks in sporadic AD brains. Elevated expression and activity of β-secretase 1 (BACE1), the rate-limiting enzyme responsible for the β-cleavage of amyloid precursor proteins to Aβ peptides, are also observed in sporadic AD brains. Previous studies have suggested that there is therapeutic potential for retinoic acid in treating neurodegeneration based on decreased Aβ. Here we discovered that BACE1 expression is elevated in the brains of both Tg2576 transgenic mice and mice on high fat diets. These conditions are associated with a neuroinflammatory response. We found that administration of all-trans-retinoic acid (atRA) down-regulated the expression of BACE1 in the brains of Tg2576 mice and in mice fed a high fat diet. Moreover, in LPS-treated mice and cultured neurons, BACE1 expression was repressed by the addition of atRA, correlating with the anti-inflammatory efficacy of atRA. Mutations of the NFκB binding site in BACE1 promoter abolished the suppressive effect of atRA. Furthermore, atRA disrupted LPS-induced nuclear translocation of NFκB and its binding to BACE1 promoter as well as promoting the recruitment of the corepressor NCoR. Our findings indicate that atRA represses BACE1 gene expression under inflammatory conditions via the modulation of NFκB signaling.
Introduction
Compelling evidence suggests that in Alzheimer disease (AD),4 amyloid-β (Aβ) deposition is associated with marked inflammatory response via activated microglias and the recruitment of astrocytes (1). Aβ peptides are derived from amyloid precursor protein (APP) by sequential proteolytic cleavage via β-secretase (BACE1) and γ-secretase (2). In AD brains, the specific regions affected by Aβ deposition are correlated with elevated levels of BACE1 expression and activity. Together with the observation of diminished amyloid pathology in BACE1-deficient mice (3), these findings define a central role of BACE1 in APP processing and Aβ generation/deposition, which makes it one of the most important therapeutic targets of AD.
BACE1 expression is tightly regulated at multiple levels from transcription to translation (3, 4). At the transcriptional level, BACE1 expression is positively or negatively regulated by a number of transcription factors under basal and cell-stressed conditions (5). We recently reported that that BACE1 transcription is up-regulated by metabolic risk factors and that the key metabolic regulatory pathway of AMPK-SIRT1-PGC-1α (where AMPK is AMP-activated protein kinase) suppressed BACE1 transcription by directly acting on the first PPAR-responsive element (PPRE) in the BACE1 gene both in cultured neurons and in vivo settings (calorie restriction, etc.) (6, 7). Although the identification of several putative binding sites for NFκB and PPARγ in BACE1 promoter strongly implicates its regulation under inflammatory condition, compelling evidence is lacking.
Retinoic acid (RA) is a metabolic product of vitamin A and is involved in several essential biological processes including proliferation, differentiation, survival, and apoptosis (8–12). In general, RA modulates its effects by binding to nuclear retinoic acid receptors (RARs) and retinoid X receptors (RXRs). RARs are ligand-dependent transcription factors that bind response elements as heterodimers with RXRs to regulate a large number of target genes (over 500 so far). RA exists in three main stereoisomeric forms, all-trans-retinoic acid (atRA), 13-cis RA, and 9-cis RA, among which atRA is the primary biologically active form in brain. RA also exerts anti-inflammatory efficacy via repressing the expression of inflammatory factors such as IL-6, IL-12, and TNFα (13–16) and modulating NFκB signaling (17, 18).
The RAR/RXR nuclear receptors are being pursued as major drug targets for various diseases such as cancer and metabolic syndrome. The importance of retinoid signaling in AD has recently emerged. Clinical and experimental studies strongly suggest that disruption of RA signaling correlates with AD pathogenesis (19). When compared with normal aged controls, AD patients have lower serum concentrations of vitamin A and β-carotene (20, 21). Defective RA transport and function are also observed in AD brains (22, 23). In vitamin A-deprived rodents, RAR expression is down-regulated, and most strikingly, this is accompanied by AD-like hallmarks including accumulation of Aβ peptides, loss of hippocampal long-term potentiation, and memory deficits (24–28). These effects were largely reversed by the administration of RA (29, 30). Taken together, these data provide strong rationale investigating RA-based therapy in AD (31–34).
Although atRA has been reported to attenuate Aβ deposition in APP/PS1 double-transgenic mice (30), the mechanism remains unclear. It has been reported that RA can reduce Aβ production via activating non-amyloidogenic α-secretase (35, 36). Here we aim to investigate the potential modulation of BACE1 expression by RA and its underlying mechanism. Based on the known potent modulatory role of RA in immune response (13–16), we speculated that RA might be able to suppress BACE1 expression under inflammatory conditions. It should be stressed that in comparison with the peripheral system, the anti-inflammatory role of RA in CNS remains largely elusive. To test our hypothesis, we examined the expression of BACE1 in atRA-treated Tg2576, high fat diet, and LPS-challenged mice. These conditions have all been reported to induce inflammatory events in the brain (34, 37, 38). We further investigated the underlying molecular mechanism using in vitro cultured rat primary neurons.
Experimental Procedures
Animals
All animal care protocols and procedures were performed in accordance with the Animal Scientific Procedures Act and with approval of the University of Tennessee Animal Care and Use Committee. High fat diet (HFD) was given to C57BL/6 mice as described (39). atRA (R2625, Sigma) dissolved in 5% DMSO/corn oil and LPS from Escherichia coli 055:B5 (L2880, Sigma) in PBS were given to C57BL/6 mice by intraperitoneal injection at 20 and 5 mg/kg, respectively. For Tg2576 transgenic mice (6–8 months) and HFD mice (8–9 months), atRA was given every 2 days for 12 and 8 weeks, respectively. For the acute inflammatory model, C57BL/6 mice were administrated atRA every other day for 3 days and then treated with LPS for 6 h or overnight.
Cell Culture and Drug Treatment
Rat primary cortical neurons were prepared as described (40) and maintained at 37 °C in a humidified incubator with 9.5% CO2. HEK 293 and Neuro-2a (N2a) cells were cultured in DMEM (Cellgro) with 10% FBS (HyClone) and 50% DMEM/50% Opti-MEM I (Gibco) with 5% FBS, respectively, and maintained at 37 °C in a humidified incubator with 5% CO2. atRA stocks in DMSO and LPS stocks in PBS were stored at −80 °C. For each experiment, atRA or LPS was diluted from its stock solution and added to the growth medium at the indicated concentrations; 0.1% (v/v) solvent was used as vehicle control.
Western Blot Analysis
Cell and cortical tissue lysates were prepared in radioimmunoprecipitation assay lysis buffer. N2a nuclear extracts and cytoplasmic extracts were prepared using the nuclear extract kit (Active Motif). The same amount of total proteins was resolved on Novex® 4–20% Tris-glycine gel (Life Technologies), and the subsequent Western blot was performed as described (Wang et al. (7)) using the following antibodies: rabbit anti-BACE1 (1:250; AB5940, Millipore), mouse anti-β-actin (1:5000; A2228, Sigma), mouse anti-tubulin (1:5000; T6074, Sigma), rabbit anti-phospho-IκB (1:205; 2859P, Cell Signaling), mouse anti-IκB (1:250; 4814P, Cell Signaling), mouse anti-NFκB p65 (1:1000; sc-71676, Santa Cruz Biotechnology), and rabbit anti-phospho-NFκB p65 (1:250; 3031S, Cell Signaling).
Immunohistochemistry and Immunocytochemistry
After perfusion with 4% paraformaldehyde/PBS, mouse brains were cryopreserved in 30% (w/v) sucrose/PBS at 4 °C overnight. Brain samples embedded in OCT were cut into coronal 20-μm sections and mounted on a glass. Brain sections were blocked at room temperature for 1 h in normal goat serum buffer (10% normal goat serum, 0.2% Triton X-100, and 0.02% NaN3 in TBS) and incubated with primary antibodies at 4 °C overnight. The subsequent incubation of fluorescence-conjugated secondary antibodies was performed at room temperature for 2 h. To visualize nucleus, sections were counterstained with DAPI (Invitrogen). After incubation with antibodies, all sections were washed three times with PBS, 10 min each time. The fluorescence was examined with microscopy.
For immunocytochemistry, drug-treated rat primary neurons were briefly rinsed with PBS and fixed with 4% paraformaldehyde, 0.12 m sucrose in PBS. Neurons were permeabilized in 0.3% Triton X-100 for 5 min. Fixed neurons were blocked in 10% BSA in PBS for 1 h at room temperature and incubated in primary antibodies at 4 °C overnight. Neurons were then incubated in fluorescence-conjugated secondary antibodies at room temperature for 1 h and counterstained with DAPI to visualize nucleus. The antibodies used were rabbit anti-BACE1 (1:50; AB5940, Millipore), mouse anti-NeuN (1:30; MAB377, Chemicon), mouse anti-NFκB p65 (1:100; sc-71676, Santa Cruz Biotechnology), and Alexa Fluor® 488 and 594 secondary antibody (1:500; Invitrogen).
Quantitative Real-time RT-PCR
RNA was prepared using TRIzol (Invitrogen) according to the manufacturer's instructions. Single-stranded cDNA was synthesized from 1 μg of total RNA using High Capacity cDNA reverse transcription kits (Applied Biosystems). Quantitative real-time PCR was performed with RealMasterMix SYBR ROX (5 Prime) according to the manufacturer's protocols using the same rat and mouse GAPDH primers, as well as rat BACE1 primers. Primers for TNFα (forward 5′-CCCTCACACTCAGATCATCTTCT-3′ and reverse 5′-GCTACGACGTGGGCTACAG-3′), IL-6 (forward 5′-GCTACCAAACTGGATATAATCAGGA-3′ and reverse 5′-CCAGGTAGCTATGGTACTCCAGAA-3′), and Toll-like receptor-4 (TLR4) (rat forward 5′-GGATGATGCCTCTCTTGCAT-3′ and reverse primer 5′-TGATCCATGCATTGGTAGGTAA-3′; mouse forward 5′-GGACTCTGATCATGGCACTG-3′ and reverse 5′-CTGATCCATGCATTGGTAGGT-3′) were used in the present studies.
Transient Transfection and Luciferase Assay
HEK 293 cells were seeded at 4–5 × 104 cells/well in a 24-well plate the day before transfection. Transient transfection was conducted using Lipofectamine 2000 (Invitrogen) following the procedure provided by the manufacturer. Deletion constructs and mutants of rat BACE1 promoter were described (Wang et al. (7)); the expression vectors of RXRα, RARα, RARβ, and RARγ were kind gifts from Dr. Xiao-kun Zhang (Sanford-Burnham Medical Research Institute) and Dr. Philippe Lefebvre (INSERM, UMR1011, F-59000, Lille, France). Drug treatment was performed 5 h after transfection. Luciferase assays were conducted with the Dual-Luciferase® reporter assay system (E1910, Promega) in a TD-20/20 luminometer.
ChIP Assay
ChIP assays were performed using the Chromatin Immunoprecipitation kit (17-295, Upstate® EMD Millipore) according to the manufacturer's instructions. Chromatin fragments from mouse cortex were prepared as described (7). 30 mg of tissues were used for each ChIP/antibody with mouse anti-NFκB p65 (sc-71676, Santa Cruz Biotechnology), goat anti-NCoR (sc-1609, Santa Cruz Biotechnology), mouse anti-PPARγ (81B8) (2443, Cell Signaling), and rabbit anti-RXRα (sc-553, Santa Cruz Biotechnology). The following primers were used in PCR assays: for mouse BACE1 promoter NFκB site, forward 5′-GAGAGCCCCTCAGCTTCTTT-3′ and reverse 5′-GGGGCCCTGATAGCTTAAAA-3′; for rat BACE1 NFκB site, forward 5′-ATGGTGGCTGGATTTTATCGAGG-3′ and reverse 5′-GTGAATTTGTCTGTGCCTTTG-3′; for rat BACE1 PPRE site, 5′-GAGTAATGTTGGTATGCCTC-3′ and reverse 5′-GGGATGAGAGTATGTCAGTC-3′.
Statistical Analysis
All quantitative data are presented as means ± S.D. Comparisons between groups were analyzed by Student's t test.
Results
atRA Treatment Reduces Cortical BACE1 Expression in Tg2576 and HFD Mice
We tested the effect of systemic administration of atRA on BACE1 expression in symptomatic Tg2576 mice overexpressing the transgene of human Swedish mutant of APP695. These mice expressed much higher BACE1 levels as compared with non-transgenic mice at the same age (Fig. 1, A and B). Continuous atRA treatment for 12 weeks significantly reduced BACE1 protein levels by 30% in the cortex of Tg2576 mice as compared with the vehicle-treated group (Fig. 1, A and B). Notably, BACE1 expression was only slightly decreased in atRA-treated non-transgenic mice, although there was no statistical significance due to the individual variation. These data suggest that atRA is more effective in inhibiting BACE1 under inflammatory conditions.
FIGURE 1.

atRA suppresses BACE1 expression in Tg2576 and HFD mouse hippocampi, correlating with its anti-inflammatory role. A, Western blot analysis of hippocampal BACE1 in atRA administrated Tg2576 and non-transgenic (Non-Tg) mice. B, quantification of BACE1 protein levels in panel A. Data represent the relative BACE1 protein levels as compared with vehicle-treated on-transgenic group; means ± S.D. n = 3–4, *, p < 0.05 by Student's t test. C, Western blot analysis of hippocampal BACE1 in HFD mice treated with or without atRA. D, quantification of BACE1 protein levels in panel C. For quantification, BACE1 protein levels relative to β-actin in the control group were set to 1; values are means ± S.D. n = 4, *, p < 0.05 by Student's t test. E and F, the mRNA levels of TNFα and IL-6 in HFD mice treated with or without atRA were determined by RT-qPCR, and the data are presented as expression levels relative to GAPDH × 1000; means ± S.D. n = 4, *, p < 0.05 by Student's t test.
In a previous study (39), neuroinflammation and a cognitive deficit in young WT C57BL/6 (B6) mice were induced by a high fat, high cholesterol diet (21% fat from milk with enriched cholesterol at 1.25%, HFD). We also found that young C57BL/6 mice consuming this HFD for 8 weeks demonstrated increased BACE1 expression (7), neuroinflammation, and learning/memory deficits (data not shown). Therefore, we tested the effect of atRA on the elevated BACE1 expression in HFD mice. As shown in Fig. 1, C and D, atRA administration for 8 weeks dramatically reduced BACE1 expression levels in the cortex of HFD mice to basal level in the control mice on the chow diet. Furthermore, quantitative RT-PCR analysis revealed a significant decrease in the expression of the proinflammatory factors TNFα and IL-6 in atRA-treated HFD mice (Fig. 1, E and F), suggesting that the suppressive effect of atRA on BACE1 expression in Tg2576 and HFD mouse brains is correlated with its anti-inflammatory role.
atRA Represses LPS-induced BACE1 Expression in Vitro and in Vivo
Although the Tg2576 and HFD mice develop a chronic inflammatory condition in the CNS, systemic LPS administration induces acute brain inflammation, which also leads to impaired memory (41). We injected LPS intraperitoneally into C57BL/6 mice and observed dramatically increased TNFα and IL-6 expression in the cortex of LPS-treated mice several hours later (Fig. 2, A and B). The increased expression of TNFα and IL-6 was significantly inhibited by atRA pretreatment (Fig. 2, A and B), suggesting that atRA blocks LPS-induced acute neuroinflammation. Therefore, we tested the effect of atRA on BACE1 expression using this model. In a Western blot analysis with cortical lysates, a complete correction of BACE1 expression was observed in the atRA-LPS-treated mice as compared with the LPS-treated mice (Fig. 2, C and D). Because astrocytes and microglias are the major sources of neuroinflammation in vivo, we sought to investigate whether LPS-induced BACE1 regulation is primarily a neuronal or glial event. Immunohistochemical analysis (Fig. 2E) revealed that the LPS-induced BACE1 co-localized with NeuN (a neuron-specific nuclear protein) and that this BACE1 was inhibited by atRA treatment. Neurons may directly respond to inflammatory agents due to neuronal expression of cytokine receptors such as the LPS and TNFα receptors (42, 43).
FIGURE 2.

atRA inhibits LPS-stimulated BACE1 expression in C57BL/6 mouse hippocampi. A and B, RT-qPCR analysis of TNFα and IL-6 mRNA levels in LPS-stimulated C57BL/6 mouse hippocampi with or without atRA administration, 6 h after of 1 mg/kg of LPS (intraperitoneally). Data represent TNFα and IL-6 mRNA expression levels relative to GAPDH; means ± S.D. n = 4, *, p < 0.05, **, p < 0.01 by Student's t test. C, Western blot analysis of hippocampal BACE1 protein level in LPS-treated mice with or without atRA administration. D, abundance of BACE1 protein level in panel C. BACE1 protein level relative to β-actin in the vehicle-treated group was set to 1; values are means ± S.D. n = 4, *, p < 0.05 by Student's t test. E, double immunostaining of BACE1 (red) and glial fibrillary acidic protein (green) in the hippocampal CA1 region and frontal cortex (CTX) in LPS-treated mice with or without atRA administration. Nuclei were counterstained with DAPI. Scale bar = 20 μm.
atRA Represses BACE1 Expression at the Transcriptional Level
Given the suppressive effects of atRA in vivo, we speculated that atRA would modulate BACE1 gene transcription in vitro. Indeed, in cultured neurons, atRA (0.01–1 μm) not only reduced BACE1 protein levels in a dose-dependent manner (Fig. 3, A and B), but also decreased BACE1 mRNA expression in neurons (Fig. 3C). Moreover, when HEK 293 cells were transfected with a vector containing the BACE1 promoter driving the luciferase reporter gene, atRA treatment caused an ∼50% reduction in BACE1 promoter activity (Fig. 3D). Together, these data suggest that atRA represses basal BACE1 expression at the transcriptional level.
FIGURE 3.
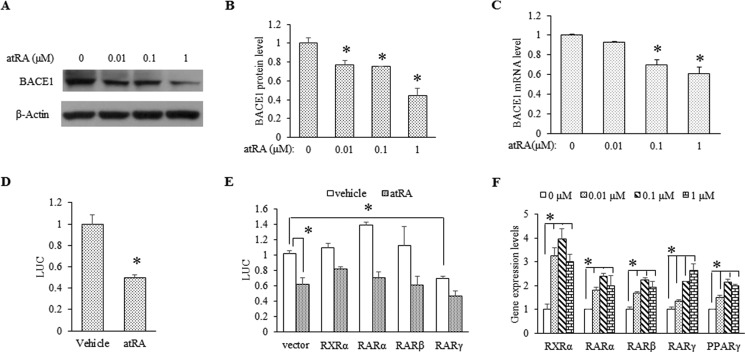
atRA down-regulates BACE1 expression at transcriptional level. A, Western blot analysis of BACE1 protein expression in rat primary neurons (DIV15) treated with atRA at the indicated concentrations for 24 h. B, quantification of the BACE1 protein level from three independent experiments. Data represent means ± S.D. n = 3, *, p < 0.05 by Student's t test as compared with vehicle-treated neurons. C, BACE1 mRNA levels determined by RT-qPCR. Cultured neurons (DIV15) were treated with atRA at the indicated concentrations for 6 h. Data are presented as BACE1 mRNA expression levels relative to β-actin × 1000; means ± S.D. n = 3, *, p < 0.05 by Student's t test as compared with vehicle-treated neurons. D, rat BACE1 promoter activity assayed in atRA-treated HEK 293 cells. HEK 293 cells were cotransfected with a rat BACE1 promoter driving the luciferase (LUC) reporter and pRL-SV40 vector. Cells were exposed to 10 μm atRA for 24 h. Luciferase values were normalized to Renilla luciferase values. Mean values of vehicle-treated cells were set to 1, n ≥ 3, *, p < 0.05 by Student's t test. E, the effect of retinoid receptors (RXR and RAR) on BACE1 promoter activity. HEK 293 cells were cotransfected with rat BACE1 promoter-luciferase vector and either retinoid receptor expression vectors or empty control vector. Cells were treated overnight with 1 μm atRA. *, p < 0.05 by Student's t test. F, the effect of atRA at various concentrations on the mRNA levels of nuclear receptor molecules. Data represent means ± S.D. n = 3, *, p < 0.05 by Student's t test.
There are four potential RAR-PPAR-RXR-responsive elements predicted in BACE1 promoter (7, 40). To determine whether the repressive effect of atRA on BACE1 promoter depends on retinoid receptors, we first tested the impact of overexpressed receptors on the co-expressed BACE1-luciferase construct. In cotransfection experiments with the expression vectors for human RXRα, RARα, RARβ, and RARγ isoforms and the rat BACE1 promoter-luciferase reporter in HEK 293 cells, we found that none of these receptors significantly affected basal BACE1 promoter activity except for RARγ (∼30% reduction) (Fig. 3E). In addition, overexpression of these receptors did not increase the repressive action of atRA on BACE1 promoter activity. Moreover, atRA treatment significantly induced expression of all RAR isoforms in cultured neurons (Fig. 3F). Interestingly, the combination of PGC-1α overexpression with atRA treatment did not cause further BACE1 suppression, nor did the overexpression of PPARγ (data not shown), suggesting that the expression levels of these receptors/cofactors are not a limiting factor for the effect of atRA. To further investigate the potential involvement of these factors, we then tested several agonists/antagonists including SR1664 and GW9662, a PPARγ agonist and antagonist, respectively; bexarotene, an RXR agonist; and AGN 194310, an RAR pan-antagonist. Consistent with the overexpression data, none of these compounds exerted significant effect on atRA-repressed BACE1 repression (data not shown). Thus, the abundance of retinoid receptors does not appear to directly contribute to the suppressive effect of atRA on BACE1 transcription. We used 1 μm atRA in all of our subsequent in vitro studies because it is equivalent to the in vivo RA concentration in the brain (∼2–5 ng/g) as measured by LC-MS (44, 45).
Because BACE1 was reported to be up-regulated in PPARγ−/− mouse embryonic fibroblast cells and PPARγ agonists were able to suppress BACE1 transcription under inflammatory conditions (46), we tested the effect of atRA in neurons challenged with TNFα (10, 30 ng/ml) and LPS (1 μg/ml) for 4 h. We found that atRA completely reduced BACE1 expression induced by these cytokines to levels even lower than the untreated control (Fig. 4A), which correlated with its effect on BACE1 mRNA (Fig. 4B). Because TNFα induction/release is usually a result of activated NFκB in microglial cells, we predicted that atRA would suppress the NFκB activation, which may partially mediate its repressive effect on BACE1 gene transcription.
FIGURE 4.
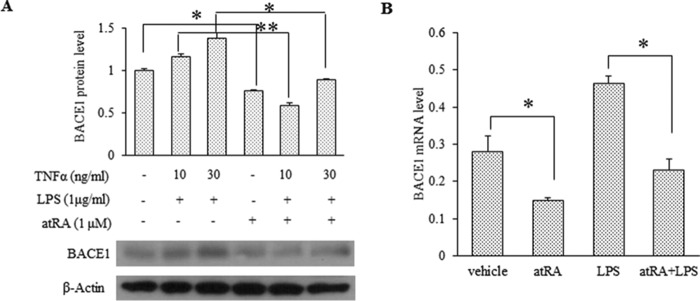
atRA inhibits LPS-stimulated BACE1 expression in rat primary neurons. A, Western blot analysis of BACE1 expression in LPS-stimulated rat primary neurons (DIV15) in the presence or absence of atRA for 24 h. The relative BACE1 protein level was quantified from three independent experiments. Values are means ± S.D. n = 3, *, p < 0.05 and **, p < 0.01 by Student's t test. B, BACE1 mRNA level determined by RT-qPCR. Neurons were stimulated with 1 μg/ml LPS for 6 h in the presence of or in the absence of 1 μm atRA. Data represent expression levels relative to GAPDH × 1000; means ± S.D. n = 3, *, p < 0.05 by Student's t test.
NFκB Mediates the Suppressive Effect of atRA on BACE1 Transcription
Both the NFκB and the PPRE site are adjacently located in the 5′ end of the BACE1 promoter. To identify the atRA targeting region in BACE1 promoter, we generated a series of BACE1 deletion mutants (Fig. 5A) and compared their promoter activity in the presence of atRA (Fig. 5B). The atRA did not suppress F2-F7 promoters, suggesting that the −1541 to −1327 region contains elements that mediate the suppressive effect of atRA. Of note, regulatory elements for NFκB and PPAR have been found in this region that repress BACE1 expression under basal and inflammatory conditions, respectively (47). Indeed, disruption of NFκB element via mutagenesis not only led to increased BACE1 transcription but also abolished the suppressive effect of atRA under basal conditions (Fig. 5C). These data suggest that NFκB plays a critical role in atRA-mediated BACE1 repression. On the other hand, atRA still repressed the promoter activity of BACE1 PPRE mutant, although the effect was reduced by 20% (Fig. 5C), suggesting a minor contribution through the PPRE to atRA-mediated BACE1 repression. In addition, we tested whether atRA would inhibit the LPS induction of the BACE1 promoter. As shown in Fig. 5D, atRA blocked the LPS induction of the wild type but not the NFκB mutant promoter. In N2a cells, atRA eliminated the LPS stimulation of BACE1 mRNA. Furthermore, an IκB kinase (IKK) inhibitor, EF-24, abolished the suppressive effect of atRA on BACE1 mRNA (Fig. 5E); surprisingly, the inhibitor by itself also inhibited BACE1 mRNA.
FIGURE 5.
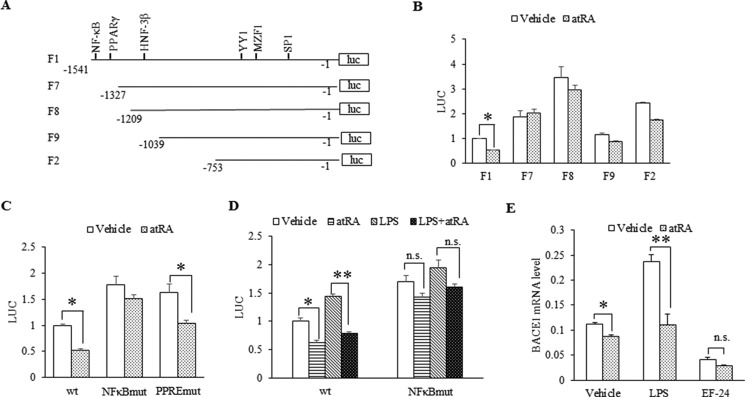
NFκB mediates the suppressive effect of atRA on BACE1 expression. A, schematic diagram of rat BACE1 promoter (1.54 kb) in pGL3-Basic. F7, F8, F9, and F2 represent four BACE1 promoter deletions. luc, luciferase. B, atRA effect on BACE1 promoter and its deletions. Rat BACE1 promoter and its deletions were transfected into HEK 293 cells, and then cells were exposed to 1 μm atRA overnight. C, atRA effect on the activity of BACE1 promoter mutants based on luciferase activity. HEK 293 cells were transfected with rat BACE1 promoter vector and its mutants (NFκBmut and PPREmut) and treated overnight with 1 μm atRA. D, atRA effect on the activity of BACE1 promoter and its mutant NFκBmut with or without LPS stimulation. HEK 293 cells were transfected with rat BACE1 promoter vector or NFκB mutant and then treated overnight with 1 μg/ml LPA in the presence of or in the absence of atRA. n.s. indicates non-statistically significant by Student's t test. E, atRA effect on BACE1 mRNA expression with LPS or EF-24 treatment. N2a cells were treated with 1 μg/ml LPS for 6 h or 5 μm EF-24 for 30 min in the presence of or in the absence of atRA. BACE1 mRNA level was determined by RT-qPCR. Data represent expression levels relative to GAPDH × 1000; means ± S.D. n = 3, *, p < 0.05 and **, p < 0.01 by Student's t test. For luciferase assay, luciferase values were normalized to Renilla luciferase values expressed by co-transfected pRL-SV40 vector. Mean values of vehicle-treated cells were set to 1, means ± S.D. n ≥ 3, *, p < 0.05 and **, p < 0.01 by Student's t test.
atRA Inhibits Inflammatory NFκB Activation
We further tested whether atRA modulates NFκB activity. The IκB kinase enzyme complex is a crucial part of the upstream NFκB signal transduction cascade. The IκBα protein inactivates the NFκB transcription factor by masking the nuclear localization signals of NFκB proteins and keeping them sequestered in an inactive state in the cytoplasm. IκB kinase specifically phosphorylates the inhibitory IκBα protein, resulting in the dissociation of IκBα from NFκB, in turn freeing NFκB to migrate into the nucleus and activate the expression of target genes. In a Western blot analysis with cortical lysates (Fig. 6, A and B), LPS-induced phosphorylation of IκBα was abolished by atRA treatment, which was accompanied by the inhibition of the phosphorylation of NFκB p65 subunit, indicating that atRA inhibits LPS-induced NFκB activation. Moreover, the nuclear localization of NFκB p65 subunit induced by LPS was also inhibited by atRA treatment in N2a cell fraction analysis (Fig. 6C) and immunofluorescence staining assay (Fig. 6D).
FIGURE 6.

atRA inhibits inflammatory NFκB activation. A, C57BL/6 mice were treated with LPS for 6 h with or without atRA administration as described under “Experimental Procedures.” Hippocampal proteins, BACE1, NFκB p65, phosphorylated NFκB p65 (p-NFκB p65), IκB-α, and phosphorylated IκB-α (p-IκB-α) were detected by Western blot, with β-actin as loading control. B, quantification of Western blots from panel A based on three independent experiments *, p < 0.05 and **, p < 0.01 by Student's t test. C and D, atRA effect on the nuclear translocation of NFκB p65. N2a cells were exposed to 1 μg/ml LPS for 1 h in the presence of or in the absence of 10 μm atRA. C, NFκB in both nuclear and cytoplasmic extracts was detected by Western blot. Cytoplasmic α-tubulin was used as loading control. D, NFκB p65 unit was stained in green, and nuclei were visualized by DAPI counterstain. Scale bar = 20 μm.
atRA Disrupts NFκB Binding to the BACE1 Promoter
To test whether atRA influences the binding of NFκB to BACE1 promoter, we performed in vitro and in vivo ChIP assays. As shown in Fig. 7A, in a ChIP assay with rat primary neurons (Fig. 7A), atRA inhibited LPS-induced NFκB p65 binding to BACE1 promoter. Meanwhile, the corepressor NCoR was recruited to the NFκB site in BACE1 promoter. Consistent with the data described earlier, binding of RXRα, PPARγ, and PGC-1α to the PPRE-RXR site was not significantly affected by atRA treatment (Fig. 7B). Most importantly, similar effects of atRA were detected in vivo by ChIP analysis with mouse cortex (Fig. 7C), confirming recruitment of NCoR to the NFκB site.
FIGURE 7.

atRA disrupts NFκB binding to BACE1 promoter. A and B, ChIP assays with cultured neurons. Rat primary neurons were stimulated with 1 μg/ml LPS for 1 h in the presence or absence of 1 μm atRA. PCR with primers for rat BACE1 promoter NFκB site (−1526 to −1507) was shown in panel A and for the PPRE site (−1357 to −1333) in panel B. TFs, transcription factors. C, ChIP assay with mouse cortex. C57L/6 mice were treated for 6 h with 1 mg/kg of LPS following 3 days of atRA pretreatment. PCR was performed with primers for mouse BACE1 promoter NFκB site (−1945 to −1942). ChIP assays were performed with indicated antibodies; IgG was used as negative control. PCR products were analyzed on 2% agarose gel. Quantification was performed based on three independent experiments *, p < 0.05 by paired t test.
Discussion
Nuclear receptors and their ligands are important regulators of gene expression. In our previous work (7), we showed that PPARγ and the coactivator PGC-1α inhibited the expression of BACE1. In this study, we explored the regulation of the BACE1 gene by atRA. Although a large number of genes are induced by atRA (48–50), we found that atRA can suppress BACE1 gene transcription. In addition, we discovered that the mechanism by which atRA suppressed BACE1 gene expression involved the inhibition of NFκB activity. To our knowledge, this is the first report of retinoids blunting the expression of the BACE1 gene. BACE1 is the most important rate-limiting enzyme in AD development, and its expression is modulated by the interplay of the retinoid and NFκB pathways.
RA Negatively Regulates the BACE1 Gene
Although more than 500 target genes have been found to be up-regulated by RA, only ∼3–5 genes were reported to be down-regulated, and only one of these negatively regulated genes was studied at a molecular level (51). The effect of RA on AP-1-responsive genes was reported to involve inhibition of AP-1 binding to the response element (52). A second group also found that RA disrupted the recruitment of AP-1 and coactivators to AP1-regulated promoters (53). In both studies, the direct binding of RAR to the AP-1 site was not required. Other studies have suggested that RA activated PKC to down-regulate the MMP (matrix metalloproteinase) gene (54). Our data indicate that the abundance of the RARs is not limiting with respect to inhibiting BACE1 expression, and we were not able to identify a specific RAR isoform that mediates this effect. Overexpression of RARγ appeared to modestly enhance the suppressive effect of atRA on BACE1 promoter activity (Fig. 3E). However, this isoform was reported to be nearly undetectable in the brain (55) and raises the question of how relevant this specific isoform is to BACE1 regulation. On the other hand, a more recent study indicates a crucial role of this isoform in activity-dependent protein synthesis at synapses pertinent to synaptic plasticity (56).
RA-mediated Regulation of NFκB Signaling
Elegant work from Bromhoff's group (17) using a transgenic murine NFκB-luciferase reporter model demonstrated an over 2-fold increase in NFκB activity in immune organs (lymph node and thymus) in vitamin A-deficient mice. This elevation of NFκB activity could be reversed by a single administration of atRA. Interestingly, in the same model system, a single injection of the RAR pan-antagonist, AGN 194310, also resulted in a marked, transient induction of whole-body luminescence. It remains to be clarified which RARs are involved in mediating RA's inhibition of LPS-induced NFκB activity in our models of neuroinflammation. Further studies from the Bromhoff laboratory (57) showed that atRA repressed LPS-induced whole-body luminescence.
Regarding the mechanism by which atRA modulates NFκB activity, our compelling evidence based on in vitro and in vivo biochemical and immunocytochemical approaches demonstrated that atRA activates and stabilizes IκBα via reducing its phosphorylation status. In addition, atRA blocks the specific p65 subunit of NFκB and its nuclear translocation. Although we did not directly measure NFκB-IκBα complexes, our data suggest an enhanced IκBα protein stability and association with the p65/50 upon atRA treatment. We did not detect significant effect of atRA on the gene expression of p65 (data not shown). Furthermore, atRA modulates the expression of proinflammatory factors and their receptors. The expression of TNFα and IL-6 was significantly decreased in atRA-treated HFD mice and LPS-treated mice (Figs. 1, E and F, and 2, A and B). Besides inducing the expression of proinflammatory factors, systemic LPS administration elevates pulmonary TLR4 expression (58). By quantitative RT-PCR analysis, we observed a significant increase of TLR4 expression in LPS-treated neurons and N2a cells. The LPS-induced TLR4 expression was dramatically suppressed in response to atRA treatment (Fig. 8). These results suggest that atRA likely suppresses LPS-induced BACE1 expression through LPS receptor. Consistent with this observation, sequence analysis reveals NFκB and PPARγ/α-RXRα binding sites in TLR4 promoter.
FIGURE 8.
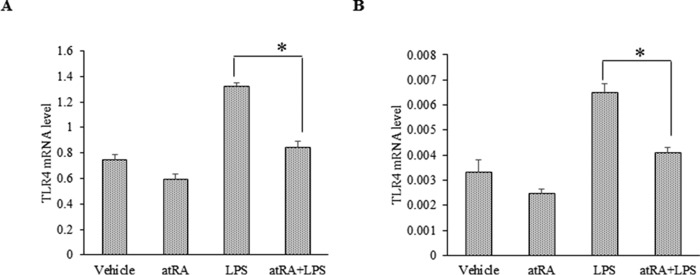
atRA suppresses BACE1 via down-regulating LPS receptor TLR4. A and B, RT-qPCR analysis of TLR4 mRNAs in N2a (A) and rat primary neurons (B). Cells were treated with LPS (1 μg/ml) for 6 h with and without atRA (1 μm). Data represent expression levels relative to GAPDH × 1000 (for N2a) and to β-actin × 1000 (neurons); means ± S.D. n = 3, *, p < 0.05 by Student's t test.
It is well known that cytoplasmic localization of inactive NFκB-IκBα complexes is controlled by both the masking of the nuclear import sequence of NFκB as well as its active nuclear export. There was an additional mechanism reported (59) in which the cytoplasmic protein RasGAP SH3-binding protein 2 (G3BP2), which interacts with both IκBα and NFκB-IκB complexes, anchored IκBα or NFκB-IκBα in the cytoplasm. This novel mechanism has not been confirmed in neurons following its initial discovery in cancer cells. Interestingly, our pilot Affymetrix gene array study reveals a 3-fold up-regulation of G3BP2 in HFD C57BL/6 mouse brains, which was completely reversed by atRA to levels of the control diet mice.5 Although we did not see altered expression of this G3BP2 gene by RA under basal conditions or with LPS addition in neurons, we cannot rule out the possibility of altered subcellular protein distribution of G3BP2 protein by atRA. The potential role of this scaffolding protein on modulation of NFκB signaling in neurons under inflammatory conditions warrants further investigation.
Does atRA Mediate Differential Mechanisms of NFκB Regulation under Basal Versus Inflammatory Conditions?
Our data clearly showed that atRA treatment suppressed BACE1 expression under both normal (basal) and inflammatory conditions, in vitro and in vivo. A 27% reduction in BACE1 protein expression was observed in atRA-treated C57BL/6 mice as compared with the vehicle-treated group. Moreover, we also demonstrated opposing effects of disrupting the NFκB binding site on BACE1 promoter activity under basal and inflammatory conditions (Fig. 5), consistent with previous studies (47). Conflicting data have been published on the inhibitory or stimulatory effects of NFκB on BACE1 expression (48, 60–62). Our work confirms the previous study (47) showing that NFκB is a repressor of BACE1 under unstressed neurons but stimulates BACE1 in activated astrocytes. Of note, although we observed a blunting effect of atRA on activated astrocytes (glial fibrillary acidic protein (GFAP)-positive cells, Fig. 2E), especially in the hippocampal CA1 region, we found the most dominant effects of atRA on blunting neuronal BACE1 expression in both hippocampi and cortex (i.e. pyramidal neurons).
RA in CNS Inflammation
The anti-inflammatory effect of RA has been investigated for several decades, primarily in cancer, diabetes, and rheumatoid arthritis. However, it has not been studied in the CNS. There was only one study showing RA counteracting the LPS-induced inflammatory regulation of cyclooxygenase (COX)-2 mRNA and protein in astrocytes and thereby reducing the synthesis of prostaglandin E2 (PGE2) by ∼60%. This work suggests that RA might be effective in suppressing inflammatory processes in the brain by inhibiting prostaglandin synthesis (63). In the present study, we demonstrated that atRA down-regulates BACE1 expression under inflammatory conditions in Tg2576, HFD, and LPS-inflamed mouse brains. Together with its effects on blunting inflammatory cytokines, it raises the possibility of using atRA or other RA analogs in treatment of neuroinflammation.
Retinoid Therapy for Sporadic AD
Neuroinflammation is a local and chronic feature in Alzheimer brains that might result from and further cause the Aβ deposits (i.e. forming vicious cycles). Hence, anti-inflammatory activity and inhibition of Aβ generation are both crucial for the treatment of AD. Our current work demonstrated the dual effects of atRA on blunting BACE1 expression and neuroinflammation, suggesting a double-edged sword of atRA therapy for AD. The regulation of NFκB signaling by atRA also provides a molecular link between these two pathogenic processes. It should be stressed that although the currently available AD mouse models are largely based on overexpressing one or more familial mutants of APP and/or presenilin genes, the majority of late-onset sporadic patients (> 99%) do not have a mutation that causes an increase in APP processing. Therefore, our data on atRA therapy of the HFD mice not only show reduced BACE1 expression but also have benefits in preventing weight gain, insulin resistance, and neuroinflammation and enhancing cognitive performance. Together, this work displays promise for using retinoid therapy for sporadic AD, in particular those cases with complication of diabetes.
Author Contributions
R. W., S. C., Y. L., S. D., Y. X., and Q. X. performed experiments, and R. W., E. A. P., and F. F. L. analyzed data and wrote the paper.
Acknowledgment
We thank Dr. Xiaokun Zhang for the RXRα- and RAR-expressing plasmids.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 AG031893 and R21 AG041934 (to F. -F. L.) and R21NS083908 (to F. -F. L. and E. A. P.), and by Alzheimer Association Grant IIRG-11-204030 (to F. -F. L.). The authors declare that they have no conflicts of interest with the contents of this article.
F.-F. Liao, unpublished data.
- AD
- Alzheimer disease
- Aβ
- amyloid-β
- APP
- amyloid precursor protein
- RA
- retinoic acid
- atRA
- all-trans-retinoic acid
- PPRE
- PPAR-responsive element
- PPAR
- peroxisome proliferator-activated receptor
- PGC-1α
- PPARγ coactivator 1-α
- RAR
- retinoic acid receptor
- RXR
- retinoid X receptor
- TLR4
- Toll-like receptor-4
- DMSO
- dimethyl sulfoxide
- HFD
- high fat diet
- N2a
- Neuro-2a
- RT-qPCR
- quantitative real-time PCR
- DIV15
- days in vitro 15.
References
- 1. Sastre M., Klockgether T., Heneka M. T. (2006) Contribution of inflammatory processes to Alzheimer's disease: molecular mechanisms. Int. J. Dev. Neurosci. 24, 167–176 [DOI] [PubMed] [Google Scholar]
- 2. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 3. Vassar R., Kovacs D. M., Yan R., Wong P. C. (2009) The β-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J. Neurosci. 29, 12787–12794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rossner S., Sastre M., Bourne K., Lichtenthaler S. F. (2006) Transcriptional and translational regulation of BACE1 expression: implications for Alzheimer's disease. Prog. Neurobiol. 79, 95–111 [DOI] [PubMed] [Google Scholar]
- 5. Guglielmotto M., Aragno M., Tamagno E., Vercellinatto I., Visentin S., Medana C., Catalano M. G., Smith M. A., Perry G., Danni O., Boccuzzi G., Tabaton M. (2012) AGEs/RAGE complex upregulates BACE1 via NFκB pathway activation. Neurobiol. Aging 33, 196.e13–27, 10.1016/j.neurobiolaging.2010.05.026 [DOI] [PubMed] [Google Scholar]
- 6. Kwak Y. D., Wang R., Li J. J., Zhang Y. W., Xu H., Liao F. F. (2011) Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol. Neurodegener. 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang R., Li J. J., Diao S., Kwak Y. D., Liu L., Zhi L., Büeler H., Bhat N. R., Williams R. W., Park E. A., Liao F. F. (2013) Metabolic stress modulates Alzheimer's β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab. 17, 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mangelsdorf D. J., Evans R. M. (1995) The RXR heterodimers and orphan receptors. Cell 83, 841–850 [DOI] [PubMed] [Google Scholar]
- 9. Chambon P. (1996) A decade of molecular biology of retinoid acid receptors. FASEB J. 10, 940–954 [PubMed] [Google Scholar]
- 10. Lane M. A., Bailey S. J. (2005) Role of retinoid signaling in the adult brain. Prog. Neurobiol. 75, 275–293 [DOI] [PubMed] [Google Scholar]
- 11. Blomhoff R., Blomhoff H. K. (2006) Overview of retinoid metabolism and function. J. Neurobiol. 66, 606–630 [DOI] [PubMed] [Google Scholar]
- 12. Maden M. (2007) Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 8, 755–765 [DOI] [PubMed] [Google Scholar]
- 13. Kirchmeyer M., Koufany M., Sebillaud S., Netter P., Jouzeau J. Y., Bianchi A. (2008) All-trans-retinoic acid suppresses interleukin-6 expression in interleukin-1-stimulated synovial fibroblasts by inhibition of ERK1/2 pathway independently of RAR activation. Arthritis Res. Ther. 10, R141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mehta K., McQueen T., Tucker S., Pandita R., Aggarwal B. B. (1994) Inhibition by all-trans-retinoic acid of tumor necrosis factor and nitric oxide production by peritoneal macrophages. J. Leukoc. Biol. 55, 336–342 [DOI] [PubMed] [Google Scholar]
- 15. Nozaki Y., Yamagata T., Sugiyama M., Ikoma S., Kinoshita K., Funauchi M. (2006) Anti-inflammatory effect of all-trans-retinoic acid in inflammatory arthritis. Clin. Immunol. 119, 272–279 [DOI] [PubMed] [Google Scholar]
- 16. Zitnik R. J., Kotloff R. M., Latifpour J., Zheng T., Whiting N. L., Schwalb J., Elias J. A. (1994) Retinoic acid inhibition of IL-1-induced IL-6 production by human lung fibroblasts. J. Immunol. 152, 1419–1427 [PubMed] [Google Scholar]
- 17. Austenaa L. M., Carlsen H., Ertesvag A., Alexander G., Blomhoff H. K., Blomhoff R. (2004) Vitamin A status significantly alters nuclear factor-κB activity assessed by in vivo imaging. FASEB J. 18, 1255–1257 [DOI] [PubMed] [Google Scholar]
- 18. Hong K., Zhang Y., Guo Y., Xie J., Wang J., He X., Lu N., Bai A. (2014) All-trans-retinoic acid attenuates experimental colitis through inhibition of NFκB signaling. Immunol. Lett. 162, 34–40 [DOI] [PubMed] [Google Scholar]
- 19. Ono K., Yamada M. (2012) Vitamin A and Alzheimer's disease. Geriatr. Gerontol. Int. 12, 180–188 [DOI] [PubMed] [Google Scholar]
- 20. Jiménez-Jiménez F. J., Molina J. A., de Bustos F., Ortí-Pareja M., Benito-León J., Tallón-Barranco A., Gasalla T., Porta J., Arenas J. (1999) Serum levels of β-carotene, α-carotene and vitamin A in patients with Alzheimer's disease. Eur. J. Neurol. 6, 495–497 [DOI] [PubMed] [Google Scholar]
- 21. Zaman Z., Roche S., Fielden P., Frost P. G., Niriella D. C., Cayley A. C. (1992) Plasma concentrations of vitamins A and E and carotenoids in Alzheimer's disease. Age Ageing 21, 91–94 [DOI] [PubMed] [Google Scholar]
- 22. Goodman A. B., Pardee A. B. (2003) Evidence for defective retinoid transport and function in late onset Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 2901–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goodman A. B. (2006) Retinoid receptors, transporters, and metabolizers as therapeutic targets in late onset Alzheimer disease. J. Cell. Physiol. 209, 598–603 [DOI] [PubMed] [Google Scholar]
- 24. Enderlin V., Pallet V., Alfos S., Dargelos E., Jaffard R., Garcin H., Higueret P. (1997) Age-related decreases in mRNA for brain nuclear receptors and target genes are reversed by retinoic acid treatment. Neurosci. Lett. 229, 125–129 [DOI] [PubMed] [Google Scholar]
- 25. Misner D. L., Jacobs S., Shimizu Y., de Urquiza A. M., Solomin L., Perlmann T., De Luca L. M., Stevens C. F., Evans R. M. (2001) Vitamin A deprivation results in reversible loss of hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 98, 11714–11719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cocco S., Diaz G., Stancampiano R., Diana A., Carta M., Curreli R., Sarais L., Fadda F. (2002) Vitamin A deficiency produces spatial learning and memory impairment in rats. Neuroscience 115, 475–482 [DOI] [PubMed] [Google Scholar]
- 27. Etchamendy N., Enderlin V., Marighetto A., Pallet V., Higueret P., Jaffard R. (2003) Vitamin A deficiency and relational memory deficit in adult mice: relationships with changes in brain retinoid signalling. Behav. Brain Res. 145, 37–49 [DOI] [PubMed] [Google Scholar]
- 28. Corcoran J. P., So P. L., Maden M. (2004) Disruption of the retinoid signaling pathway causes a deposition of amyloid β in the adult rat brain. Eur. J. Neurosci. 20, 896–902 [DOI] [PubMed] [Google Scholar]
- 29. Etchamendy N., Enderlin V., Marighetto A., Vouimba R. M., Pallet V., Jaffard R., Higueret P. (2001) Alleviation of a selective age-related relational memory deficit in mice by pharmacologically induced normalization of brain retinoid signaling. J. Neurosci. 21, 6423–6429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ding Y., Qiao A., Wang Z., Goodwin J. S., Lee E. S., Block M. L., Allsbrook M., McDonald M. P., Fan G. H. (2008) Retinoic acid attenuates β-amyloid deposition and rescues memory deficits in an Alzheimer's disease transgenic mouse model. J. Neurosci. 28, 11622–11634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Malaspina A., Michael-Titus A. T. (2008) Is the modulation of retinoid and retinoid-associated signaling a future therapeutic strategy in neurological trauma and neurodegeneration? J. Neurochem. 104, 584–595 [DOI] [PubMed] [Google Scholar]
- 32. Lee H. P., Casadesus G., Zhu X., Lee H. G., Perry G., Smith M. A., Gustaw-Rothenberg K., Lerner A. (2009) All-trans-retinoic acid as a novel therapeutic strategy for Alzheimer's disease. Expert Rev. Neurother. 9, 1615–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shudo K., Fukasawa H., Nakagomi M., Yamagata N. (2009) Towards retinoid therapy for Alzheimer's disease. Curr. Alzheimer Res. 6, 302–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lerner A. J., Gustaw-Rothenberg K., Smyth S., Casadesus G. (2012) Retinoids for treatment of Alzheimer's disease. Biofactors 38, 84–89 [DOI] [PubMed] [Google Scholar]
- 35. Koryakina A., Aeberhard J., Kiefer S., Hamburger M., Küenzi P. (2009) Regulation of secretases by all-trans-retinoic acid. FEBS J. 276, 2645–2655 [DOI] [PubMed] [Google Scholar]
- 36. Tippmann F., Hundt J., Schneider A., Endres K., Fahrenholz F. (2009) Up-regulation of the α-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 23, 1643–1654 [DOI] [PubMed] [Google Scholar]
- 37. Lee J. W., Lee Y. K., Yuk D. Y., Choi D. Y., Ban S. B., Oh K. W., Hong J. T. (2008) Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of β-amyloid generation. J. Neuroinflammation 5, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Espinosa-Oliva A. M., de Pablos R. M., Herrera A. J. (2013) Intracranial injection of LPS in rat as animal model of neuroinflammation. Methods Mol. Biol. 1041, 295–305 [DOI] [PubMed] [Google Scholar]
- 39. Thirumangalakudi L., Prakasam A., Zhang R., Bimonte-Nelson H., Sambamurti K., Kindy M. S., Bhat N. R. (2008) High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 106, 475–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen Y., Zhou K., Wang R., Liu Y., Kwak Y. D., Ma T., Thompson R. C., Zhao Y., Smith L., Gasparini L., Luo Z., Xu H., Liao FF. (2009) Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer's amyloid peptides via up-regulating BACE1 transcription. Proc. Natl. Acad. Sci. U.S.A. 106, 3907–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valero J., Mastrella G., Neiva I., Sánchez S., Malva J. O. (2014) Long-term effects of an acute and systemic administration of LPS on adult neurogenesis and spatial memory. Front. Neurosci. 8, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Culpan D., Cram D., Chalmers K., Cornish A., Palmer L., Palmer J., Hughes A., Passmore P., Craig D., Wilcock G. K., Kehoe P. G., Love S. (2009) TNFR-associated factor-2 (TRAF-2) in Alzheimer's disease. Neurobiol. Aging 30, 1052–1060 [DOI] [PubMed] [Google Scholar]
- 43. He P., Zhong Z., Lindholm K., Berning L., Lee W., Lemere C., Staufenbiel M., Li R., Shen Y. (2007) Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer's mice. J. Cell Biol. 178, 829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmidt C. K., Brouwer A., Nau H. (2003) Chromatographic analysis of endogenous retinoids in tissues and serum. Anal. Biochem. 315, 36–48 [DOI] [PubMed] [Google Scholar]
- 45. Kane M. A., Chen N., Sparks S., Napoli J. L. (2005) Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem. J. 388, 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sastre M., Dewachter I., Rossner S., Bogdanovic N., Rosen E., Borghgraef P., Evert B. O., Dumitrescu-Ozimek L., Thal D. R., Landreth G., Walter J., Klockgether T., van Leuven F., Heneka M. T. (2006) Nonsteroidal anti-inflammatory drugs repress β-secretase gene promoter activity by the activation of PPARγ. Proc. Natl. Acad. Sci. U.S.A. 103, 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bourne K. Z., Ferrari D. C., Lange-Dohna C., Rossner S., Wood T. G., Perez-Polo J. R. (2007) Differential regulation of BACE1 promoter activity by nuclear factor-κB in neurons and glia upon exposure to β-amyloid peptides. J. Neurosci. Res. 85, 1194–1204 [DOI] [PubMed] [Google Scholar]
- 48. Balmer J. E., Blomhoff R. (2002) Gene expression regulation by retinoid acid. J. Lipid Res. 43, 1773–1808 [DOI] [PubMed] [Google Scholar]
- 49. Wallen-Mackenzie A., Mata de Urquiza A., Petersson S., Rodriguez F. J., Friling S., Wagner J., Ordentlich P., Lengqvist J., Heyman R. A., Arenas E., Perlmann T. (2003) Nurr-RXR heterodimers mediate RXR ligand-induced signaling in neuronal cells. Genes Dev. 17, 3036–3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bastien J., Rochette-Egly C. (2004) Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 328, 1–16 [DOI] [PubMed] [Google Scholar]
- 51. le Maire A., Alvarez S., Shankaranarayanan P., Lera A. R., Bourguet W., Gronemeyer H. (2012) Retinoid receptors and therapeutic applications of RAR/RXR modulators. Curr. Top. Med. Chem. 12, 505–527 [DOI] [PubMed] [Google Scholar]
- 52. Schüle R., Rangarajan P., Yang N., Kliewer S., Ransone L. J., Bolado J., Verma I. M., Evans R. M. (1991) Retinoic acid is a negative regulator of AP-1-responsive genes. Proc. Natl. Acad. Sci. U.S.A. 88, 6092–6096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Benkoussa M., Brand C., Delmotte M. H., Formstecher P., Lefebvre P. (2002) Retinoic acid receptors inhibit AP1 activation by regulating extracellular signal-regulated kinase and CBP recruitment to an AP1-responsive promoter. Mol. Cell. Biol. 22, 4522–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dutta A., Sen T., Chatterjee A. (2010) All-trans-retinoid acid (ATRA) downregulates MMP-9 by modulating its regulatory molecules. Cell Adh. Migr. 4, 409–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zetterström R. H., Lindqvist E., Mata de Urquiza A., Tomac A., Eriksson U., Perlmann T., Olson L. (1999) Role of retinoids in the CNS: differential expression of retinoid binding proteins and receptors and evidence for presence of retinoic acid. Eur. J. Neurosci. 11, 407–416 [DOI] [PubMed] [Google Scholar]
- 56. Poon M. M., Chen L. (2008) Retinoic acid-gated sequence-specific translational control by RARα. Proc. Natl. Acad. Sci. U.S.A. 105, 20303–20308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Austenaa L. M., Carlsen H., Hollung K., Blomhoff H. K., Blomhoff R. (2009) Retinoic acid dampens LPS-induced NFκB activity: results from human monoblasts and in vivo imaging of NFκB reporter mice. J. Nutr. Biochem. 20, 726–734 [DOI] [PubMed] [Google Scholar]
- 58. Janardhan K. S., McIsaac M., Fowlie J., Shrivastav A., Caldwell S., Sharma R. K., Singh B. (2006) Toll like receptor-4 expression in lipopolysaccharide induced lung inflammation. Histol. Histopathol. 21, 687–696 [DOI] [PubMed] [Google Scholar]
- 59. Prigent M., Barlat I., Langen H., Dargemont C. (2000) IκBα and IκBα/NFκ B complexes are retained in the cytoplasm through interaction with a novel partner, RasGAP SH3-binding protein 2. J. Biol. Chem. 275, 36441–36449 [DOI] [PubMed] [Google Scholar]
- 60. Buggia-Prevot V., Sevalle J., Rossner S., Checler F. (2008) NFκB-dependent control of BACE1 promoter transactivation by Aβ42. J. Biol. Chem. 283, 10037–10047 [DOI] [PubMed] [Google Scholar]
- 61. Chen C. H., Zhou W., Liu S., Deng Y., Cai F., Tone M., Tone Y., Tong Y., Song W. (2012) Increased NFκB signaling up-regulates BACE1 expression and its therapeutic potential in Alzheimer's disease. Int. J. Neuropsychopharmacol. 15, 77–90 [DOI] [PubMed] [Google Scholar]
- 62. Marwarha G., Raza S., Meiers C., Ghribi O. (2014) Leptin attenuates BACE1 expression and amyloid-β genesis via the activation of SIRT1 signaling pathway. Biochim. Biophys. Acta 1842, 1587–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kampmann E., Johann S., van Neerven S., Beyer C., Mey J. (2008) Anti-inflammatory effect of retinoic acid on prostaglandin synthesis in cultured cortical astrocytes. J. Neurochem. 106, 320–332 [DOI] [PubMed] [Google Scholar]