
FIGURE 8.
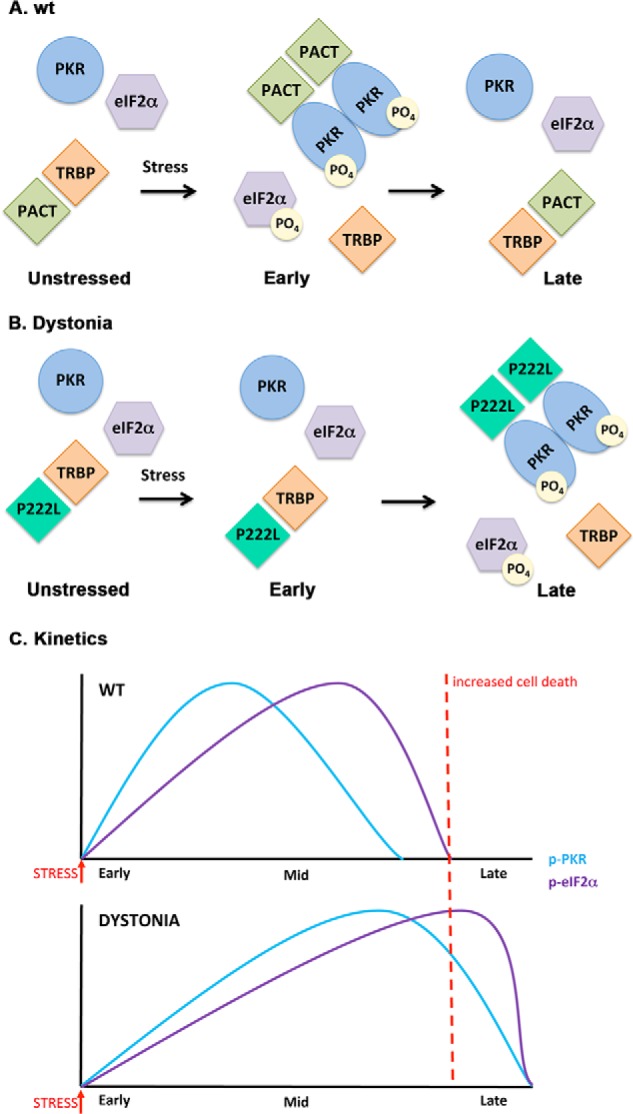
A schematic model of PKR activation in WT and dystonia cells. A, WT cells. As previously established (Refs. 19, 21, 22), in the absence of stress, PACT heterodimerizes with TRBP, PKR is catalytically inactive, and eIF2α is not phosphorylated. At early time points after ER stress, PACT dissociates from TRBP due to its phosphorylation, forms homodimers that bind to PKR with high affinity, and activates its kinase activity leading to eIF2α phosphorylation. At late time points after ER stress, cells recover by forming TRBP-PACT heterodimers and turning off PKR and eIF2α phosphorylation. B, dystonia cells. In the absence of stress, P222L mutant forms heterodimers with TRBP, PKR is catalytically inactive and eIF2α is not phosphorylated. At early time points after ER stress, P222L remains bound to TRBP, and PKR and eIF2α phosphorylation is inhibited. At late time points after ER stress, P222L dissociates from TRBP, forms homodimers that bind to PKR with high affinity and activate its kinase activity leading to eIF2α phosphorylation. Thus, at late time point cells are unable to recover efficiently from ER stress because PKR and eIF2α remain phosphorylated. C, schematic representation of the altered PKR activation and eIF2α phosphorylation kinetics in dystonia cells. Blue line, PKR phosphorylation; purple line, eIF2α phosphorylation. Red dotted line, a threshold time point. If PKR and eIF2α remain in their phosphorylated state beyond this time point, recovery from ER stress is prevented, resulting in increased apoptosis.