

Abstract
Background
A peptide derived from Antrum Mucosal Protein (AMP)-18 (gastrokine-1) reduces the extent of mucosal erosions and clinical severity in mice with dextran sulfate sodium (DSS)-induced colonic injury. The present study set out to determine if AMP peptide was also therapeutic for immune- and cytokine-mediated mouse models of intestinal injury and inflammatory bowel diseases (IBD) by enhancing and stabilizing tight junctions (TJs).
Methods
Therapeutic effects of AMP peptide were examined in interleukin-10 deficient and a T cell adoptive transfer models of colitis in immunodeficient recombinase activating gene-1 knock-out (RAG-1−/−) mice. Mechanisms by which AMP peptide enhances barrier function and structure were studied ex vivo using intestine and colon from mice given lipopolysaccharide (LPS), and in AMP-18 deficient mice given DSS.
Results
In interleukin-10 deficient mice given piroxicam, AMP peptide enhanced recovery after weight loss, protected against colon shortening and segmental dilation, and reduced the colitis activity score. In the T cell transfer model, treatment with the peptide protected against colon shortening. In mice given LPS in vivo to induce gut injury, AMP peptide prevented the onset of, and reversed established intestinal hyperpermeability by targeting TJ proteins and perijunctional actin. AMP-18-deficient mice challenged with DSS exhibited increased mortality, developed erosions in the colon, and had lower levels of ZO-1 in TJs than heterozygous littermates or wild-type mice.
Conclusions
The results indicate that AMP-18/peptide may serve a protective role against injury along the GI mucosal barrier, and recommend further development of AMP peptide as a novel agent to treat patients with IBD.
Keywords: Antrum Mucosal Protein (AMP)-18, gastrokine-1, IBD, Tight Junctions, ZO-1
INTRODUCTION
Crohn’s disease and ulcerative colitis are diseases of unknown etiology that cause substantial morbidity throughout the world. The inflammatory damage to the bowel mucosa that occurs in these patients results in many serious complications, including an increased incidence of colon cancer. Current therapeutic strategies are directed at suppressing the humoral and cellular aspects of immune and inflammatory responses in patients with IBD. These regimens require local or systemic administration of agents which, in general, are incompletely effective and cause significant morbidities that often preclude their long-term use. Side effects include infections, risk of neoplasia, teratogenicity, and numerous metabolic derangements such as hypertension, diabetes, obesity and bone disease. Clearly new agents are needed to treat this condition.
We have characterized a novel 18 kDa protein synthesized by mucosal cells of the gastric antrum called Antrum Mucosal Protein (AMP)-18, or gastrokine-1 (1). This protein, and a synthetic 21-mer AMP peptide derived from its central domain, have actions in cell culture and mice with colonic injury (2, 3) that could be developed into a new agent to treat inflammatory bowel diseases (IBD).
AMP-18 and AMP peptide were each found to function as a mitogen, motogen, and cell protective agent for intestinal epithelial cells in culture (1–3). Treatment with AMP peptide protected against disruption of barrier function and structure in monolayer cultures of intestinal epithelial cells subjected to oxidant-, indomethacin-, dextran sulfate sodium (DSS)-, or low calcium-mediated injury. Increasing accumulation of specific tight and adherens junction proteins, preventing their loss during injury, and facilitating assembly of new tight junctions (TJs) following barrier disruption were found to be some of the possible mechanisms by which AMP peptide exerted its cell protective effects in these cultures used to model the mucosal barrier (4, 5). The peptide also enhanced accumulation of specific TJ proteins (occludin and ZO-1) in the colonic mucosa when given to normal mice. The possibility of AMP peptide having therapeutic efficacy was entertained when we observed that administration of DSS reduced the amount of these two TJ proteins in mouse colonic tissue before the appearance of histological erosions (2, 3). As predicted by these observations, AMP peptide treatment of DSS-induced colonic injury in mice delayed the appearance of bloody stool, and reduced the extent of weight loss, colon shortening and mucosal erosions (6). These biological actions in the colon following systemic administration by either the subcutaneous (s.c.) or intraperitoneal (i.p.) route, without any side effects, suggest that AMP peptide could be developed into a therapeutic agent to treat IBD.
The objectives of this study are to determine: (1) if the protective and regenerative effects of AMP peptide that target TJs in the colonic mucosal barrier in DSS-mediated injury in mice extend to other (immune, LPS) models of human IBD, and (2) if AMP-18 expression in normal animals constitutively protects the GI tract against injury distal to the pyloric valve.
MATERICALS AND METHODS
Materials
Chemically synthesized AMP peptide (LDALVKEKKLQGKGPGGPPPK) and a scrambled peptide (GKPLGQPGKVPKLDGKEPLAK) were prepared at GenScript (Piscataway, NJ) as described previously (7). AMP peptide and full-length recombinant AMP-18 protein were found to be equally effective in triggering cellular responses, as previously reported (6–8). Occludin and ZO-1 antibodies were obtained from Life Technologies (Gaithersburg, MD). LPS and other reagents were purchased from Sigma-Aldrich unless otherwise specified. Mice were purchased from Jackson Laboratory (Bar Harbor, ME). Approval for use of mice and the protocols was obtained from the Animal Care and Use Committee of the University of Chicago.
Methods
Colitis model in IL-10 deficient mice exposed to piroxicam and treated with AMP peptide
Exposure of IL-10 deficient (IL-10−/−) mice on a C57/BL6 background to the non-steroidal anti-inflammatory drug (NSAID) piroxicam results in development of moderate to severe colitis within 2 weeks that is more uniform and exhibits greater disease penetrance than in IL-10−/− mice not given the drug (9–11). The NSAID-induced colitis is characterized by marked infiltration of the colon with CD4+ T cells and macrophages that exhibit a Th1-type phenotype. Twenty-two C57/BL6 IL-10−/− (age 5–6 weeks) mice were given piroxicam (200 ppm) mixed into the chow and pelleted, for 11 days. Mice were then switched to normal diet and divided into 2 groups of 11 animals randomly; one group was given daily s.c. injections with AMP peptide (25 mg/kg) in phosphate-buffered saline (PBS, vehicle), and the other group, PBS alone (control). In one experiment, mice were treated with AMP peptide or vehicle for 9 days, and in a second experiment, for 18 days. After treatment, animals were anesthetized, killed, and the colons were harvested. The length of the entire colon and length of dilated colonic segments with a diameter ≥ 3.5 mm were measured. The colons were then fixed in 10% phosphate-buffered formalin and embedded in paraffin. Sections were cut at 4 μm and stained with hematoxylin and eosin for examination by light microscopy. The slides were reviewed in a blinded fashion, using a colitis activity scoring scheme outlined by Berg et al. (9), which includes measurements of the severity, number of submucosal lesions, and the extent of mononuclear cell infiltration in the lamina propria.
T cell dependent murine model of colitis
An adoptive transfer model of colitis was created in immunodeficient recombinase activating gene-1 knock-out (RAG-1−/−) mice (on C57BL/6 background) that were depleted of regulatory T cells and reconstituted with splenic CD45RBhigh CD4+ T cells. Colonic inflammation usually starts 4–6 weeks after transfer, has a predictable time course, is reproducible, and has a similar inflammatory mediator profile as some forms of the disease in humans [17–19]. CD4+ T cells were isolated from the spleens of 5- to 8-week old female pathogen-free C57BL/6 mice (MACS® CD4+ T cell isolation kit, Miltenyi Biotec, San Diego, CA) and sorted by flow cytometry into fractions containing the 35% of CD4+ cells with the brightest CD45RB-staining (CD4+CD45RBhigh), and the 35% with the lowest CD45RB-staining (CD4+CD45RBlow) cells. Sorted CD4+CD45RBhigh cells (4 X 105) (95% pure) were transferred i.p. into recipient 5-to 8-week old female RAG-1−/− mice. RAG-1−/− mice that did not receive cells served as controls. Development of colitis in the mice was followed by documenting progressive weight loss and disease activity (12, 13) by monitoring hunching and wasting of the mouse, colon thickening, stool consistency, and the appearance of gross blood in the stool. In addition, colonoscopy was used on mice to determine the extent and severity of IBD without sacrificing the animal. When the animals given CD45RBhigh CD4+ T cells lost 5% body weight, or exhibited diarrhea or hematochezia, treatment with AMP peptide or vehicle was initiated.
Ex vivo permeability assay of colon and jejunum
Lipopolysaccharide (LPS) has been used experimentally to induce barrier disruption and gut hyperpermeability in vivo, probably by activating innate immune cell cytokine production of TNF-α, a major mediator in the pathogenesis of IBD (14). Increased permeability of the GI tract was induced in C57BL/10 mice with a single dose of E. coli LPS (data not shown). To determine if AMP peptide could protect against the effects of LPS, AMP peptide in PBS (25 mg/kg, s.c.) was administered for 5 days, 1 day, or 6 hr before an injection of LPS (100 μg, i.p.) (Fig. 4). Thirty minutes later the intestines or colons were quickly removed, 2-cm long segments of colon or jejunum were cut, and the bowel lumen was washed out. A sac filled with a solution of fluorescein-isothiocyanate (FITC)-dextran (Mr 4 kD) was created and incubated in buffered saline at 37°C. Mucosal to serosal permeability was assessed by measuring the appearance of fluorescence in the buffer during the next 75 minutes (15).
Fig. 4. AMP peptide protects against LPS-induced gut hyperpermeability of mouse colon and jejunum in vivo.

C57BL/10 mice (n=3 in each group) were treated with AMP peptide (25 mg/kg, s.c.) and then injected with LPS (100 μg, i.p.), or injected with LPS alone. Animals were killed 30 min after LPS injection. Segments (2-cm long) of colon or jejunum were used for permeability assay by filling these sacs with FITC-dextran (Mr 4 kDa). Flux of FITC-dextran from mucosa to serosa during the next 75 minutes was measured by fluorometry. Left panel, colon segments of mice given LPS alone (red line), or AMP peptide given 5 days before LPS (blue line). Right panel, jejunum segments of mice given LPS alone (red line), a single dose of AMP peptide 1 day (blue line) or 6 h (green line) before LPS, or no treatment (broken black line; control). Values are FITC flux per cm of intestine for three experiments.
To determine the effects of AMP peptide on established gut hyperpermeability, mice were given LPS, and 15 hr later, when permeability was markedly increased compared to control animals (14), a single s.c. injection of AMP peptide (25 mg/kg, in PBS) was given. Six hr later the mice were killed, the gut was harvested, and FITC-dextran flux in colon or jejunum sacs was assayed as described above. In addition, the intestinal tissues were processed, embedded in paraffin wax, cut into 4-μm sections and subjected to immunohistochemical (IHC) staining to examine distribution of the TJ junction proteins, occludin and ZO-1, as well as perijunctional actin, which were subsequently visualized by immunofluorescence conjugated secondary antibodies. The slides were then observed and imaged with laser scanning confocal microscopy (LSCM).
ZO-1 expression in AMP-18 knockout mice treated with DSS
AMP-18 knockout (AMP-18−/−) mice were generated by replacing the AMP-18 gene with a BAC-based vector expressing β-galactosidase from the E. coli lacZ gene and neomycin phosphotransferase coding sequences. Heterozygous (AMP-18+/−) embryonic stem cells from C57BL/6 mice were implanted into C57BL/6 embryos to create AMP-18+/− mice. The growth and development of the first generation of AMP-18−/− mice was observed over 24 weeks. AMP-18−/− mice grew and developed normally compared to AMP-18+/+ or AMP-18+/− littermates, showing no spontaneous gastroenteritis.
To assess the effects of deletion of the AMP-18 gene on colonic barrier function and structure, wild type (WT), AMP-18+/−, and AMP-18−/− mice (6-week old) were given drinking water containing 2% DSS for 5 days, then switched back to water, and followed until day13. Animals were sacrificed by cervical dislocation after euthanasia with CO2, and colons were collected and immediately fixed overnight with 4% formaldehyde in PBS (pH 7.2). The tissues were then processed, embedded in paraffin wax, cut into 4-μm sections and subjected to hematoxylin and eosin staining. To ascertain ZO-1 levels in colonic tissues, immunohistochemistry staining was carried out with an anti-ZO-1 antibody (Life Technologies, Inc.) and horseradish peroxidase-conjugated second antibody. Antigens were then visualized with 3, 3′-diaminobenzidine substrate (Sigma-Aldrich) and imaged under a light microscope at 63X magnification. ZO-1 intensity was scored semi-quantitatively from 0 to 3, and Student’s t-test was used to determine differences in expression between tissue groups. Statistical significance was taken as P ≤ 0.05.
RESULTS
Therapeutic effects of AMP peptide in interleukin-10 gene-deficient mouse model of human IBD
Chronic patchy colitis, similar to human Crohn’s disease, developed in all IL-10−/− mice eating chow containing the anti-inflammatory drug, piroxicam for one week; colitis did not require continued administration of piroxicam (9–11). As expected, all 22 mice demonstrated marked weight loss (26.5%) after being fed on a diet containing 200 ppm piroxicam for 11 days (Fig. 1), with the appearance of soft, but not bloody stools. When piroxicam-containing chow was replaced with normal chow, all mice given AMP peptide or PBS (vehicle) started to recover and gain weight. In one experiment, mice were treated with AMP peptide or vehicle for 9 days, and in a separate experiment, mice were treated for 18 days. The weight of the animals given AMP peptide increased progressively for 18 days so they returned to their starting weight, whereas mice injected daily with PBS did not (P=0.011). In addition, a change in stool consistency from soft to hard was observed in most mice after 11–13 days of AMP peptide treatment, but took longer in control animals.
Fig. 1. Antrum mucosal protein (AMP)-18 peptide enhances body weight recovery from colitis in IL-10−/− mice.

After eating piroxicam-containing chow for 11 days, mice (n = 22) lost 26.5 ± 1.4% of their initial body weight (●). Diet was switched to standard chow, and the next day AMP peptide (25 mg/kg) in phosphate-buffered saline (▼) or vehicle (○) was injected s.c. once daily. AMP peptide-treated mice recovered to their starting weight, whereas mice given vehicle did not (P=0.011).
In summary, a systemic benefit of AMP peptide was the complete recovery of body weight in AMP peptide-treated but not in vehicle-treated mice. AMP peptide reduced the extent of colon shortening and length of the dilated colon segment, as well as the colitis activity score (Fig. 2). Myeloperoxidase immunohistochemistry (not shown) used as an index of inflammation in colonic tissue, confirmed the histological analysis and grading.
Fig. 2. Effect of AMP peptide on colitis in IL-10−/− mice.
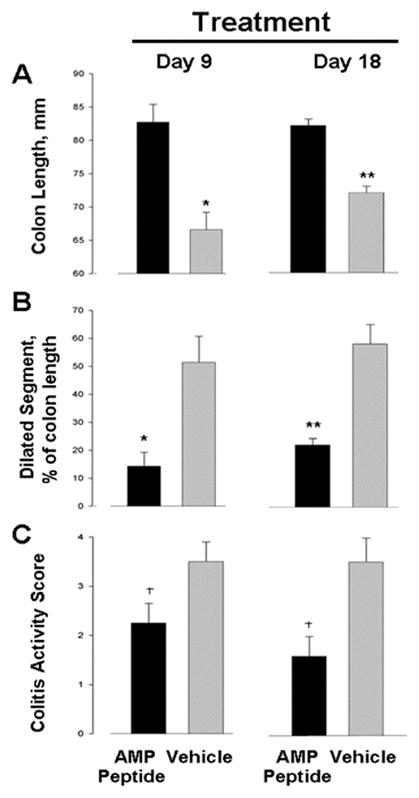
In two separate experiments IL-10−/− mice (n = 22) were fed piroxicam-containing chow for 11 days and then switched to standard chow. The following day mice were given a daily injection (s.c.) of either AMP peptide in PBS or PBS alone. After 9 or 18 days of treatment the animals were killed, and measurements were made of both the entire colon and a dilated colon segment (diameter ≥ 3.5 mm). The colon was prepared for histological examination to assess the colitis activity score. (A) Treatment with AMP peptide significantly reduced the extent of colon shortening after 9 (*P ≤ 0.005) and 18 days (**P < 0.001). (B) The length of the dilated segment was significantly shorter in peptide-treated animals than in vehicle-treated mice when expressed as a % of colon length after both 9 (*P ≤ 0.005) and 18 days (**P < 0.001) of treatment. (C) Treatment with AMP peptide reduced the colitis activity score throughout the entire colon after 9 and 18 days compared to vehicle. †P ≤ 0.043, AMP peptide vs. vehicle. Each value is mean ± standard error for 5–6 mice.
Therapeutic effects of AMP peptide in the T cell transfer model of IBD
The therapeutic efficacy of AMP peptide was also tested in a T-cell mediated “adoptive transfer” model of colitis. RAG-1−/− immunodeficient mice (on a C57BL/6J background; 5–8-week old females) depleted of regulatory T cells were reconstituted with splenic CD45RBhigh CD4+ T cells (4 X 105 cells, 95% pure). The transferred T cells mediate colonic inflammation in the recipients, causing chronic colitis with a clinical syndrome that resembles human IBD (16–18). Therapeutic efficacy of AMP peptide was assessed in age- and weight-matched mice, given either AMP peptide or vehicle.
As shown in Fig. 3, the colons of mice treated with AMP peptide for 10 or 21 days were about 15% longer than in animals treated with the vehicle, suggesting that the peptide protected against inflammation. In mice treated with AMP peptide, serosal adhesions between the colon and overlying loops of small bowel were much less extensive, there was less dilation of the proximal colon, hard stools compared to soft stools appeared sooner, and the animals were clearly physically stronger.
Fig. 3. AMP peptide reduces colonic shortening in T-cell transfer model of IBD.
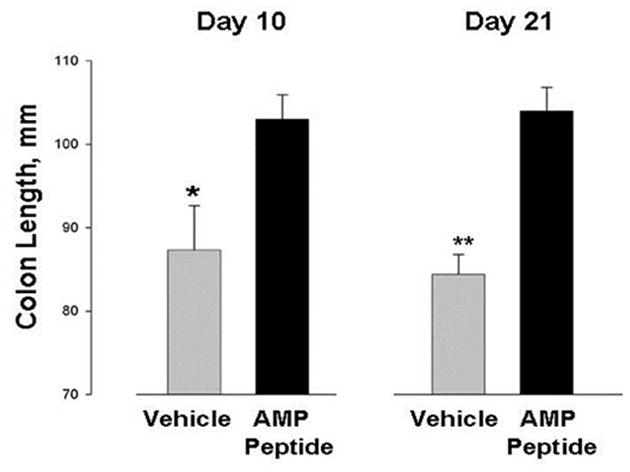
RAG-1−/− mice were reconstituted with splenic CD45RBhigh CD4+ T cells (4 X 105 cells, 95% pure). Treatment with AMP peptide (25 mg peptide/kg, s.c.) or vehicle began when the animals lost 5% body weight, or exhibited diarrhea or hematochezia. Whole colons were harvested and lengths were measured on days 10 or 21 (n=20; * P=0.032, ** P=0.00005).
AMP peptide prevents and reverses gut hyperpermeability induced by LPS
Then we asked if AMP peptide could alter intestinal permeability which is often increased in patients with IBD (19–21). Colon segments from mice treated with AMP peptide for 5 days before administration of LPS (left panel, Fig. 4) showed that FITC-dextran flux was considerably lower than in segments from animals treated with LPS alone. FITC-dextran flux in jejunum segments is shown in the right panel of Fig. 4. LPS-induced hyperpermeability was compared to untreated control mice. Pretreatment with AMP peptide for 1 day or 6 hr before injection of LPS prevented the increase in FITC-dextran flux induced by LPS, reducing permeability to the level seen in control animals. Therefore, pretreatment with AMP peptide prevents LPS-induced gut mucosal barrier disruption.
An agent that could reverse established gut hyperpermeability would have great therapeutic value in clinical practice. So we asked if AMP peptide could prevent LPS-induced hyperpermeability if given after LPS administration. LPS significantly increased the mucosal to serosal flux as expected (14) when compared to control, indicating that decreased barrier function induced by LPS can last for at least 21 hr when left untreated. Flux in mice treated with a single dose of AMP peptide 15 hr after LPS was given was similar to flux in control animals indicating that peptide treatment reversed gut hyperpermeability. This result provides evidence of a therapeutic effect of AMP peptide to reverse ongoing gut hyperpermeability in the setting of established LPS-induced barrier disruption.
To determine if this therapeutic effect of AMP peptide on mucosal structure and function could be a consequence of its known capacity to enhance accumulation of TJ proteins in the mucosal barrier, we used immunofluorescent staining and LSCM to assess occludin, ZO-1, and perijunctional actin in jejunum tissue from mice whose flux is presented in Fig. 5. The typical “chicken wire” appearance of TJ proteins and perijunctional actin in jejunum tissue from control mice is shown (Fig. 5, top three panels). Injection of LPS disrupted the ordered array of occludin (left), ZO-1 (middle), and perijunctional actin (right) (middle panels). When mice given LPS were treated with a single injection of AMP peptide 15 hr later, jejunal tissue examined 6 hr afterwards (bottom panels) demonstrated that that occludin and ZO-1 were again localized to TJs together with perijunctional actin. These observations show that AMP peptide restored the normal distribution of these proteins at TJs, an event that could reverse LPS-mediated disruption of mucosal barrier function (hyperpermeability) (Fig. 5) and structure (Fig. 6.)
Fig. 5. AMP peptide can therapeutically reverse barrier disruption induced by LPS.

Mice (n=3) were given LPS (100 μg, i.p.) (red line), or no LPS (vehicle control) (n=3) (broken black line), and killed 21 hr later. Another group of mice (n=3) received LPS, and 15 hr later (when gut permeability was markedly increased compared to control animals) a single injection of AMP peptide (25 mg/kg, s.c.) was given (blue line). Six hr later the gut was harvested and permeability in jejunum sacs was assayed as in Fig. 1. Values are FITC flux per cm of intestine, and are representative of three experiments.
Fig. 6. Treatment with AMP peptide reverses LPS-induced disruption of occludin, ZO-1, and perijunctional actin in mouse jejunum mucosal tissue.
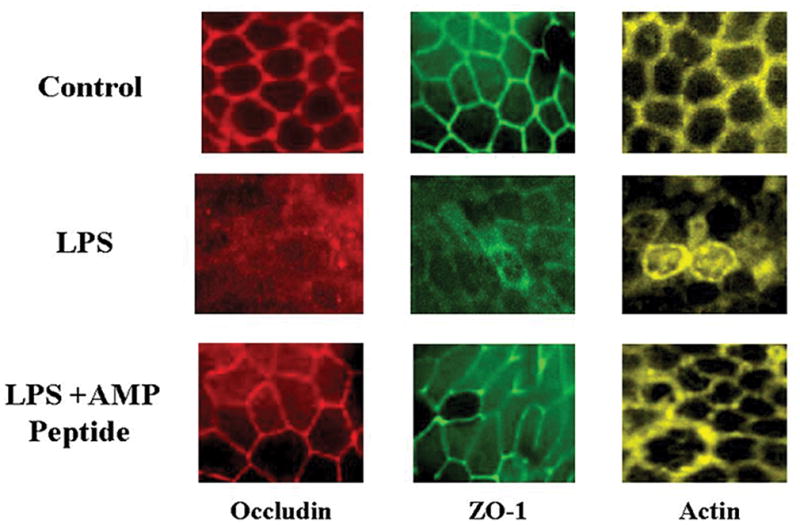
Tissue from mice with LPS-induced gut hyperpermeability were mounted on coverslips and subjected to IHC staining and LSCM. Tissue was taken for study 6 hr after administration of LPS; 21 hr after the experiment was initiated. Localization of occludin, ZO-1, and perijunctional actin in jejunum mucosal tissue from control animals is displayed from left to right in the top panels. Exposure to LPS disrupted TJs and diminished the amount of occludin (red), ZO-1 (green), and perijunctional actin (yellow) (middle panels). Treatment with AMP peptide restored the physiological distribution of occludin, ZO-1, and perijunctional actin in mice given LPS (bottom panels). Displayed are maximal projections of 5-μm Z-stacks through the area of TJs.
AMP-18 deficiency is associated with increased mortality and decreased colonic mucosa ZO-1 level upon exposure to DSS
The results in Fig. 6 show that TJs play a significant role in maintaining colonic barrier function and that AMP peptide can repair disruption of the barrier by targeting TJs. These beneficial effects of AMP peptide in reversing structural and functional injuries led us to ask if expression of AMP-18 is required for normal maintenance of the GI colonic barrier. To accomplish this we created an AMP-18 knockout mouse, and assessed its vulnerability to DSS-mediated injury at the level of TJs.
WT animals and AMP-18+/− mice exhibited a 25- and 30%-mortality, respectively, 12 and 13 days following exposure to 2% DSS for 5 days and then switched back to water. When AMP-18−/− mice (6-weeks old) were followed until day13, they demonstrated a significantly higher mortality (70%) compared to AMP-18+/+ and AMP-18+/− littermates (P<0.02) (Fig. 7). Histological study performed on day 4 revealed that after DSS treatment, severe erosions were evident in the AMP-18−/− mice, but not in WT or AMP-18+/− animals (Fig. 8). These observations suggest that the absence of AMP-18 gene expression makes the animals more vulnerable to the toxic effects of DSS as manifested by structural changes in the colonic mucosa and increased mortality.
Fig. 7. Increased mortality of AMP-18−/− mice with DSS-induced colonic injury.

WT (n=8), AMP-18+/− (n=16) and AMP-18−/− (n=14) mice were given water containing 2% DSS for 5 days, then switched back to regular water, and followed until day 13. Mortality of AMP-18−/− mice (70%) was more than 2-fold greater than in WT (25%) or AMP-18+/− (30%) littermates at that time.
Fig. 8. Colonic mucosal barrier structural injury is more marked in AMP-18−/− mice exposed to DSS.

Colon histology on day 4 in animals described in Fig. 7 revealed erosions in AMP-18−/− mice (bottom panel), whereas the mucosa of WT (top panel) and AMP-18+/− animals (middle panel) appeared normal; 10X. Representative images from 38 mice are shown.
To determine if the absence of AMP-18 in knockout animals is associated with increased mortality by an effect on TJs, we performed IHC to study ZO-1 levels in colonic tissues. TJ ZO-1 intensity was compared in heterozygous and homozygous mice given DSS. Fig. 9 shows control heterozygous mice not given DSS (panel A) exhibited strong TJ ZO-1 expression as did 3 heterozygous animals given DSS (panels B–D). In contrast, ZO-1 staining in homozygous mice was reduced after DSS treatment (panels F–H) compared to heterozygous littermates. Semi-quantitative analysis revealed a 38±13% decrease in ZO-1 staining (P<0.05) in the homozygous DSS-treated mice.
Fig. 9. Decreased ZO-1 levels in colonic mucosa of DSS-treated AMP-18−/− mice compared to AMP-18+/−.

AMP-18+/−, AMP-18−/− mice were exposed to 2% DSS for 5 days then switched back to water. Immunohistochemistry staining was carried out with an anti-ZO-1 antibody and imaged; 63X. Each panel represents a different animal. ZO-1 staining is less intense in the 3 AMP-18−/− compared to the 3 AMP-18+/− mice given DSS.
DISCUSSION
The results of this study indicate that AMP peptide served as an effective therapeutic in two immune-mediated murine models of colonic mucosal barrier structural and functional injury relevant to human IBD; NSAID-induced colonic injury in IL-10 deficient animals genetically programmed to develop IBD (Figs. 1, 2), and the T-cell mediated “adoptive transfer” model of colitis (Fig. 3). AMP peptide was also effective in reversing LPS induced barrier functional injury by targeting TJ proteins (Figs. 4–6). AMP-18 gene expression appears to be required for optimal defense against colonic mucosal injury because AMP-18−/− animals exhibit increased sensitivity to DSS that may be a function of decreased ZO-1 levels in colonic mucosa TJs (Figs. 7–9). Deficiency of AMP-18 in mice was associated with increased mortality, more marked colonic tissue injury, and decreased ZO-1 levels when the animals were exposed to DSS. These observations suggest that AMP-18 plays a significant role in maintaining colonic barrier structure and function by targeting TJs, and could serve as a therapeutic agent in patients with IBD.
Although the pathogenesis of IBD is not fully understood, dysregulation of the mucosal immune response to constituents of the normal microflora, that is in part genetically determined, is a major hypothesis (22). Several investigators, however, have provided experimental evidence pointing to a primary defect in epithelial barrier function that is under complex genetic control (23–25). Whatever the underlying mechanisms that mediate mucosal injury, administration of AMP peptide could stabilize colonic epithelial TJs, reseal mucosal injuries, and speed wound healing to restore barrier structure and function. In patients with IBD, immune dysregulation in the colonic mucosa could cause barrier dysfunction, thereby intensifying immune dysregulation and setting in motion a vicious cycle that exacerbates disease (12, 26, 27), as summarized below.

This cycle could be broken by the capacity of AMP peptide to protect TJs, stabilize barrier function and structure, stimulate restitution and cell proliferation, and thereby restore epithelial integrity. AMP peptide represents a potential new class of therapeutic agents and a different strategy than the current biologics and immunomodulating agents used to treat IBD that is not expected to compromise the innate immune system as do agents such as corticosteroids, azathioprine, cyclosporine, and infliximab.
TJs are a critical component of the intrinsic barrier that seals the paracellular spaces and establishes the mucosal barrier (28). These intercellular bridges are comprised of multiple proteins including integral membrane proteins such as occludin, claudins, junctional adhesion molecules (JAMs), and multiple cytoplasmic proteins such as zonula occludens (ZO)-1, ZO-2, cingulin, and others that form the terminal plaque (29–34). The TJ transmembrane protein occludin is linked via ZO-1 to the apical perijunctional F-actin ring as well (35–37). TJs and their interactions with perijunctional F-actin, undergoing dynamic homeostasis in both physiological and pathological conditions, regulate paracellular permeability across monolayer cell cultures and mucosal epithelial barrier in vivo. In addition, the TJs are important mediators of “outside –in” and “inside-out” signaling pathways that regulate epithelial proliferation and differentiation. In IBD, structure and function of TJs have been shown to be disrupted through multiple agents and mechanisms including cytokines and apoptosis. Cytokines such as TNF-α, interleukin (IL)-1β and IL-6 have been identified as major mediators of mucosal injury (9). Thus a therapeutic agent that stabilizes TJs and maintains the mucosal barrier would be of great value to treat patients with IBD.
Previously we observed that AMP peptide is an effective therapeutic in mice with DSS-induced colonic mucosal injury (3, 6). Treatment with AMP peptide also reduced mortality, preserved colonic mucosal integrity, and prevented transmural inflammation in trinitrobenzenesulfonic acid (TNBS)-induced colitis in vitamin D receptor (VDR)-deficient mice (38). In this project set out to determine if the peptide was also effective in two additional murine models of immune-mediated intestinal injury relevant to IBD: piroxicam-triggered colitis in IL-10-deficient animals, and a T-cell mediated “adoptive transfer” model of colitis. The capacity of AMP peptide to stimulate accumulation of specific TJ proteins, increase transepithelial electrical resistance, and facilitate TJ assembly by overriding the conditions that lead to disassembly of TJs suggest a protective effect against disruption of the epithelial barrier during flares of IBD, stabilization of the colonic mucosa, and acceleration of repair after injury (1–4, 6). We believe that AMP-18 is unique because it is the only agent of which we are aware that appears to facilitate assembly of specific proteins into new TJs. The actions of AMP peptide are predicted to re-establish intact intestinal barrier function in vivo after injury, thereby protecting patients against bacterial invasion, submucosal inflammation, and possible sepsis. In addition, re-sealing breaks in the epithelial barrier would attenuate immune responses to foreign antigens that are thought to initiate or sustain immune reactivity to self antigens. We hypothesize that administration of AMP peptide would limit mucosal injury by protecting TJs during cell stress, facilitate assembly of proteins to form new TJs after barrier injury, and thereby protect the colonic mucosa of patients before, during, and after exacerbations of IBD. In addition, some family members of patients with Crohn’s disease exhibit increased intestinal permeability (20, 21) suggesting a defect in the colonic mucosal barrier that could be amenable to treatment with AMP peptide. From this perspective, stabilizing TJs can be viewed as a novel approach to preventing or treating IBD (39). Having recently identified CCKBR as an AMP-18 receptor by which AMP peptide exerts its diverse effects could enable us to increase understanding of how its activation protects and repairs the mucosal barrier after injury and also identify signaling targets by which the ligand operates on GI epithelial cells (7). These observations suggest for the first time that AMP-18 plays a significant role in protecting colonic epithelium against immune-mediated injury, apparently by targeting TJs (Figs. 1–3).
Our results suggest that treatment with AMP peptide reversed established gut hyperpermeability (Fig. 5), possibly in part, by rapidly recruiting pre-existing cytosolic proteins to assemble new TJs (Fig. 6), thereby repairing the structure and function of the disrupted mucosal barrier. Previous studies carried out in monolayer cultures of colonic epithelial cells (Caco-2/bbe line) suggest the capacity of AMP peptide to repair the injured mucosal barrier is mediated by facilitating translocation of pre-formed cytosolic proteins into nascent TJs, rather than by initiating synthesis of new TJ proteins because these beneficial effects of AMP peptide occurred in the presence of cycloheximide (3–5).
AMP-18-deficient mice exhibited increased mortality when exposed to DSS, as well as greater structural injury in the distal colon, and reduced levels of ZO-1 in colonic mucosal TJs than their heterozygous littermates (Figs. 7–9). These observations suggest that AMP-18 can protect mucosal barrier structure and function in the murine GI tract, and are consistent with our findings that AMP peptide can serve a therapeutic role by protecting intestinal tissue against injury and speeding healing in normal mice given DSS (3, 6), IL-10 knockout animals given piroxicam (Figs. 1, 2), RAG-1−/− mice subjected to T cell adoptive transfer (Fig. 3), and animals with LPS-induced hyperpermeability (Figs. 4–6). How AMP-18, a protein synthesized in gastric epithelial cells and not found in the circulation, protects the mucosal barrier remains to be determined, although its protease-resistance (40, 41) raises the possibility that at least a functional portion of undigested AMP-18 protein can reach the distal intestine and colon and exert its biological effects at these sites distant from the stomach.
Administration of AMP peptide to patients with active colitis could enhance repair of the mucosal barrier by reducing cell detachment, apoptosis and necrosis, speeding restitution of surviving cells at the edges of mucosal wounds, and stimulating cell proliferation followed by formation of TJs to regenerate a structurally repaired functional colonic epithelium. These pleiotropic effects on cells of the colonic mucosal barrier and clinical beneficial effects in immune and non-immune models of colonic inflammation and injury suggest that further development of AMP peptide as a therapeutic agent could lead to a new agent to promote regeneration and repair of the injured digestive tract and healing in patients with IBD.
Acknowledgments
Grant Support: This work was supported by National Institutes of Health (NIH) grant R21 NCI 176032, the National Center For Advancing Translational Sciences of the NIH award UL1 TR000430, a grant from the Crohn’s and Colitis Foundation of America, and from the Broad Medical Research Foundation to F. G. Toback; Rainin Foundation Grant 13H16 to D. L. Boone; and NIH grants, K08 DK088953 and R03 DK103007 to C. R. Weber.
References
- 1.Martin TE, Powell CT, Wang Z, et al. A novel mitogenic protein that is highly expressed in cells of the gastric antrum mucosa. Am J Physiol Gastrointest Liver Physiol. 2003;285:G332–343. doi: 10.1152/ajpgi.00453.2002. [DOI] [PubMed] [Google Scholar]
- 2.Toback FG, Walsh-Reitz MM, Musch MW, et al. Peptide fragments of AMP-18, a novel secreted gastric antrum mucosal protein, are mitogenic and motogenic. Am J Physiol Gastrointest Liver Physiol. 2003;285:G344–353. doi: 10.1152/ajpgi.00455.2002. [DOI] [PubMed] [Google Scholar]
- 3.Walsh-Reitz MM, Huang EF, Musch MW, et al. AMP-18 protects barrier function of colonic epithelial cells: role of tight junction proteins. Am J Physiol Gastrointest Liver Physiol. 2005;289:G163–171. doi: 10.1152/ajpgi.00013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh-Reitz MM, Kartha S, Bissonnette M, et al. Antrum mucosal protein-18 activates multiple pathways to assemble and stabilize tight junctions. Gastroenterology. 2006;130:A-41. [Google Scholar]
- 5.Kartha S, Walsh-Reitz MM, Bissonnette M, et al. Antrum mucosal protein-18 facilitates assembly of tight junction proteins by activating protein kinase C-zeta. Gastroenterology. 2005;128:A-538–A-539. [Google Scholar]
- 6.Chen P, Kartha S, Bissonnette M, et al. AMP-18 facilitates assembly and stabilization of tight junctions to protect the colonic mucosal barrier. Inflamm Bowel Dis. 2012 doi: 10.1002/ibd.22886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen P, Lingen M, Sonis ST, et al. Role of AMP-18 in oral mucositis. Oral Oncol. 2011;47:831–839. doi: 10.1016/j.oraloncology.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu X, Chen P, Sonis ST, et al. A Novel Peptide to Treat Oral Mucositis Blocks Endothelial and Epithelial Cell Apoptosis. Int J Radiat Oncol Biol Phys. 2012:83, e409–415. doi: 10.1016/j.ijrobp.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berg DJ, Zhang J, Weinstock JV, et al. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123:1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
- 10.Zhao J, de Vera J, Narushima S, et al. R-spondin1, a novel intestinotrophic mitogen, ameliorates experimental colitis in mice. Gastroenterology. 2007;132:1331–1343. doi: 10.1053/j.gastro.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Berg DJ, Davidson N, Kuhn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gassler N, Rohr C, Schneider A, et al. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281:G216–228. doi: 10.1152/ajpgi.2001.281.1.G216. [DOI] [PubMed] [Google Scholar]
- 13.Xu D, Liu H, Komai-Koma M, et al. CD4+CD25+ regulatory T cells suppress differentiation and functions of Th1 and Th2 cells, Leishmania major infection, and colitis in mice. J Immunol. 2003;170:394–399. doi: 10.4049/jimmunol.170.1.394. [DOI] [PubMed] [Google Scholar]
- 14.Han X, Fink MP, Yang R, et al. Increased iNOS activity is essential for intestinal epithelial tight junction dysfunction in endotoxemic mice. Shock. 2004;21:261–270. doi: 10.1097/01.shk.0000112346.38599.10. [DOI] [PubMed] [Google Scholar]
- 15.Fujiya M, Musch MW, Nakagawa Y, et al. The Bacillus subtilis quorum-sensing molecule CSF contributes to intestinal homeostasis via OCTN2, a host cell membrane transporter. Cell Host Microbe. 2007;1:299–308. doi: 10.1016/j.chom.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Mombaerts P, Mizoguchi E, Grusby MJ, et al. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 1993;75:274–282. doi: 10.1016/0092-8674(93)80069-q. [DOI] [PubMed] [Google Scholar]
- 17.Powrie F, Leach MW, Mauze S, et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 18.De Winter H, Cheroutre H, Kronenberg M. Mucosal immunity and inflammation. II. The yin and yang of T cells in intestinal inflammation: pathogenic and protective roles in a mouse colitis model. Am J Physiol. 1999;276:G1317–1321. doi: 10.1152/ajpgi.1999.276.6.G1317. [DOI] [PubMed] [Google Scholar]
- 19.Hollander D, Vadheim CM, Brettholz E, et al. Increased intestinal permeability in patients with Crohn’s disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–885. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- 20.Hollander D. Intestinal permeability, leaky gut, and intestinal disorders. Curr Gastroenterol Rep. 1999;1:410–416. doi: 10.1007/s11894-999-0023-5. [DOI] [PubMed] [Google Scholar]
- 21.Hollander D. Intestinal permeability in patients with Crohn’s disease and their relatives. Dig Liver Dis. 2001;33:649–651. doi: 10.1016/s1590-8658(01)80038-4. [DOI] [PubMed] [Google Scholar]
- 22.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olson TS, Reuter BK, Scott KG, et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med. 2006;203:541–552. doi: 10.1084/jem.20050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Resta-Lenert S, Smitham J, Barrett KE. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/− mice. Am J Physiol Gastrointest Liver Physiol. 2005;289:G153–162. doi: 10.1152/ajpgi.00395.2004. [DOI] [PubMed] [Google Scholar]
- 25.Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270:1203–1207. doi: 10.1126/science.270.5239.1203. [DOI] [PubMed] [Google Scholar]
- 26.Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84:282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- 27.Schmitz H, Barmeyer C, Fromm M, et al. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. 1999;116:301–309. doi: 10.1016/s0016-5085(99)70126-5. [DOI] [PubMed] [Google Scholar]
- 28.Madara JL. Regulation of the movement of solutes across tight junctions. Annu Rev Physiol. 1998;60:143–159. doi: 10.1146/annurev.physiol.60.1.143. [DOI] [PubMed] [Google Scholar]
- 29.Byers SW, Citi S, Anderson JM, et al. Polarized functions and permeability properties of rat epididymal epithelial cells in vitro. J Reprod Fertil. 1992;95:385–396. doi: 10.1530/jrf.0.0950385. [DOI] [PubMed] [Google Scholar]
- 30.Martin-Padura I, Lostaglio S, Schneemann M, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furuse M, Fujita K, Hiiragi T, et al. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jesaitis LA, Goodenough DA. Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J Cell Biol. 1994;124:949–961. doi: 10.1083/jcb.124.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keon BH, Schafer S, Kuhn C, et al. Symplekin, a novel type of tight junction plaque protein. J Cell Biol. 1996;134:1003–1018. doi: 10.1083/jcb.134.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byers S, Graham R, Dai HN, et al. Development of Sertoli cell junctional specializations and the distribution of the tight-junction-associated protein ZO-1 in the mouse testis. Am J Anat. 1991;191:35–47. doi: 10.1002/aja.1001910104. [DOI] [PubMed] [Google Scholar]
- 35.McCarthy KM, Skare IB, Stankewich MC, et al. Occludin is a functional component of the tight junction. J Cell Sci. 1996;109 (Pt 9):2287–2298. doi: 10.1242/jcs.109.9.2287. [DOI] [PubMed] [Google Scholar]
- 36.Wong V, Gumbiner BM. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997;136:399–409. doi: 10.1083/jcb.136.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitic LL, Van Itallie CM, Anderson JM. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: lessons from mutant animals and proteins. Am J Physiol Gastrointest Liver Physiol. 2000;279:G250–254. doi: 10.1152/ajpgi.2000.279.2.G250. [DOI] [PubMed] [Google Scholar]
- 38.Chen P, Li YC, Toback FG. AMP-18 targets p21 to maintain epithelial homeostasis. PloS one. 2015 doi: 10.1371/journal.pone.0125490. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mannon P. Gut permeability and colitis. Gastroenterology. 2009;137:732–734. doi: 10.1053/j.gastro.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 40.Hnia K, Notarnicola C, de Santa Barbara P, et al. Biochemical properties of gastrokine-1 purified from chicken gizzard smooth muscle. PloS one. 2008;3:e3854. doi: 10.1371/journal.pone.0003854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavone LM, Del Vecchio P, Mallardo P, et al. Structural characterization and biological properties of human gastrokine 1. Molecular bioSystems. 2013;9:412–421. doi: 10.1039/c2mb25308a. [DOI] [PubMed] [Google Scholar]