

Significance
Staphylococcus aureus secretes a range of virulence factors to evade immune recognition. One of these, staphylococcal superantigen-like protein 3 (SSL3), disrupts an important component of our innate immune system: activation of Toll-like receptor 2 (TLR2) by bacterial lipopeptides. The crystal structure of the SSL3–TLR2 complex now provides the structural basis for a unique mechanism of full TLR2 antagonism in which SSL3 interferes with both ligand binding and receptor dimerization. Our novel insights on the host–pathogen interaction may contribute to vaccine development and form a starting point for the design of structure-based mimics to inhibit aberrant TLR2 activation in several inflammatory diseases and disease states.
Keywords: S. aureus, Toll-like receptor, immune evasion, innate immunity, crystal structure
Abstract
Toll-like receptors (TLRs) are crucial in innate recognition of invading micro-organisms and their subsequent clearance. Bacteria are not passive bystanders and have evolved complex evasion mechanisms. Staphylococcus aureus secretes a potent TLR2 antagonist, staphylococcal superantigen-like protein 3 (SSL3), which prevents receptor stimulation by pathogen-associated lipopeptides. Here, we present crystal structures of SSL3 and its complex with TLR2. The structure reveals that formation of the specific inhibitory complex is predominantly mediated by hydrophobic contacts between SSL3 and TLR2 and does not involve interaction of TLR2–glycans with the conserved LewisX binding site of SSL3. In the complex, SSL3 partially covers the entrance to the lipopeptide binding pocket in TLR2, reducing its size by ∼50%. We show that this is sufficient to inhibit binding of agonist Pam2CSK4 effectively, yet allows SSL3 to bind to an already formed TLR2–Pam2CSK4 complex. The binding site of SSL3 overlaps those of TLR2 dimerization partners TLR1 and TLR6 extensively. Combined, our data reveal a robust dual mechanism in which SSL3 interferes with TLR2 activation at two stages: by binding to TLR2, it blocks ligand binding and thus inhibits activation. Second, by interacting with an already formed TLR2–lipopeptide complex, it prevents TLR heterodimerization and downstream signaling.
In recent years, Staphylococcus aureus has become a major health threat to both humans and domestic animals. It is found as a commensal bacterium in ∼30% of the human population, but when it becomes infectious it can cause a wide diversity of diseases, ranging from mild skin infections to life-threatening invasive conditions such as pneumonia and sepsis (1). Increased antibiotic resistance and a high amount of virulence factors secreted by S. aureus contribute to its emergence as a pathogen. Among these secreted virulence factors are the staphylococcal superantigen-like proteins (SSLs), a family of 14 proteins located on two genomic clusters (2–4). Recently, we and others identified SSL3 as a potent inhibitor of Toll-like receptor 2 (TLR2) (5, 6), an innate immunity receptor that is a dominant factor in immune recognition of S. aureus (7–10).
TLR2 belongs to a family of 10 homologous innate immunity receptors that are activated by pathogen-associated molecular patterns (PAMPs) (11). TLR2 binds bacterial lipopeptides and lipoproteins. Subsequent formation of heterodimers with TLR1 or TLR6 leads to MyD88-dependent activation of the NF-κB pathway (12). TLR2 has dual ligand specificity that is determined by its dimerization partner; stimulation by diacyl lipopeptides from Gram-positive bacteria, including S. aureus, induces the formation of heterodimers with TLR6 (13), whereas triacyl lipopeptides from Gram-negative bacteria initiate formation of TLR2–TLR1 dimers (14). The structural basis for lipopeptide specificity was revealed by crystal structures of TLR2–TLR1 and TLR2–TLR6 complexes with their respective lipopeptide analogs Pam3CSK4 and Pam2CSK4: TLR2 binds two lipid tails in a large hydrophobic pocket, whereas the third lipid tail of triacyl lipopeptides is accommodated by a smaller pocket present in TLR1, but not in TLR6 (15, 16).
The family of SSL proteins, including SSL3, share structural similarities to superantigens, but lack superantigenic activity. Interestingly, the functions that have been discovered for SSLs so far have all been linked to immune evasion. SSL5 inhibits neutrophil extravasation (17, 18) and phagocyte function (19, 20), SSL7 binds IgA and inhibits complement (21), and SSL10 inhibits IgG1-mediated phagocytosis (22, 23), blood coagulation (24), and the chemokine receptor CXCR4 (25). In addition to SSL3, also weak TLR2 inhibitory activity was observed for SSL4 (5), but it remains unknown whether that is its dominant function. This variety of immunomodulatory molecules and functions reflects the importance of the different components of our innate immune system in the defense against S. aureus (26).
In this study we determined the crystal structures of SSL3 and the SSL3–TLR2 complex. In combination with mutagenesis and binding studies, our data provide a novel working mechanism of a functional TLR2 antagonist.
Results
Structure of SSL3ΔN.
To study the structural basis for inhibition of TLR2 activation by virulence factor SSL3, we expressed and purified SSL3ΔN, which lacks 133 N-terminal residues. Deletion of the N-terminal region proved essential to obtain crystals, but does not affect its activity toward TLR2 (Fig. S1A). The crystal structure of SSL3ΔN, with two molecules in the asymmetric unit, was solved at 1.94 Å resolution (Fig. S2A and Table S1) by molecular replacement. SSL3 exhibits the characteristic two-domain fold of superantigens and other SSLs (27, 28). The C-terminal β-grasp domain (residues 228–326) contains a V-shaped binding site for sialyl LewisX, which is conserved in SSL2-6 and -11 (Fig. S3 A and B) (28). The N-terminal OB domain (residues 134–227) displays well-defined but markedly different conformations for loops β1–β2 and α3–β4 (Fig. S2 B and C). These conformational differences likely arise from crystal contacts, and suggest considerable flexibility of these loops in solution.
Fig. S1.
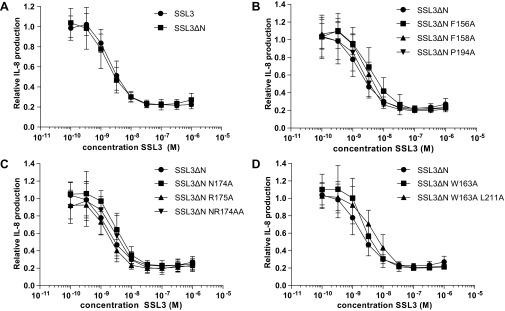
TLR2 inhibition by SSL3 WT, SSL3ΔN, and SSL3ΔN point mutants. HEK TLR2/6 cells were stimulated with MALP-2, and IL-8 production was measured in the presence of increasing concentrations of SSL3 or SSL3∆N. IL-8 production is relative to cells not treated with SSL3. Data points represent at least three independent experiments, and error bars show mean ± SD. (A) SSL3 and SSL3∆N, which lacks amino-terminal residues 1–133, have equal TLR2 inhibitory capacities. (B–D) IL-8 production in the presence of SSL3ΔN mutants that did not have a significant effect on TLR2 inhibitory capacity.
Fig. S2.

Structure of SSL3ΔN shows structural variation in loops of the β-grasp domain. (A) Cartoon visualization of the SSL3ΔN asymmetric unit that contains two molecules SSL3, both comprising residues 134–326. Two SSL3 molecules contact via three loops (β1–β2, β2–β3, and α3–β4) in the OB domain (orange), an arrangement that is clearly different from the previously observed SSL dimers formed by antiparallel packing of two β-grasp domains (yellow) (28). (B) Cartoon visualization of the SSL3ΔN structure showing the N-terminal OB domain (residues 134–227, orange) and the C-terminal β-grasp domain (residues 228–326, yellow). The OB domain comprises a five-stranded β-barrel and two short α-helices, and two conformations observed for the β1–β2 and α3–β4 loops are indicated in red. The β-grasp domain contains a central α-helix packed against a mixed five-stranded β-sheet. Residues in strand β10 and helix α5 contribute to a V-shaped binding site for sialyl LewisX containing glycans. (C) Two conformations of loop β1–β2 (panels 1 and 2) and loop α3–β4 (panels 3 and 4) that are observed in the crystal structure of SSL3 shown in stick representation in their corresponding 2Fo–Fc maps contoured at 1.2 σ above the average density. Except for the Lys191 side chain, these loops are well defined in the electron density maps, and differences likely arise from their involvement in crystal packing. (D) Structural variation of the β1–β2, α3–β4, and β4–β5 loops in superimposed structures of “free” SSL3 (SSL3A and B, yellow), SSL4 (blue; PDB ID code 4DXF) (38) and the SSL3–TLR2 complex (orange). Whereas free SSL3 differs substantially from TLR2-bound SSL3, the structure of SSL4 shows much smaller differences. (E) Structural variation of the TLR2 binding site as observed in the SSL3–TLR2 complex (orange; Lower Left) and the corresponding region in free SSL3A (yellow, Upper Left), SSL3B (yellow, Upper Right), and SSL4 (blue, Lower Right), all shown in stick representation in their corresponding 2Fo–Fc maps contoured at 1.2 σ above the average density. The orientation of the displayed regions is equal to Fig. 2C.
Table S1.
Data collection and refinement statistics
| Data collection and refinement | SSL3 | SSL3–TLR2 |
| Data collection | ||
| Wavelength (Å) | 0.9999 | 1.0000 |
| Space group | P212121 | P61 |
| Cell dimensions | ||
| a, b, c (Å) | 58.00, 79.22, 96.38 | 104.20, 104.20, 153.63 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 120 |
| Resolution range (Å)* | 61.2–1.94 (1.99–1.94) | 52.1–3.2 (3.31–3.2) |
| Total no. reflections | 223,907 (16,027) | 36,297 (7,265) |
| No. unique reflections | 33,556 (2,419) | 14,687 (3,088) |
| Rmerge | 0.093 (0.73) | 0.225 (1.08) |
| I/σI | 11.1 (2.7) | 5.6 (2.0) |
| Redundancy | 6.7 (6.6) | 2.5 (2.4) |
| Completeness (%) | 99.8 (99.9) | 94.1 (96.7) |
| CC(1/2) | 0.99 (0.63) | 0.92 (0.41) |
| Refinement | ||
| Rwork/Rfree | 0.177/0.224 | 0.231/0.271 |
| No. atoms | 3,521 | 5,976 |
| Protein | 3,266 | 5,890 |
| Water/other ligands | 214/9 | 0/86 |
| Average B/Wilson B (Å2) | 37.5/32.2 | 57.4/44.6 |
| Rmsd | ||
| Bond lengths (Å) | 0.0079 | 0.0070 |
| Bond angles (°) | 1.21 | 1.23 |
| Ramachandran plot | ||
| Favored (%) | 97.0 | 92.8 |
| Allowed (%) | 3.0 | 6.7 |
| Outliers (%) | 0 | 0.5 |
Numbers between brackets refer to the outer resolution shell.
Fig. S3.

Nanomolar binding of SSL3 to TLR2 does not involve sialyl LewisX interactions. (A) Stick representation of residues that are observed to bind sialyl LewisX in SSL4 (Right) (PDB ID code 4DXG) (38) and the corresponding region of SSL3 (Left). Sialyl LewisX sugar moieties are labeled: sialic acid (S), galactose (G), N-acetyl glucosamine (N), and fucose (F). Hydrogen bonds between SSL4 residues and sialyl LewisX are indicated with dashed lines. (B) Sequence alignment of SSL3, SSL4, SSL5, and SSL11 shows the conserved sialyl LewisX binding pattern (TxExxKxxQx[N/H]RxxD; purple) (27, 28, 38). Residues highlighted in gray only contribute to the hydrogen bonding network through backbone atoms, and therefore these interactions are sequence independent. (C) SSL3 and TLR2 have nanomolar affinity. In an AlphaScreen competitive binding assay, unlabeled SSL3 was titrated in and competes with biotin-labeled SSL3 for TLR2-Fc binding. The IC50 value corresponds to a Kd of 0.6 ± 0.4 nM. Data are expressed relative to binding of SSL3–biotin to TLR2 with no unlabeled SSL3 present. Data points represent the mean ± SD of at least three independent experiments. (D) Geometric analysis of the feasibility of sialyl LewisX interactions in the SSL3–TLR2 complex. (Left) Sialyl LewisX was positioned in the SSL3–TLR2 complex by superimposing the LewisX binding sites of SSL3 and the SSL4–sialyl LewisX complex (PDB ID code 4DXG) (38). Indicated are the shortest distance between the LewisX glycan and the two nearest glycosylated asparagine residues in TLR2 (shown in blue sticks). (Right) Chemical structure of a N-linked glycan including a mannose-linked sialyl LewisX [GlcNac(-Fuc)-Gal-Sia]. The linker between the asparagine and LewisX consists of two N-acetylglucosamine and two mannose units that can span a distance of at most 21 Å in a fully extended conformation, as was estimated on the basis of crystal structures in the protein data bank that contain this glycan motif. This distance is twofold smaller than the distances to the nearest N-linked asparagines in TLR2, excluding the involvement of sialyl LewisX binding in the observed SSL3–TLR2 complex.
Structure of the SSL3ΔN–mTLR2 Complex.
To facilitate expression and crystallization of TLR2, previous structural studies used constructs in which the C-terminal cap domain (LRRCT) together with one leucine rich repeat (LRR) had been replaced by a fragment of a hagfish variable lymphocyte receptor (VLR) (15, 29). We successfully produced a mouse TLR2 (mTLR2) construct covering the entire extracellular region of the protein and crystallized it in a 1:1 complex with SSL3ΔN. The structure was solved to 3.2 Å resolution (Fig. 1 and Table S1) using molecular replacement with the structures of SSL3ΔN and the mTLR2–VLR fusion (PDB ID code 2Z81) (15).
Fig. 1.

Crystal structure of the SSL3ΔN–mTLR2 complex. The SSL3ΔN OB and β-grasp domains are shown in orange and yellow, respectively, mTLR2 in green, and the mTLR2 LRRCT domain in a darker shade of green. Odd-numbered LRRs, helices H1–H6 of TLR2, and SSL3 loops that contact TLR2 are labeled.
Overall, the structures of TLR2 and SSL3 are well-defined (Fig. S4 A and B); the N- and C-terminal regions of TLR2, however, display increased average temperature factors. The LRRCT domain of TLR2 is structurally similar to that of TLR3 (Fig. S4 C–F), although 22 C-terminal residues appear disordered and could not be modeled satisfactorily. The observed flexibility of this region might, at least in part, account for the success of the VLR fusion approach.
Fig. S4.

The SSL3–TLR2 complex. (A and B) Cα traces of TLR2 (A) and SSL3 (B) in the SSL3–TLR2 complex colored according to their atomic B-factor on a scale between 20 Å2 (yellow) and 100 Å2 (red). (C and D) Superpositions of mouse TLR2 from the SSL3–TLR2 complex (green) on the TLR2–VLR6 fusion proteins used for crystallization of the TLR2–PE–DTPA complex (blue, C) (16) and the TLR2–TLR6–Pam2CSK4 complex (blue, D) (16). The structures are displayed as Cα traces in stereoview. Overall the structures are very similar. The native mTLR2 LRRCT domain, however, deviates significantly from the chimeric VLR6 LRRCT domain, which share 11% sequence identity. The TLR2 structures also show conformational variations in LRR10, and the extended loop of LRR11, two regions that determine the size and shape of the lipopeptide pocket entrance. The conformation of these loops in the SSL3–TLR2 complex is very similar to those observed for TLR2 in complex with nonactivating ligand PE–DTPA (C), and clearly different from the TLR2–TLR6–Pam2CSK4 complex (D). (E) Overlay of the LRRCT domains of mTLR2 (green and gray) and hTLR3 (blue), shown in stereoview with the conserved cysteine pattern highlighted in yellow. The TLR2 region that was modeled as polyalanine is shown in gray. The overall rmsd difference between the two LRRCT domains, which have 25% sequence identity, is 1.1 Å. The largest observed difference comprises the 13 residue α-helix in TLR3 (residues 651–663), which is split in a shorter nine-residue α-helix (residues 539–547) and a five-residue 310 helix (residues 549–553) in TLR2. Residues 576–587 are absent in the TLR2 model, including the disulfide bridge between Cys539 and Cys585, which suggest that this region suffered from radiation damage or is intrinsically flexible. (F) Sequence alignment of LRR19 and the LRRCT domain from mTLR2 and hTLR3. Also shown is the secondary structure as observed in the SSL3–TLR2 complex and hTLR3 (PDB ID code 2A0Z) (40). Residues in the SSL3–TLR2 complex that were modeled as polyalanine are indicated by a dashed line. Identical residues between TLR2 and TLR3 are highlighted in blue, and the conserved cysteines in yellow.
After refinement of the TLR2 and SSL3 structures, residual electron density in the lipid binding pocket located between LRR11 and LRR12 suggested the presence of a phospholipid (Fig. S5A). Subsequent native mass spectrometry analysis of TLR2 detected a mixture of phosphatidylcholine (PC) lipids with acyl chain lengths varying between 12 and 20 (Fig. S5 B–H). Apparently, PC binds sufficiently tightly as to remain associated with TLR2 during the purification process. The residual density in the lipid-binding pocket was subsequently modeled as PC, with its phosphoglycerol moiety positioned just inside, and its choline head group outside, the pocket.
Fig. S5.

Phosphatidylcholine lipids in the TLR2 lipopeptide binding pocket. (A) Stereoview of the 2Fo–Fc map calculated using phases derived from the SSL3–TLR2 structure without ligand, and contoured at 0.7σ. Also shown are stick representations of the PC lipid ligand with C12 and C13 acyl chains and a chloride ion that were modeled in the electron density. Carbon, oxygen, nitrogen, and phosphorus atoms are respectively colored gray, red, blue, and orange; the chloride ion is shown as a gray sphere. (B) Baseline-corrected mass spectrum of denatured TLR2 with native glycans. TLR2 forms a broad charge state envelope, based on which an average molecular weight of 75.2 ± 0.1 kDa was calculated. Charge states are indicated above the peaks. (C) Native MS analysis of TLR2. Broad-range mass spectrum of native TLR2 (red) with charge states indicated above the peaks. Due to the existence of different TLR2 glycoforms and/or binding of different phospholipid species, the m/z signals are relatively broad, allowing only an approximate molecular weight calculation (79.1 ± 0.1 kDa). Free phospholipids (cyan) are visible in the low m/z region. (D–F) Native tandem MS analysis of TLR2. The region around m/z 4378 was mass selected in the quadrupole mass analyzer and subsequently fragmented by collision-induced dissociation in the collision cell. Shown are the resulting tandem mass spectra. (D) At low collisional activation, only phospholipid-bound TLR2 is detected. (E) Increasing the collision to 40 V leads to dissociation of phospholipids from TLR2. (F) Applying a collision voltage of 100 V results in enhanced phospholipid dissociation and fragmentation. The fragment ion at m/z 184 (orange) is characteristic for fragmentation of the phosphatidylcholine head group. (Inset) Zoom-in of the m/z region where phospholipids are detected. (G) List of the observed m/z peaks (660–830 m/z) measured in the tandem MS analysis showing a range of TLR2-bound PC lipid species with two acyl chains that have a total of 28–38 carbon atoms, 0, 1, or 2 unsaturations, and a [M+H]+ mass for the single charged ions as depicted in the last column. Lipid chain abbreviations used: La, lauroyl (12:0); M, myristoyl (14:0); P, palmitoyl (16:0); Po, palmitoleoyl (16:1); S, stearoyl (18:0); O, oleoyl (18:1); L, linoleoyl (18:2); A, arachidoyl (20:0); G, gadoleoyl (20:1). (H) Chemical structures of the phosphatidylcholine fragment ion (orange) and the TLR2-bound PC lipids with the consensus part (blue) and variable lipid tail length (gray). For ready comparison, all phosphatidylcholine structures and masses depicted here correspond to single charge (+1) ions.
In the crystal structure of the SSL3–TLR2 complex, SSL3 binds with its OB domain on the convex face of the characteristic horseshoe-like structure of TLR2 and partially covers the entrance of the lipopeptide binding pocket. Quantitative assessment of the SSL3–TLR2 interaction using the AlphaScreen assay (30) yields a binding affinity of 0.6 ± 0.4 nM (Fig. S3C). The β-grasp domain of SSL3 does not contact TLR2; its LewisX binding site is located more than 50 Å away from the nearest N-glycosylated asparagine in TLR2, a distance that cannot be bridged by a glycan antenna (Fig. S3D). Formation of the TLR2–SSL3 complex does therefore not involve binding of TLR2 glycans to the LewisX binding site of SSL3, but is mediated by protein–protein interactions only.
The SSL3–TLR2 Binding Interface.
The interface between SSL3 and TLR2 buries 1640 Å2 of solvent accessible surface and is predominantly hydrophobic in nature; it consists of TLR2 residues located in LRR11–LRR13, including helices H2–H4 and SSL3 residues in four loops of the OB domain as indicated in Fig. 1. Three of these SSL3 loops differ in conformation compared with the structure of SSL3 alone (Fig. S2D), suggesting that TLR2 binding is accompanied by considerable conformational changes in SSL3 (Fig. S2E).
The SSL3 footprint on TLR2 is arc shaped and surrounds three sides of the entrance to the lipopeptide binding pocket (Fig. 2A). At one end of the arc, near helix H5, a continuous hydrophobic patch comprising SSL3 residues Phe156, Phe158, Leu160, and Pro194 interacts with TLR2 residues Phe349, Leu350, Gln375, Tyr376, and Asn379. In the center of the arc, a stretch of residues from the β2–β3 loop is positioned on top of TLR2 helices H3 and H4. Besides many hydrophobic interactions, this region contains the only hydrophilic interactions observed in the interface: Arg175 forms a salt bridge with Asp327, whereas hydrogen bonds are present between Arg175 and Ser329, and between Asn174 and His358. At the other end of the arc Trp163 stacks on Tyr323 in TLR2, whereas Leu211 and Lys213 have interactions with TLR2 residues Leu324 and Tyr326, respectively. TLR2 residues that contact SSL3 in the crystal structure are conserved between mouse and human TLR2 (hTLR2), except for a single Ser354Leu substitution at the periphery of the binding site. Therefore, the structures of the human and mouse SSL3–TLR2 complexes are likely very similar.
Fig. 2.
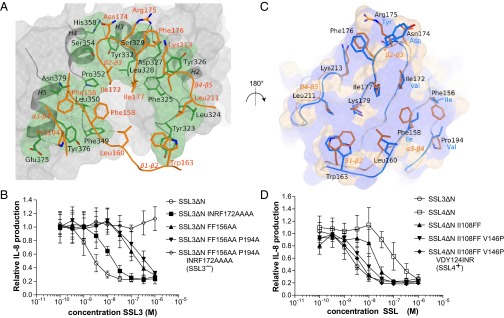
The SSL3–TLR2 interface and characterization of the TLR2 binding sites in SSL3 and SSL4. (A) Footprint of SSL3 (green) on the van der Waals surface of TLR2 (gray). Residues of SSL3 and TLR2 that are within 5 Å of its binding partner are shown in orange and green sticks, respectively. Van der Waals interactions are shown as dashed lines; hydrogen bonds and salt bridges as solid lines. (B) TLR2 inhibitory activity of SSL3 mutants. IL-8 production was measured after 6 h of MALP-2 (3 ng/mL) stimulation of HEK TLR2/6 cells and is expressed relative to cells not treated with SSL3. Data points represent the mean ± SD of at least three independent experiments. (C) Comparison of the TLR2 binding site of SSL3 and the corresponding region of SSL4. Residues of SSL3 and SSL4 are shown in orange and blue sticks, respectively. Black labels refer to the SSL3 sequence; substitutions in SSL4 are labeled in blue. Also shown is the van der Waals surface of SSL3 with hydrophobic regions colored purple and hydrophilic regions colored wheat, emphasizing the hydrophobic nature of the TLR2 binding site. (D) TLR2 inhibitory activity of SSL4 mutants. Indicated amino acids of SSL4 were replaced by amino acids of SSL3. Data points represent the mean ± SD of at least three independent experiments.
Mutagenesis of SSL3 and SSL4.
To confirm the binding site observed in the crystal structure, we mutated SSL3 residues located in the interface to alanines (Fig. 2A). The effect of mutation on inhibitory capacity was measured through IL-8 production after MALP-2 stimulation of HEK cells stably expressing human TLR2–TLR6. Single mutants showed no or only minor effects, with at most a twofold decrease in SSL3 activity (Fig. S1 B–D). Mutation of both Phe156 and Phe158 gave a 100-fold reduction (Fig. 2B). If, in addition to Phe156 and Phe158, nearby residue Pro194 was also mutated, a further small decrease in activity was observed. Mutating a stretch of residues in loop β2–β3, Ile172, Asn174, Arg175, and Phe176 resulted in a moderate 10-fold decrease in activity. Complete loss of SSL3 function could be achieved by combining mutations of the Phe156/Phe158/Pro194 patch and the β2–β3 stretch (SSL3− in Fig. 2B). Mutation of Trp163 and nearby residue Leu211 had no effect on SSL3 activity (Fig. S1D), suggesting that this region of the interaction surface does not contribute significantly to TLR2 binding. It appears that strong SSL3–TLR2 binding is the sum of many—mainly hydrophobic—interactions in which residues Phe156 and Phe158 play a prominent role.
SSL3 and SSL4 show high sequential and structural homology, but substantially differ in their capacity to inhibit TLR2 (5). SSL3 residues important for TLR2 binding are poorly conserved in SSL4 (Fig. 2C), which may explain the 100-fold less potency of SSL4 as a TLR2 inhibitor. The equivalent SSL4 residues in these OB domain loops, however, are also predominantly hydrophobic, and suggest that TLR2 binding involves the same site in SSL4. Additionally, the main-chain conformation of these loops in SSL4 is more similar to TLR2-bound SSL3 than free SSL3 itself (Fig. S2 D and E). To investigate the difference in inhibitory capacity between the two proteins, we replaced amino acids in SSL4 by their counterparts in SSL3. Replacement of both Ile108 and Ile110 by phenylalanines results in a fivefold increased TLR2 inhibition (Fig. 2D). Additional replacement of Val146 by proline enhances its function 20-fold compared with SSL4. Replacement of the β2–β3 stretch (Val124Ile, Asp125Asn, Tyr126Arg) on top of this has a minor additional effect, and generates an SSL4 mutant with the potency of SSL3 (SSL4+ in Fig. 2D). The observed gradual increase of SSL4 potency upon progressive introduction of SSL3 residues confirms that the TLR2 binding sites of SSL3 and SSL4 are located at equivalent sites.
SSL3 Inhibits TLR Dimerization and Lipopeptide Binding.
TLR2 activation vitally depends on the binding of bacterial lipopeptides and subsequent formation of TLR2–TLR1 or TLR2–TLR6 heterodimers. The mechanism of TLR2 inhibition by SSL3 could involve interference in either or both of these steps. From our structural data presented here, it is directly evident that SSL3 blocks productive dimerization; SSL3 binding extensively overlaps with the region of TLR2 that is involved in dimerization with TLR6 (Fig. 3A) as well as TLR1 (Fig. S6A). Because dimerization is crucial for signaling, the functional consequence of SSL3 binding is that TLR2 stimulation by diacyl as well as triacyl lipopeptides is inhibited.
Fig. 3.

Inhibition mechanism of SSL3. (A) Hypothetical complex of TLR2 (gray surface), SSL3 (orange surface), and TLR6 (blue cartoon) as obtained by superposing SSL3–TLR2 and TLR2–TLR6 (PDB ID code 3A79) (16). (Inset) TLR2 residues involved in binding to SSL3 (orange), TLR6 (blue), or both (red). (B) Dimensions of the entrance to the TLR2 lipopeptide binding pocket in the SSL3–TLR2 complex, measured in the presence (Left) and absence (Right) of SSL3. (C) AlphaScreen assay measuring the binding of Pam2CSK4–biotin to mTLR2–Fc fusion protein preincubated with different concentrations of SSL3 or SSL3−. Data are expressed relative to binding in absence of SSL3, and data points represent the mean ± SD of at least three independent experiments.
Fig. S6.
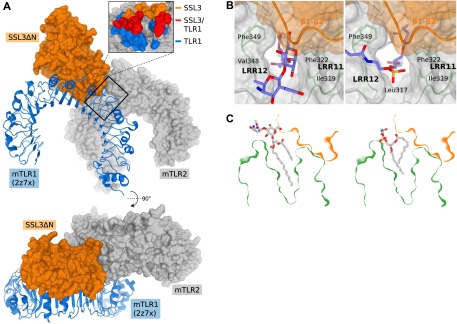
Binding of pnLTA and PE–DTPA, but not TLR1, is compatible with binding to the SSL3–TLR2 complex. (A) Surface representation of the SSL3–TLR2 complex (orange and gray) and a cartoon view of TLR1 (blue), as obtained by superposing the SSL3–TLR2 and TLR2–TLR1 (PDB ID code 2Z7X) (15) crystal structures. (Inset) TLR2 residues involved in binding to SSL3 (orange), TLR1 (blue), or both (red). Overlapping binding sites demonstrate that simultaneous binding of SSL3 and TLR1 to TLR2 is excluded. (B) Superposition of the TLR2–pnLTA (Left) and TLR2–PE–DTPA (Right) structures on the SSL3–TLR2 complex places the head groups of both lipids in the opening to the lipopeptide binding pocket. TLR2 and SSL3 are respectively shown in gray and orange surface representation, the lipids in blue stick representation. The TLR2–pnLTA (PDB ID code 3A7B) and TLR2–PE–DTPA (PDB ID code 3A7C) structures have been obtained from Kang et al. (16). Superposition of the observed conformations of the pnLTA and PE–DTPA ligands in the SSL3–TLR2 structure is possible without severe clashes, indicating that both ligands can be accommodated in a SSL3–TLR2 complex. (C) Cross-section of the SSL3–TLR2 complex, including the TLR2-bound conformations of pnLTA (Left) and PE–DTPA (Right), showing that both lipids can be placed in the SSL3–TLR2 complex without severe clashes.
The structure of the SSL3–TLR2 complex furthermore suggests that binding of lipopeptides is inhibited, because SSL3 docks over the entrance to the ligand-binding pocket. However, an opening of ∼5 × 9 Å remains in the SSL3–TLR2 interface (Fig. 3B), which is about half of the original entrance size. In our AlphaScreen assay we observed concentration-dependent inhibition of Pam2CSK4–TLR2 binding by SSL3, whereas the loss of function mutant SSL3− had no effect (Fig. 3C). These data show that the observed size reduction of the pocket entrance upon binding of SSL3 effectively inhibits lipopeptide binding to TLR2.
SSL3 Binds to the TLR2–Pam2CSK4 Complex.
Our observation of a PC molecule in the lipid binding pocket of the SSL3–TLR2 complex shows that PC does not block SSL3 binding. The conformation of bound PC is noticeably similar to the previously observed binding modes for the synthetic phosphatidylethanolamine derivative PE–DTPA (Fig. 4 A and B) and saccharolipid lipoteichoic acid (pnLTA) from Streptococcus pneumonia (16), ligands that have little or no ability to activate TLR2 (16, 31, 32). In these complexes and our structure (ignoring the presence of SSL3), the lipopeptide binding pockets display similar open conformations and the conformations of pnLTA and PE–DTPA appear to be compatible with binding in the TLR2–SSL3 complex (Fig. S6 B and C).
Fig. 4.
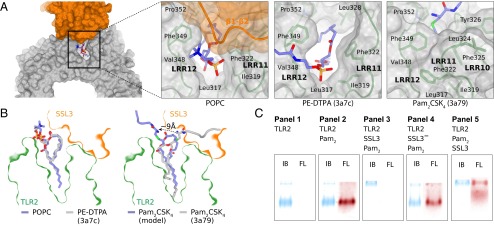
Binding of SSL3 to TLR2–lipid complexes. (A) Positioning of lipid head groups in the entrance to the TRL2 binding pocket: PC in the SSL3–TLR2 complex (Left), PE–DTPA in TLR2 (Center; PDB ID code 3A7C), Pam2CSK4 in TLR2–TLR6 complex (Right; TLR6 not shown, PDB ID code 3A79) (16). (B) Cross-sections of the SSL3–TLR2 surface near the lipopeptide pocket with ligands from A in stick representation: PC (blue, Left), PE–DTPA (gray, Left), and Pam2CSK4 (gray, Right). Binding of SSL3 in the presence of Pam2CSK4 would require a substantial conformational change of its head group as shown in the modeled Pam2CSK4 (blue, Right). (C) Native PAGE analysis of hTLR2 (panel 1) and hTLR2 complexes formed after incubation of hTLR2 (7 μM) with Pam2CSK4Rhodamine (20 μM; 18 h at 37 °C) and/or SSL3 (40 μM; 30 min at 20 °C) in the designated order (panels 2–5). Bands were visualized by rhodamine fluorescence (FL, red) and subsequent staining with Instant Blue (IB, blue).
These observations raise the question whether SSL3 can also bind if an activating ligand like Pam2CSK4 is present—a scenario that would enable SSL3 to block TLR2 signaling even after a bacterial ligand is engaged. The binding modes of nonactivating ligands in TLR2 and Pam2CSK4 in the TLR2–TLR6 complex are, however, completely different. In the latter complex the TLR2 pocket is nearly closed due to a conformational change of LRR10 and LRR11, and the glycerol moiety of the ligand is oriented differently with the head group cysteine bound in the so-called “sulfur site” (16); a conformation that would not be compatible with SSL3 binding (Fig. 4 A and B).
To establish experimentally whether SSL3 is capable of binding a preformed TLR2–Pam2CSK4 complex, we used native PAGE and visualized the presence of bound lipopeptide with fluorescent Pam2CSK4–rhodamine. Addition of Pam2CSK4–rhodamine to TLR2 generates a fluorescent band at the same height as TLR2 alone (Fig. 4C, panels 1 and 2). Incubation of TLR2 with SSL3 followed by the addition of Pam2CSK4–rhodamine results in the appearance of a more slowly migrating, nonfluorescent band containing the SSL3–TLR2 complex (Fig. 4C, panel 3) as was confirmed by in-gel digestion mass spectrometry, whereas no complex is formed with the loss of function mutant SSL3− (Fig. 4C, panel 4). If, however, Pam2CSK4–rhodamine is allowed to bind TLR2 before addition of SSL3, we observe that the band corresponding to the SSL3–TLR2 complex is fluorescent (Fig. 4C, panel 5), implying the formation of a SSL3–TLR2–Pam2CSK4 triple complex. The existence of this triple complex was confirmed by native mass spectrometry (Fig. S7 A–D). Furthermore, binding of Pam2CSK4 to TLR2 does not affect association with SSL3 (Fig. S7E). Therefore, SSL3 is indeed able to block TLR2 signaling after a bacterial ligand is engaged.
Fig. S7.
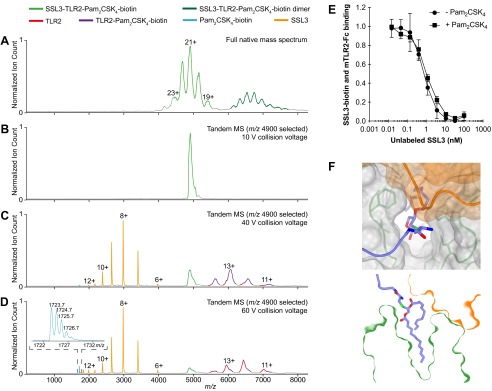
SSL3 binding to a TLR2–Pam2CSK4 complex. (A–D) Native MS analysis of the SSL3–TLR2–Pam2CSK4–biotin complex: (A) Broad-range native mass spectrum showing the ternary complex (light green, 103.1 ± 0.1 kDa) and a second species (dark green), which likely represents a dimer form of the complex (estimated molecular weight 200–209 kDa). (B–D) Tandem MS analysis of the SSL3–TLR2–Pam2CSK4–biotin complex. The region around m/z 4,900 was mass selected and the sequential disassembly of the complex was monitored by increasing the collision voltage to induce dissociation. Shown are the resulting tandem mass spectra. (B) The protein complex remains intact at 10-V collisional activation. (C) At a collision voltage of 40 V, SSL3 (orange, 23.814 ± 0.001 kDa) is expelled from the complex and Pam2CSK4–biotin (cyan) starts to dissociate. Consequently, both free TLR2 (red, 77.27 ± 0.01 kDa) and TLR2–Pam2CSK4–biotin (purple, 79.03 ± 0.05 kDa) are visible. (D) Increasing the collision voltage further to 60 V leads to almost complete dissociation of Pam2CSK4–biotin, the isotope pattern of which is depicted (Inset). The average molecular weight of the ligand was determined to 1724.7 Da (expected: 1724.4 Da). (E) Binding of Pam2CSK4 to TLR2 does not affect the binding of SSL3. Pam2CSK4 was allowed to bind mTLR2–Fc before a competitive AlphaScreen binding assay with biotinylated SSL3 and unlabeled SSL3 was performed. In the presence of Pam2CSK4 a Kd of 0.8 ± 0.2 nM was obtained, compared with the reference of 0.6 ± 0.4 nM in absence of ligand (same reference as shown in Fig. S3C). Data points represent the mean ± SD of at least three independent experiments. (F) Top view and cross-section of the SSL3–TLR2 surface near the lipopeptide pocket. Modeling of Pam2CSK4 in the SSL3–TLR2 complex in a similar conformation as was observed for PC shows that the peptide moiety of Pam2CSK4 can protrude into the solvent region through the observed pocket opening. Pam2CSK4 must adopt this conformation characteristic for nonactivating ligands (Fig. 4 A and B), because its conformation as observed in the Pam2CSK4–TLR2–TLR6 complex is not compatible with the formation of a SSL3–TLR2–Pam2CSK4 triple complex.
In view of the structural data presented above, TLR2 and Pam2CSK4 within the triple complex must adopt a conformation typically observed for TLR2 bound to nonactivating ligands. Modeling shows that it is indeed possible to accommodate Pam2CSK4 in the SSL3–TLR2 complex (Fig. 4B and Fig. S7F). Combined, our data show that SSL3 is able to interfere with TLR2 activation at two stages: first, its binding to TLR2 prevents lipopeptide binding, and second, its binding to an already formed TLR2–lipopeptide complex prevents dimerization.
Discussion
Recognition of bacterial lipopeptides by TLR2 is critical for the defense against S. aureus. From the opposite perspective, inhibition of TLR2 by SSL3 is a powerful mechanism of S. aureus to survive inside its host. The crystal structure of the SSL3–TLR2 complex presented here shows that the highly hydrophobic binding interface is critically dependent on a set of seven SSL3 residues with prominent roles for Phe156 and Phe158. This set of seven residues appears to be highly conserved among SSL3s from different S. aureus strains, but is absent in SSL4, the closest SSL3 relative within the SSL family and itself a weak TLR2 inhibitor. Introduction of these residues in SSL4 enhances its capacity to inhibit TLR2 to a similar level as SSL3 (Fig. 2D). Interestingly, in strain MRSA252(SAR0425), these residues are present in SSL4, whereas they are not conserved in SSL3 (Fig. S8), and, accordingly, SSL4 is the stronger TLR2 inhibitor (5). Possibly, this strain underwent a genetic recombination event in which its overall capacity to evade TLR2 activation has been preserved, underlining the importance of TLR2 evasion.
Fig. S8.

Alignment of SSL3 and SSL4. Sequence alignment highlighting residues found in the TLR2 binding interface of SSL3 from S. aureus strain NCTC 8325 and their conservation in SSL4 from the same strain and SSL3 and SSL4 from strain MRSA252. Nearly all of the SSL3 amino acids involved in TLR2 interactions are present in SSL4 MRSA252, explaining its enhanced TLR2 inhibitory activity with respect to SSL4 from strain NCTC 8325 (5). However, SSL3 from MRSA252 contains only one of the seven conserved amino acids, indicating that genetic recombination may have occurred in this strain that switched the activity between its SSL3 and SSL4. The length of the proteins, however, corresponds more closely to their original family members, with both SSL3s having the longer N-terminal domain. SSL3 and SSL4 have been assigned based on their gene order in the genomes of NCTC 8325 (SAOUHSC_00386 and SAOUHSC_00389) and MRSA252 (SAR_RS02110 and SAR_RS02115).
Sialyl LewisX-dependent mechanisms have been described for functional activity of multiple SSL proteins, including SSL5 and SSL11 (27, 28). The sialyl LewisX binding site is fully conserved in SSL3, but its role in TLR2 inhibition has been unclear. SSL3 residue Arg308, previously described to be crucial for sialic acid binding, was found to be involved in, yet not crucial for, binding and activity of SSL3 (5). Yokoyama et al. (6) reported that mutation of Phe297–Glu298, residues also involved in LewisX binding, results in decreased binding to cells, but has no effect on binding to TLR2 itself. Our crystallographic data show that the distance from the LewisX binding site of SSL3 to the nearest N-linked glycosylation site in both mouse and human TLR2 is too large for interaction to occur (Fig. S3D). Thus, glycan binding does not contribute directly to formation of the specific inhibitory complex, which is therefore exclusively mediated by protein–protein interactions. We hypothesize that the actual functional role of glycan binding is to increase the local SSL3 concentration on the immune cell surface, which is known to be rich in sialyl LewisX sugars (33)—a preconcentration step that would lead to more efficient TLR2 inhibition.
In this study we show that SSL3 interferes in TLR2 activation at two stages: first, SSL3 inhibits binding of bacterial lipopeptides, and, second, if a lipopeptide has already been engaged by TLR2, SSL3 prevents the formation of TLR2–TLR1 and TLR2–TLR6 heterodimers. A critical aspect of the SSL3–TLR2 complex that enables this dual mechanism is the opening to the lipopeptide binding pocket that remains after SSL3 binding. SSL3 only blocks about half of the pocket entrance, and our experiments show that this is sufficient to inhibit lipid entry, but does allow for the accommodation of the head group of a lipopeptide that is already bound to TLR2 before SSL3 binding. Whereas this provides a functional role for the opening, it remains to be seen whether binding of SSL3 to a TLR2–lipopeptide complex is a prevalent pathway in vivo. Alternatively, the opening may also serve a different purpose—namely, enabling the binding of SSL3 to TLR2–phospholipid complexes. It has not been established that TLR2 associates with phospholipids in vivo; however, the presence of copurified PC in our TLR2 preparation suggests that this may well be the case. In this scenario, an opening to the binding pocket of TLR2 is required to prevent steric hindrance of nonactivating phospholipids upon binding of SSL3.
Unraveling the mechanism of TLR2 inhibition by SSL3 gives new insights in the host–pathogen interaction and provides new tools to study TLR2 receptor biology. Aberrant TLR2 activation is linked to several diseases, including acute and chronic inflammatory conditions (34), making it an interesting therapeutic target. Our structural data provide a starting point for the development of SSL3 derivatives that could be used to block TLR2 activation in a therapeutic setting.
Materials and Methods
Expression and Purification of SSL3 and SSL4 Mutants.
The SSL3 and SSL4 genes of S. aureus strain NCTC 8325 (SAOUHSC_00386 and SAOUHSC_00389) were used for construction of truncated proteins SSL3ΔN comprising residues 134–326, SSL4ΔN (residues 79–278), and mutants of SSL3ΔN and SSL4ΔN listed in Table S2. All variants were expressed with a noncleavable N-terminal His6-tag in Escherichia coli Rosetta-gami(DE3)pLysS, refolded from insoluble fractions and purified as described (5). Proteins were stored in PBS, and protein purity was determined as >95% by SDS/PAGE.
Table S2.
List of all SSL3 and SSL4 mutants and their IC50 values
| Mutants | Mutations | IC50 values (M) |
| SSL3 | ||
| SSL3 | — | 2.2 ± 0.7 × 10−9 |
| SSL3ΔN | Δ1–133 | 1.9 ± 0.8 × 10−9 |
| SSL3ΔN N174A | N174A | 3.1 ± 1.1 × 10−9 |
| SSL3ΔN R175A | R175A | 1.5 ± 0.7 × 10−9 |
| SSL3ΔN NR174AA | N174A, R175A | 3.3 ± 2.1 × 10−9 |
| SSL3ΔN INRF172AAAA | I172A, N174A, R175A, F176A | 1.2 ± 0.3 × 10−8 |
| SSL3ΔN F156A | F156A | 4.4 ± 2.2 × 10−9 |
| SSL3ΔN F158A | F158A | 2.6 ± 0.6 × 10−9 |
| SSL3ΔN P194A | P194A | 2.0 ± 0.5 × 10−9 |
| SSL3ΔN W163A | W163A | 2.4 ± 0.5 × 10−9 |
| SSL3ΔN W163A L211A | W163A, L211A | 5.8 ± 3.4 × 10−9 |
| SSL3ΔN FF156AA | F156A, F158A | 1.3 ± 0.5 × 10−7 |
| SSL3ΔN FF156AA P194A | F156A, F158A, P194A | 7.3 ± 10.7 × 10−6 |
| SSL3ΔN FF156AA P194A INRF172AAAA | F156A, F158A, P194A, I172A, N174A, R175A, F176A | Mutant does not reach IC50 |
| SSL4 | ||
| SSL4ΔN | Δ1–78 | 1.7 ± 1.3 × 10−7 |
| SSL4ΔN II108FF | I108F, I110F | 2.3 ± 1.2 × 10−8 |
| SSL4ΔN II108FF V146P | I108F, I110F, V146P | 7.6 ± 5.9 × 10−9 |
| SSL4ΔN II108FF V146P VDY124INR | I108F, I110F, V146P, V124I, D126N, Y127R | 3.6 ± 1.7 × 10−9 |
For crystallization purposes, SSL3ΔN was expressed with a cleavable N-terminal His6-tag and isolated following the same procedure. Tobacco etch virus (TEV) protease cleavage was performed overnight in 25 mM Tris-Cl buffer (pH 8.2) and 150 mM NaCl. After addition of imidazole to a final concentration of 10 mM, TEV protease and any residual undigested SSL3 were removed by filtration through a HiTrap chelating HP column. SSL3ΔN was ultimately purified by size-exclusion chromatography over a Superdex75 column (GE Healthcare) equilibrated in 10 mM Tris-Cl buffer (pH 8.2) and 150 mM NaCl, and concentrated to 12 mg/mL.
Expression and Purification of TLR2 Ectodomains.
Ectodomains of mouse (Gln25–Ala588, NM_011905) and human (Lys19–Ala589, NM_003264) TLR2 were transiently expressed with the N-terminal His6-StrepII3-TEV tag in HEK293-EBNA1-S and HEK293-EBNA-1 cells, respectively (U-Protein Express BV) as described (5). Protein yields were optimized by plasmid titration (35), which indicated that transfections with 10-fold dilutions of expression plasmid in nonexpressing dummy plasmid improved TLR2 production approximately two- to threefold. Further improvement of protein yield was achieved by cotransfecting a PRAT4A (NM_006586) expression plasmid at a ratio of 1:40. Crystallization experiments with mTLR2 were preceded by removal of the purification tag with TEV protease as described for SSL3ΔN and gel filtration on a preequilibrated Superdex 200 column (GE Healthcare) with 10 mM Tris-Cl buffer (pH 8.2) and 150 mM NaCl.
Crystallization and Data Collection of SSL3ΔN and the SSL3ΔN–mTLR2 Complex.
SSL3ΔN crystals were grown at 292 K using sitting-drop vapor diffusion against a well solution containing 0.2 M potassium thiocyanate and 20% (wt/vol) PEG 3350. Crystals were cryoprotected in well condition containing 20% (vol/vol) glycerol before flash-freezing in liquid nitrogen. Diffraction data to 1.94 Å resolution were collected at the Swiss Light Source on the PX beamline. For crystallization of the SSL3–TLR2 complex, the individual proteins were mixed in a 1.1:1 molar ratio with final concentrations of 1.4 mg/mL and 3.8 mg/mL, respectively. Crystals were obtained through sitting-drop vapor diffusion against a well solution containing 0.1 M PCB buffer (pH 5.0; sodium propionate, sodium cacodylate, and Bis-Tris propane) (Qiagen) and 25% (wt/vol) PEG 1500. For data collection, crystals were cryoprotected in well solution containing 20% (vol/vol) glycerol before flash-freezing in liquid nitrogen. X-ray diffraction data to 3.2 Å resolution were collected at the PETRA III beamline (DESY). Details about structure determination and refinement procedures are included in SI Materials and Methods. Statistics of data processing and refinement are listed in Table S1.
Cell Lines.
HEK cells expressing TLR2 and TLR6 were obtained from InvivoGen and cultured in DMEM in the presence of 10 µg/mL blasticidin, 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% (vol/vol) FCS.
Ligand-Induced Cytokine Production.
HEK-TLR2/6 cells were seeded in 96-well culture plates. After reaching confluency, cells were incubated with the SSLs or SSL mutants for 30 min at 37 °C. MALP-2 (Santa Cruz) was then added to a final concentration of 3 ng/mL. After 6 h, culture supernatants were collected and tested for IL-8 production using specific ELISA, following manufacturer’s instructions (Sanquin).
Binding Studies.
The AlphaScreen assay (Perkin-Elmer Life Sciences) (30) was used to determine TLR2–ligand interactions. Murine TLR2–Fc (R&D systems), final concentration 9 nM, was mixed with a concentration range (0.01–100 nM) of SSL3ΔN or SSL3ΔN− (FF156AA P194A INRF172AAAA) in PBS containing 0.05% human serum albumin. After 45 min, Pam2CSK4–biotin (Tocris) was added to a final concentration of 9 nM and incubation was continued for another 45 min. Next, 20 µg/mL of streptavidin donor beads and 20 µg/mL Protein-G acceptor beads were added and incubated for 45 min. Samples were measured at 680 nm in a CLARIOstar microplate reader (BMG Labtech).
Native PAGE experiments were performed to study TLR2–ligand interactions as described (36). Purified human TLR2 (7 µM, final concentration), in some cases preincubated with SSL3ΔN or SSL3ΔN− (40 µM) for 30 min at room temperature, was mixed with Pam2CSK4Rhodamine (20 µM, InvivoGen) and incubated for 18 h at 37 °C. To examine whether ligand binding and SSL3 binding can simultaneously occur, first TLR2 and Pam2CSK4Rhodamine were allowed to bind for 18 h at 37 °C, after which SSL3 was added. Samples were loaded on 12.5% native glycine gels and run for 3 h at 200 V. Rhodamine fluorescence was detected using a LAS 4010 imaging system (GE Healthcare) equipped with a 520-nm excitation LED and a 575-20BP emission filter. Subsequently, the gel was stained with Instant Blue protein stain (Expedeon).
SI Materials and Methods
Data Collection and Refinement of the SSL3ΔN and SSL3ΔN–mTLR2 Structures.
Diffraction data to 1.94 Å obtained for crystals of SSL3ΔN was integrated with MOSFLM and further processed using CCP4 software (37). Integrated data were first scaled with Aimless. Molecular replacement was then performed with PHASER and the structure of SSL4 as initial search model (PDB ID code 4DXF) (38). The structure was refined with REFMAC alternated with manual model improvement using COOT. Rwork and Rfree had final values of 0.177 and 0.224. Statistics of data processing and refinement are listed in Table S1.
Diffraction data to 3.2 Å obtained for crystals of the SSL3ΔN–mTLR2 complex was integrated with XDS. The structure was solved by molecular replacement in PHASER using chain A of the SSL3ΔN structure and residues 27–506 of mTLR2 (PDB ID code 2Z81) (15) as initial search models. Refinement of the model was performed with Phenix (39). The initial molecular replacement solution that includes the structures of SSL3ΔN and mTLR227–506 was further optimized by performing a rigid-body refinement using separate rigid bodies comprising SSL3 residues 136–224 and 225–326 and TLR2 residues 27–250 and 251–506. Further refinement included TLS refinement using automatic TLS group identification. In view of the limited resolution of the diffraction data, external dihedral angle restraints derived from the structures of SSL3ΔN and the 1.8-Å resolution structure of mTLR227–506 were applied, except for regions where the electron density convincingly indicated a different conformation and partially modeled N-linked glycans connected to Asn414 and Asn442. One of these regions concerns loops in LRR10 and LRR11, which determine the shape and size of the lipopeptide pocket entrance. LRR11 contacts SSL3 and its conformation is well-defined. The long LRR10 loop is not involved in SSL3 binding and although it displays increased B-factors (Fig. S4A), the electron density allowed building of the entire loop.
In the mTLR2–VLR6 fusion protein, LRR19 and the LRRCT domain of mTLR2 have been replaced by a fragment of hagfish VLR. Sequence identity between this VLR fragment and mTLR2 is only 11%. Therefore, we used the corresponding region of hTLR3 (PDB ID code 2A0Z) (40), which is 25% identical (Fig. S4F), to guide building of LRR19 and the LRRCT domain. The LRRCT domain harbors a CXC motive typical for C-cap domains of LRR structures. Both cysteines are involved in disulfide bridge formation. Whereas the Cys537–Cys564 bridge could be modeled with confidence (Fig. S4E), no electron density was visible for the Cys539–Cys585 bridge. Possibly, radiation damage suffered by this bond explains the poor ordering of this region, although it cannot be excluded that it is inherently flexible. TLR2 residues on the C-terminal side of Cys564 either were modeled as polyalanine (residues 569–575) or are absent from the final model (residues 576–587).
Continuous residual density in the lipid binding pocket of TLR2 suggested the presence of a copurified diacetylated lipid (Fig. S5A). Native mass spectrometry confirmed the presence of a lipid and identified it as PC with acyl chain lengths varying form from C12 to C20 (Fig. S5 B–H). Accordingly, we modeled PC in the binding pocket with its phosphoglycerol moiety positioned in the pocket opening and the choline headgroup located just outside, interacting with a chloride ion. The available electron density allowed modeling of C13 and C12 acyl chains at the sn1 and sn2 hydroxyl groups, respectively. The size of the TLR2 pocket, however, does not exclude binding of longer acyl chains. Refinement of the complex after inclusion of PC had a negligible effect on Rwork and Rfree that have final values of 0.231 and 0.271, respectively. Statistics of data processing and the final model are listed in Table S1.
Mass Spectrometry.
hTLR2 and Pam2CSK4–biotin were allowed to bind overnight at room temperature (RT), before addition of SSL3. The final complex with equimolar concentrations of 12 μM was incubated at RT for 1 h. mTLR2 and the SSL3–hTLR2–Pam2CSK4–biotin complex were buffer-exchanged to 150 mM ammonium acetate, pH 7.0 (for native MS analysis) or 0.1% formic acid solution (for MS under denaturing conditions). Buffer exchange was performed in eight steps at 13,000 × g and 4 °C using centrifugal filter units with a 30-kDa molecular weight cutoff (Millipore). Subsequently, samples were diluted to a protein concentration of 3–5 μM and introduced into the mass spectrometer with gold-coated borosilicate needles to facilitate electrospray ionization. Denatured samples were analyzed on a modified LCT time-of-flight mass spectrometer (Waters) with the instrument settings optimized as follows: capillary voltage 1,100–1,300 V, sample cone voltage 40–100 V, extraction cone voltage 0–20 V, and source region pressure 4–6 mbar. Native MS was performed using a modified quadrupole time-of-flight instrument (Waters) (41), which was operated with the following parameters: capillary voltage 1,300–1,400 V, sample cone voltage 20–30 V, extraction cone voltage 0–10 V, collision cell voltage 10 V, source region pressure 8 mbar, collision gas pressure 0.01 mbar. For native tandem MS experiments, in-source activation was optimized by increasing sample and extraction cone voltages to 30–100 V and 10–20 V, respectively. The collision cell voltage, which is applied after mass selection of the precursor ion, was varied to monitor the respective dissociation of phospholipids, SSL3, or Pam2CSK4–biotin. All mass spectra were acquired in positive ion mode and calibrated using CsI clusters. Data analysis was performed with MassLynx v4.1 (Waters).
Construction of BAP-Tagged SSL3ΔN and in Vitro Biotinylation.
A biotin acceptor peptide (BAP-tag; GLNDIFEAQKIEWHE) was incorporated by PCR in between the His-tag and the coding sequence of SSL3ΔN. Expression and purification was performed as described in Materials and Methods.
SSL3-BAP was dialyzed to 50 mM Tris, 25 mM NaCl, pH 8.0. BAP-tagged SSL3 was biotinylated in vitro at 37 °C for 4 h using a mixture with final concentrations of 22 µM protein, 10 mM ATP, 50 µM d-biotin (Sigma-Aldrich), 10 mM MgCl2, and 0.44 µM BirA biotin–protein ligase (Avidity). Biotinylation was confirmed with Western Blot analysis using StrepTactin–HRP (1:10.000; IBA).
Binding Studies.
AlphaScreen assay was performed for Kd determinations of the SSL3–TLR2 complex using a competitive binding format (30). mTLR2–Fc (0.1 nM, final concentration) was simultaneously incubated with biotin-labeled BAP-SSL3 (1 nM) and different concentrations of unlabeled SSL3 (ranging from 0.01 nM to 100 nM) for 45 min. Binding was determined by addition of 20 µg/mL streptavidin donor beads and 20 µg/mL Protein-G acceptor beads. To examine the effect of TLR2-ligand binding on the Kd, Pam2CSK4 (1 nM) was preincubated with TLR2 for 45 min before the competitive binding assay was performed.
Acknowledgments
We thank Swiss Light Source and DESY for providing data collection facilities, and the beamline scientists for their help with data collection. This work was supported in part by Dutch Top Institute Pharma Project D1-101, ECHO Grant 700.58.006 from the Council of Chemical Sciences of the Netherlands Organization for Scientific Research (to E.G.H.), ManiFold Project 317371 from the Seventh Framework Programme of the European Union (to the Bijvoet Center), and ZonMw Grant 205200004 from the Netherlands Organization for Health Research and Development (to J.A.G.v.S.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.-O.L. is a guest editor invited by the Editorial Board.
Data deposition: The atomic coordinates and structure factors for SSL3ΔN and for the SSL3ΔN–mTLR2 complex have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 5D3D and 5D3I).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1502026112/-/DCSupplemental.
References
- 1.Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339(8):520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Williams RJ, et al. Identification of a novel gene cluster encoding staphylococcal exotoxin-like proteins: Characterization of the prototypic gene and its protein product, SET1. Infect Immun. 2000;68(8):4407–4415. doi: 10.1128/iai.68.8.4407-4415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lina G, et al. International Nomenclature Committee for Staphylococcal Superantigens Standard nomenclature for the superantigens expressed by Staphylococcus. J Infect Dis. 2004;189(12):2334–2336. doi: 10.1086/420852. [DOI] [PubMed] [Google Scholar]
- 4.Jongerius I, et al. Staphylococcal complement evasion by various convertase-blocking molecules. J Exp Med. 2007;204(10):2461–2471. doi: 10.1084/jem.20070818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bardoel BW, et al. Evasion of Toll-like receptor 2 activation by staphylococcal superantigen-like protein 3. J Mol Med (Berl) 2012;90(10):1109–1120. doi: 10.1007/s00109-012-0926-8. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama R, et al. Staphylococcal superantigen-like protein 3 binds to the Toll-like receptor 2 extracellular domain and inhibits cytokine production induced by Staphylococcus aureus, cell wall component, or lipopeptides in murine macrophages. Infect Immun. 2012;80(8):2816–2825. doi: 10.1128/IAI.00399-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bubeck Wardenburg J, Williams WA, Missiakas D. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Natl Acad Sci USA. 2006;103(37):13831–13836. doi: 10.1073/pnas.0603072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashimoto M, et al. Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int Immunol. 2006;18(2):355–362. doi: 10.1093/intimm/dxh374. [DOI] [PubMed] [Google Scholar]
- 9.Yimin KM, et al. Contribution of toll-like receptor 2 to the innate response against Staphylococcus aureus infection in mice. PLoS One. 2013;8(9):e74287. doi: 10.1371/journal.pone.0074287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165(10):5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 11.Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. 2014;14(8):546–558. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- 12.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi O, et al. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. 2001;13(7):933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi O, et al. Cutting edge: Role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169(1):10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 15.Jin MS, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130(6):1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Kang JY, et al. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity. 2009;31(6):873–884. doi: 10.1016/j.immuni.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 17.Bestebroer J, et al. Staphylococcal superantigen-like 5 binds PSGL-1 and inhibits P-selectin-mediated neutrophil rolling. Blood. 2007;109(7):2936–2943. doi: 10.1182/blood-2006-06-015461. [DOI] [PubMed] [Google Scholar]
- 18.Walenkamp AME, et al. Staphylococcal SSL5 binding to human leukemia cells inhibits cell adhesion to endothelial cells and platelets. Cell Oncol. 2010;32(1-2):1–10. doi: 10.3233/CLO-2009-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Haas CJC, et al. Staphylococcal superantigen-like 5 activates platelets and supports platelet adhesion under flow conditions, which involves glycoprotein Ibalpha and alpha IIb beta 3. J Thromb Haemost. 2009;7(11):1867–1874. doi: 10.1111/j.1538-7836.2009.03564.x. [DOI] [PubMed] [Google Scholar]
- 20.Bestebroer J, et al. Staphylococcal SSL5 inhibits leukocyte activation by chemokines and anaphylatoxins. Blood. 2009;113(2):328–337. doi: 10.1182/blood-2008-04-153882. [DOI] [PubMed] [Google Scholar]
- 21.Bestebroer J, et al. Functional basis for complement evasion by staphylococcal superantigen-like 7. Cell Microbiol. 2010;12(10):1506–1516. doi: 10.1111/j.1462-5822.2010.01486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel D, Wines BD, Langley RJ, Fraser JD. Specificity of staphylococcal superantigen-like protein 10 toward the human IgG1 Fc domain. J Immunol. 2010;184(11):6283–6292. doi: 10.4049/jimmunol.0903311. [DOI] [PubMed] [Google Scholar]
- 23.Itoh S, et al. Staphylococcal superantigen-like protein 10 (SSL10) binds to human immunoglobulin G (IgG) and inhibits complement activation via the classical pathway. Mol Immunol. 2010;47(4):932–938. doi: 10.1016/j.molimm.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 24.Itoh S, et al. Staphylococcal superantigen-like protein 10 (SSL10) inhibits blood coagulation by binding to prothrombin and factor Xa via their γ-carboxyglutamic acid (Gla) domain. J Biol Chem. 2013;288(30):21569–21580. doi: 10.1074/jbc.M113.451419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walenkamp AME, et al. Staphylococcal superantigen-like 10 inhibits CXCL12-induced human tumor cell migration. Neoplasia. 2009;11(4):333–344. doi: 10.1593/neo.81508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bardoel BW, Strijp JAG. Molecular battle between host and bacterium: Recognition in innate immunity. J Mol Recognit. 2011;24(6):1077–1086. doi: 10.1002/jmr.1156. [DOI] [PubMed] [Google Scholar]
- 27.Chung MC, et al. The crystal structure of staphylococcal superantigen-like protein 11 in complex with sialyl Lewis X reveals the mechanism for cell binding and immune inhibition. Mol Microbiol. 2007;66(6):1342–1355. doi: 10.1111/j.1365-2958.2007.05989.x. [DOI] [PubMed] [Google Scholar]
- 28.Baker HM, et al. Crystal structures of the staphylococcal toxin SSL5 in complex with sialyl Lewis X reveal a conserved binding site that shares common features with viral and bacterial sialic acid binding proteins. J Mol Biol. 2007;374(5):1298–1308. doi: 10.1016/j.jmb.2007.09.091. [DOI] [PubMed] [Google Scholar]
- 29.Kim HM, et al. Structural diversity of the hagfish variable lymphocyte receptors. J Biol Chem. 2007;282(9):6726–6732. doi: 10.1074/jbc.M608471200. [DOI] [PubMed] [Google Scholar]
- 30.Bosse R, Illy C, Chelsky D, Sciences PL. Application Note. Principles of AlphaScreen. PerkinElmer Life Sciences; Montreal: 2002. [Google Scholar]
- 31.Han SH, Kim JH, Martin M, Michalek SM, Nahm MH. Pneumococcal lipoteichoic acid (LTA) is not as potent as staphylococcal LTA in stimulating Toll-like receptor 2. Infect Immun. 2003;71(10):5541–5548. doi: 10.1128/IAI.71.10.5541-5548.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zähringer U, Lindner B, Inamura S, Heine H, Alexander C. TLR2 - promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology. 2008;213(3-4):205–224. doi: 10.1016/j.imbio.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 33.Munro JM, et al. Expression of sialyl-Lewis X, an E-selectin ligand, in inflammation, immune processes, and lymphoid tissues. Am J Pathol. 1992;141(6):1397–1408. [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Yin H, Zhao M, Lu Q. TLR2 and TLR4 in autoimmune diseases: A comprehensive review. Clin Rev Allergy Immunol. 2014;47(2):136–147. doi: 10.1007/s12016-013-8402-y. [DOI] [PubMed] [Google Scholar]
- 35.Halff EF, Versteeg M, Brondijk THC, Huizinga EG. When less becomes more: Optimization of protein expression in HEK293-EBNA1 cells using plasmid titration - a case study for NLRs. Protein Expr Purif. 2014;99:27–34. doi: 10.1016/j.pep.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 36.Jiménez-Dalmaroni MJ, et al. Soluble human TLR2 ectodomain binds diacylglycerol from microbial lipopeptides and glycolipids. Innate Immun. 2015;21(2):175–193. doi: 10.1177/1753425914524077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hermans SJ, et al. Structural and functional properties of staphylococcal superantigen-like protein 4. Infect Immun. 2012;80(11):4004–4013. doi: 10.1128/IAI.00764-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell JK, et al. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci USA. 2005;102(31):10976–10980. doi: 10.1073/pnas.0505077102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Heuvel RHH, et al. Improving the performance of a quadrupole time-of-flight instrument for macromolecular mass spectrometry. Anal Chem. 2006;78(21):7473–7483. doi: 10.1021/ac061039a. [DOI] [PubMed] [Google Scholar]