

Abstract
Vitiligo is an acquired depigmentation disorder largely caused by defective melanocyte- or autoimmunity-induced melanocyte destruction. The aryl hydrocarbon receptor (AHR) is essential for melanocyte homeostasis and immune process, and abnormal AHR was observed in vitiligo. We previously identified the T allele of AHR −129C > T variant as a protective factor against vitiligo. However, biological characterization underlying such effects is not fully certain, further validation by mechanistic research is warranted and was conducted in the present study. We showed that −129T allele promoted AHR transcriptional activity through facilitating its interaction with SP1 transcription factor (SP1) compared with −129C allele. We subsequently found reduced peripheral AHR and SP1 transcript expressions in vitiligo and a negative correlation of AHR level with disease duration. We also investigated AHR-related cytokines and observed increased serum TNF-α concentration and diminished serum levels of IL-10 and TGF-β1 in vitiligo. Further genetic analysis showed that -129T carriers possessed higher levels of AHR and IL-10 than −129C carriers. Therefore, our study indicates that the modulation of AHR transcription by a promoter variant has a profound influence on vitiligo, not only advancing our understanding on AHR function but also providing novel insight into the pathogenesis of degenerative or autoimmune diseases including vitiligo.
Vitiligo is a chronic depigmentation disorder resulting from melanocyte destruction. The incidence of vitiligo is approximately 0.5 ∼ 8% worldwide, and over 50% of the patients develop the disease between the ages of 10 and 30 years1. Vitiligo deeply affects both the physical and mental health of patients, the course and treatment response of which are highly variable2. Contributing factors for the initiation of vitiligo are unknown, although genetic susceptibility, autoimmunity, oxidative stress and melanocyte-intrinsic abnormalities have been implicated1. Accumulating data emphasize the crucial role of melanocyte-inherent defects in vitiligo, with evidence of aberrant melanogenesis pathway and impaired melanocyte development1,3. Previous studies have showed that abnormality of the rate-limiting enzymes in melanin synthesis process, including tyrosinase (TYR) and tyrosinase-related protein (TYRP), may induce excessive toxic metabolites and trigger cellular damage in vitiligo2. In addition, defection in stem cell factor/stem cell factor receptor (SCF/C-Kit) melanocyte survival pathway has been suggested to contribute to melanocyte apoptosis in vitiligo2,4,5. Besides directly inducing apoptosis, melanocyte-inherent aberrations could further initiate or amplify the autoimmune damage in vitiligo6,7,8,9.
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor and belongs to the basic-helix-loop-helix family10. Upon binding ligand, AHR translocates into the nucleus to govern target genes11. AHR is well characterized for immune regulation through mediating T-cell differentiation and cytokine milieu12,13, and more recently, scientific evidence strongly supports that AHR is vital to melanocyte homeostasis. Activation of AHR pathway stimulated melanogenesis by improving expressions of TYR and TYRP in human melanocytes14. Ahr-deficient mice showed decreased melanocyte number and hypopigmentation in the tail skin caused by reduced levels of SCF and C-Kit15. Additionally, significantly declined expressions of AHR and its target genes were observed in the epidermis of vitiligo patients16. These data imply that mutation or dysfunction of AHR might be involved in vitiligo.
In consideration of the critical role of AHR in both melanocyte and cellular immunity and aberrant AHR pathway in vitiligo, we previously evaluated the potential association between AHR polymorphisms and vitiligo susceptibility. Our data demonstrated that the T allele of −129C > T variant (rs10249788) in the AHR promoter region is associated with a protective effect on vitiligo in Han Chinese populations17, which might be a functional variation through altering AHR transcription process. The promoter of human AHR gene lacks TATA and CCAAT boxes but possesses several putative SP1 transcription factor (SP1) binding sites within a highly GC-rich region18. SP1 is a Cys2/His2-type zinc-finger transcription factor that binds to GC box elements (5′-GGGCGG-3′) within promoter region19. SP1 is particularly important to the TATA-less genes, which regulates transcription of multiple target genes involved in cell growth, differentiation, apoptosis and immune response20. Early researches have revealed that SP1 dominates the maximal constitutive activity and basal expression of AHR gene via binding to these GC-rich motifs21. The abnormal interaction between SP1 and AHR promoter is responsible for AHR down-regulation in human diseases22,23. As the −129C > T polymorphism is in the core promoter region of AHR gene and is juxtaposed 5′ to the SP1 recognition sequence, we therefore hypothesized that the AHR −129C > T polymorphism could influence its transcription and downstream effectors in melanocyte biology or immune system, which could further affect the development of vitiligo. In the present study, we performed functional research to explore the molecular mechanisms underlying such genetic marker for vitiligo.
Results
Effects of AHR −129C > T polymorphism on AHR transcriptional activity
To assess the AHR promoter activity related to −129C > T polymorphism, C or T promoter constructs were transiently transfected in human normal melanocyte PIG1 cells, human malignant melanoma LiBr cells and human embryonic kidney 293T cells. As shown in Fig. 1, the vectors with −129T allele had enhanced relative luciferase activity compared with that of those with −129C allele (P = 0.018 for PIG1 cells; P = 0.010 for LiBr cells; P = 0.023 for 293T cells). These results suggest that the AHR −129T allele possesses an increased AHR transcriptional activity.
Figure 1. Effects of the −129C > T polymorphism on AHR promoter activity.
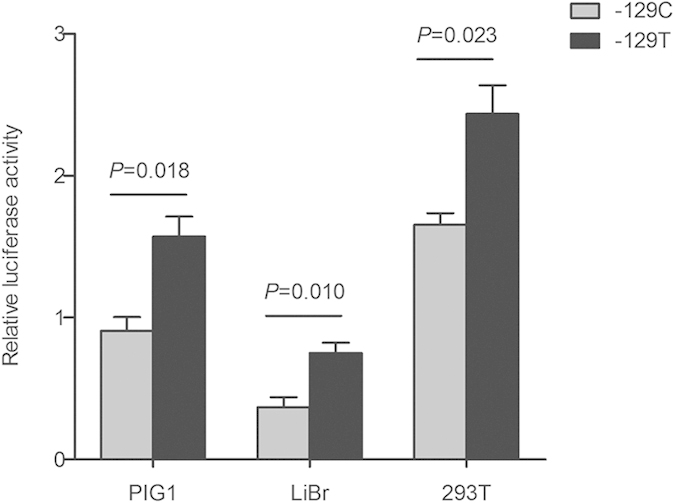
The luciferase reporter plasmids (pGL3-Basic) containing −129C or −129T allele were transiently transfected in PIG1, LiBr and 293T cells. The luciferase activity was normalized against the Renilla luciferase, compared with the construct counterpart. Each column represents Mean ± SEM of three independent experiments.
Impacts of AHR −129C > T polymorphism on SP1 binding to AHR promoter
Both nucleotide sequences and DNA shape influence specific binding of proteins to DNA24,25. The hydroxyl radical cleavage pattern embodies information on sequence-dependent variation in DNA structure, including solvent accessibility, minor groove width and electrostatic potential26,27. Where the minor groove is wide, and deoxyribose backbone hydrogens are exposed, cleavage intensity is high; where the groove is narrow, and backbone hydrogens are diminished in exposure, cleavage intensity is low27,28. Minor groove width and electrostatic potential are important for protein binding25. Specifically, narrowing of the groove is associated with more negative electrostatic potential, which is beneficial for proteins to insert side chains with positive charge into the groove28,29. To test the effect of AHR −129C > T variant on DNA local structure, bioinformatic analysis was performed using the OH Radical Cleavage Intensity Database (ORChID) and showed different hydroxyl radical cleavage patterns among variants of the AHR −129C > T polymorphism. As shown in Fig. 2c, the T allele with relatively lower score reflects narrowing of the DNA backbone, which might function as an electrostatic groove to further improve the DNA-protein interaction.
Figure 2. Analysis of SP1 binding to the AHR promoter around −129C > T polymorphism.

(a) Electrophoretic mobility shift assay with biotin-labeled either −129C or −129T probes and PIG1 cell nuclear extracts. Lanes 1 and 5, mobilities of the labeled −129C or −129T probes without nuclear extracts; lanes 2 and 6, mobilities of the labeled −129C or −129T probes with nuclear extracts in the absence of competitor; lanes 3 and 7, shifted bands were abolished by 100-fold unlabeled cold probes; lanes 4 and 8, super shift assays incubating with anti-SP1 antibody showed the supershifted protein complex. (b) Quantification of bands in Fig. 2a (lanes 2 and 6) was done using ImageJ software (National Institutes of Health), with the C allele binding being defined as 100%. Results are from three independent experiments with similar results. (c) Allelic difference in DNA surface structure was detected at AHR −129C > T polymorphism by using predicted hydroxyl radical cleavage patterns. (d) Western blots showed that PIG1 and LiBr cells expressed relative levels of SP1 protein. (e) Chromatin immunoprecipitation assay with PIG1 cells (−129CC genotype) and LiBr cells (−129TT genotype) in the presence of anti-SP1 (lane 3), anti-RNA Polymerase II (positive control; lane 7) antibodies, or control IgG (negative control; lane 2 and 6). (f) The SP1 inhibitor mithramycin A suppressed the expression of AHR protein in PIG1 and LiBr cells, measured by western bolts. These data confirm the biological interaction between SP1 and AHR promoter influenced by the −129 C > T polymorphism.
To detect the role of −129C > T polymorphism in the interplay between AHR promoter and transcriptional regulators, we carried out electrophoretic mobility shift assays (EMSA) and found the DNA-protein complex with stronger intensity for the −129T allele than −129C allele (Fig. 2a, lanes 2 and 6; P = 0.008, Fig. 2b), which was abolished by excessive unlabeled probes (Fig. 2a, lanes 3 and 7). Since the AHR −129C > T polymorphism is close to the binding site of SP1, we then conducted supershift assays and detected that anti-SP1 antibody supershifted the C and T allele-specific bands (Fig. 2a, lanes 4 and 8), implying that this promoter variant could modify allele-specific binding affinity of SP1. We next performed chromatin immunoprecipitation assays (ChIP) to verify these observations within PIG1 cells (−129CC) and LiBr cells (−129TT). Both cell lines expressed comparable levels of SP1 protein (Fig. 2d). Similarly, −129TT genotype had higher abundance in the interaction between SP1 and AHR promoter than that of −129CC genotype (Fig. 2e, lane 3). To affirm the role of SP1 activation in AHR expression, PIG1 and LiBr cells were treated with 100 nM mithramycin A, a specific SP1 inhibitor that competitively binds to the SP1-binding sites30. The basal expression of AHR protein was relatively higher in LiBr cells (−129TT) than PIG1 cells (−129CC), and mithramycin A treatment suppressed AHR protein expression in PIG1 and LiBr cells (Fig. 2f). Taken together, these data reveal that SP1 prefers to bind to the AHR promoter containing −129T allele rather than −129C allele.
AHR and SP1 transcript levels in vitiligo patients and controls with different genotypes for the AHR −129C > T polymorphism
To investigate the impact of −129C > T polymorphism on AHR transcript level and the correlation between SP1 and AHR expressions, we measured the AHR and SP1 mRNA levels in the peripheral blood mononuclear cells (PBMCs) from 23 vitiligo patients and 23 age- and sex-matched controls harboring different AHR −129 genotypes (n = 16 for CC, n = 6 for CT and n = 1 for TT in vitiligo patients; n = 17 for CC, n = 5 for CT and n = 1 for TT in controls; Hardy–Weinberg equilibrium test: χ2 = 0.571, P = 0.450). We observed positive correlation between AHR and SP1 expressions (R = 0.805, P < 0.001, Fig. 3d), both of which were obviously diminished in vitiligo patients compared with controls (P = 0.007 for AHR, Fig. 3a; P < 0.001 for SP1, Fig. 3b). Moreover, the AHR transcript expression was negatively correlated with total disease duration (R = −0.556, P = 0.020, Fig. 3e), but no significant relevance with disease onset age and body surface area (BSA) involvement (Fig. 3f,g). As expected, individuals with protective −129TT/CT genotypes had higher AHR level than those with −129CC genotype (6.059 ± 1.467 vs. 2.476 ± 0.356, P = 0.017, Fig. 3c). These findings implicate that the AHR −129C > T polymorphism reflects on the level of AHR transcript expression.
Figure 3. Differential mRNA levels of AHR and SP1 in individuals carrying different AHR −129 genotypes.

(a) Transcript level of AHR was significantly declined in vitiligo patients compared with controls. (b) Significant difference in SP1 mRNA level between vitiligo patients and controls was detected. (c) Individuals with −129CT/TT genotypes expressed higher level of AHR mRNA than those in the −129CC group. (d) AHR transcript expression was positively correlated with SP1 mRNA level. Spearman’s correlation analysis of AHR mRNA expression to disease duration (month; e), onset age (year; f) and body surface area involvement (BSA, %; g) was conducted in 17 vitiligo patients with integrity data of clinical characteristics (n = 13 for −129CC and n = 4 for −129CT + TT in vitiligo patients).
Association between the AHR −129C > T polymorphism and serum cytokine levels in vitiligo
Because T cell-mediated autoimmunity and altered cytokines are involved in vitiligo as well as AHR plays a pivotal role in T cell development and cytokine production, mutation or abnormal expression of AHR may influence cytokine levels associated with vitiligo. We therefore examined the major cytokines of T helper and regulatory T cells in 16 vitiligo patients and 16 age- and sex-matched controls carrying various AHR −129 genotypes (n = 10 for CC, n = 4 for CT and n = 2 for TT in vitiligo patients; n = 11 for CC, n = 4 for CT and n = 1 for TT in controls; Hardy–Weinberg equilibrium test: χ2 = 0.515, P = 0.473). We detected significantly increased TNF-α concentration and decreased levels of IL-10 and TGF-β1 in the serum of vitiligo patients compared with controls (29.451 ± 4.748 ng/ml vs. 66.500 ± 17.805 ng/ml, P = 0.022 for TNF-α; 20.649 ± 9.514 vs. 6.375 ± 1.885 ng/ml, P = 0.019 for IL-10; 7258.105 ± 687.912 ng/ml vs. 5895.036 ± 365.716 ng/ml, P = 0.004 for TGF-β1; Fig. 4a,e,g). No difference was observed between the cases and controls in the levels of IFN-γ, IL-17A and IL-22 (Fig. 4c,i,k). Furthermore, individuals with −129TT/CT genotypes had greatly enhanced IL-10 expression compared with the group with −129CC genotype (7.118 ± 1.869 ng/ml vs. 25.719 ± 13.565 ng/ml, P = 0.023; Fig. 4f). However, the AHR −129C > T polymorphism seems not to be related to other cytokines (Fig. 4b,d,h,j,l). The evidence supports that the AHR −129C > T polymorphism might be associated with IL-10 production.
Figure 4. Impacts of the AHR −129C > T polymorphism on serum cytokine levels in vitiligo.

The amounts of TNF-α (a), IFN-γ (c), IL-10 (e), TGF-β1 (g), IL-17A (i) and IL-22 (k) in the serum from vitiligo patients and controls were determined by ELISA. To compare cytokine expressions between −129CC risk genotype group and −129CT + TT protective genotype group, genetic analysis data were acquired from cases and controls together (b,d,f,h,j and l). The y axis is in log10 scale.
Discussion
In the current study, we investigated the molecular mechanisms of AHR functional variation (−129C > T polymorphism) underlying its association with vitiligo. Our data demonstrated that the protective T allele of this polymorphism dramatically enhanced AHR promoter activity through promoting the binding activity of SP1. We also found decreased AHR transcript expression in the PBMCs of vitiligo patients, which was negatively correlated with disease duration. Further genetic analysis showed that −129T carriers had higher AHR expression than −129C carriers. We further detected AHR related cytokines in vitiligo patients and observed declined serum IL-10 level and its association with the AHR −129C > T variant. Thus, our scientific evidence strengthens our previous finding17 and further clarifies that the AHR −129C > T polymorphism is indeed a functional genetic marker for vitiligo.
In this report, we first proved the contribution of −129C > T variation to AHR promoter function, which might influence AHR transcription by altering its interaction with transcriptional regulators. Human AHR gene contains features of a housekeeping gene as possessing GC-rich regions31. The core promoter region (−1 ∼ −250) has a GC content of 70%, containing several putative binding sites for SP132. Deletion of the SP1 recognition site reduced basal promoter activity of human AHR gene to 20%21. In addition, impaired interaction between SP1 and AHR promoter caused by SP1 inhibitor or AHR hypermethylation led to AHR downregulation22,23,33. Consistent with previous study, we also found that SP1 inhibitor down-regulated AHR expression in PIG1 and LiBr cells. These results suggest that the SP1-binding motif is sufficient for maximal constitutive expression of AHR genes. Given the pivotal role of SP1 in AHR expression and AHR −129C > T variation located nearby the core binding site for SP1, we then performed EMSA and ChIP assays and observed the −129T allele with higher SP1 binding affinity than the −129C allele. Li D. et al. previously reported that the −129C > T variant interfered the binding activity of nuclear factor 1-C, which might down-regulate AHR transcription via antagonizing SP1 occupancy34. Although the mutual effect of SP1 and other regulators on AHR transcription remains unclear, it is conceivable that the AHR −129C > T variant may affect the binding of SP1 to the AHR promoter in a direct or indirect manner.
The association between AHR expression and vitiligo remains to be elucidated. Gene expression analysis of PBMCs from independent cases and controls showed apparently decreased AHR mRNA level in vitiligo, which was clearly negatively correlated with disease duration of vitiligo patients, suggesting that down-regulation of AHR transcription is relative to vitiligo. We also found that declined SP1 mRNA level in vitiligo patients and a strongly positive correlation between SP1 and AHR expressions. It has been reported that the expression levels of SP1 and AHR vary greatly among different tissues21, but highest mRNA levels of SP1 and AHR were observed in the same tissues, such as thymus, lung, and spleen35,36. Differential SP1 expression and abnormal SP1 binding activity were demonstrated to mediate reduced expressions of AHR and its target genes in human breast cancer cells22,37. In order to investigate whether the regulatory role of −129C > T mutation in SP1 binding affinity and AHR promoter activity could eventually influence AHR transcript expression, we performed genetic analysis showing that the protective −129T allele is associated with elevated expression of AHR mRNA. Both in-vitro and in-vivo transcriptional analyses provided consistent results, indicating the potential effect of −129C > T mutation on AHR expression, which might in turn influence its function in regulating melanogenic factors including TYR, TYRP, SCF and C-Kit14,15. Preliminary studies have implicated that abnormalities of TYR and TYRP, such as genetic mutation, decreased expression, altered folding or maturation and aberrant retention, may lead to increased toxic metabolites and subsequent melanocyte apoptosis in vitiligo38,39. TYRP could prevent premature melanocyte death in animals39. Moreover, Mutation of SCF or C-Kit caused hair hypopigmentation in mice40, and aberrant SCF/C-Kit pathway contributed to vitiligo and piebaldism in human41,42,43,44. SCF could protect primary human melanocytes against apoptosis and promote repigmentation in vitiligo mouse model45,46. Accordingly, it could be speculate that the AHR −129C > T polymorphism might be associated with vitiligo through influencing AHR expression and its downstream melanogenic factors.
To further understand another significance of AHR −129C > T polymorphism, we assessed the impact of this functional variant on the immune cytokines in vitiligo. Consistent with previous findings47,48, we found increased TNF-α concentration and declined levels of IL-10 and TGF-β1 in the serum of vitiligo patients. Further genetic analysis showed that IL-10 was strikingly higher in subjects carrying AHR −129T allele than −129C allele. Recent studies have demonstrated that AHR could affect IL-10 expression via mediating the differentiation of Treg cells49,50. AHR activation promotes Treg cells development by diverse mechanisms, including transcription regulation13, epigenetic modification51, cooperation with transcription factors Smad1 (SMAD family member 1) and Aiolos (IKAROS family zinc finger 3)50, inhibition of IL-252 and limitation of STAT1 (signal transducer and activator of transcription 1) activation53. These Treg cells induced by AHR pathway were shown to ameliorate autoimmune diseases such as type I diabetes54, multiple sclerosis13, colitis51 and graft versus host disease55. Further, naive T cells from Ahr null mice inefficiently generated Tregs with decreased IL-10 levels53,56, indicating a specific role of AHR in Treg generation and IL-10 production. Recently, abnormal Tregs were identified in vitiligo, with evidence of reduced number, impaired skin homing, defective function and declined IL-10 expression48,57,58,59. Consequently, our data of genetic analysis imply that the AHR −129C > T mutation might be related to vitiligo partially by affecting IL-10 production, which further supports previous findings about AHR function in immune regulation. Nevertheless, immune system contains many biological structures and complex processes, which could be modulated by mutual effort of multiple factors including AHR. Thus, functional studies may be needed to explore the regulatory mechanisms of AHR pathway in the immune pathology of vitiligo further.
There are several limitations within our study. First, owing to absence of melanocytes in vitligo epidermis and Koebner phenomenon exiting in vitiligo, it is hard to acquire primary melanocytes or tissue samples from vitiligo patients and we therefore used PBMCs instead of melanocytes to confirm these findings. Second, multiple genes and various environmental factors make collaborative contribution to vitiligo. These highlight the need to explore the gene-gene or gene-environment interaction in vitiligo in our future genetic studies.
In summary, we provide evidence that AHR –129C > T variant could lead to allele-specific binding of SP1 to AHR promoter and further modify its transcription and downstream effectors in vitiligo. Therefore, our findings have not only advanced our understanding on AHR physiological function but also provided significant insight into the pathogenesis of degenerative disorders or autoimmune diseases including vitiligo.
Methods
In silico AHR promoter analysis
ORChID (dna.bu.edu/orchid) was used to predict the hydroxyl radical cleavage pattern of local variation in solvent-accessible surface area of duplex DNA, and provided information on the local structural DNA profiles from nucleotides sequence containing the AHR −129C > T polymorphism.
Cell culture
Human normal melanocyte PIG1 cell line (a gift from Dr Caroline Le Poole, Loyola University Chicago, Maywood, IL, USA) was cultured in Medium 254 (Cascade Biologics/Invitrogen, Portland, OR, USA) supplemented with Human Melanocyte Growth Supplement (Cascade Biologics/Invitrogen, Portland, OR, USA) and 5% Fetal Bovine Serum (FBS; Invitrogen, Carlsbad, CA, USA). Human embryonic kidney 293T cell line and human malignant melanoma LiBr cell line were maintained in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS. For SP1 inhibition, PIG1 and LiBr cells were treated with 100 nM chemical inhibitor mithramycin A (Enzo Life Sciences, Farmingdale, NY) or control vehicle DMSO (Sigma-Aldrich Corp., St. Louis, MO) for 48 h.
Blood samples
For assays of gene expression, 23 patients with vitiligo and 23 sex- and age-matched control subjects were enrolled in the present study. We obtained 10 ml of peripheral blood from each participant for PBMCs isolation as described58. For cytokines measurement, we used 4 ml of peripheral blood from 16 vitiligo patients and 16 sex- and age-matched health subjects for serum collection. The detailed methods of recruiting subjects and genotyping of AHR −129C > T variant have been described previously17. In particular, information on demographics and other characteristics, including disease duration, onset age and BSA, was obtained using questionnaires (details shown in Supplementary Table S1 online). All the subjects gave written informed consent. The study was approved by the ethics review board of the Fourth Military Medical University and was conducted according to the Declaration of Helsinki Principles.
Construction of reporter plasmids
A 534-bp DNA fragment (−534 to −1) of human AHR promoter region was directly synthesized with added KpnI and MluI sites at 5′ ends in forward and reverse sequences, respectively (Cnservice Invitrogen, Shanghai, China). The DNA sequence was inserted between KpnI and MluI sites in the firefly luciferase reporter vector pGL3-Basic (Promega, Madison, WI, USA). The −129C > T polymorphism was introduced into the recombinant pGL3 reporter vector by performing site-directed mutagenesis. The sequence of constructs was confirmed by direct DNA sequencing.
Transient transfections and dual-luciferase assay
For transfections, PIG1, LiBr and 293T cells were plated onto 24-well plates, respectively. The cells were transfected with 0.8 μg of the recombinant pGL3 reporter vector with either −129C or −129T allele using Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA). The pGL3-Basic vector without an insert and pGL3-Control vector (Promega, Madison, WI, USA) were used as a negative control and a positive control, respectively. All plasmids were cotransfected with 0.02 μg of pRL-SV40 vector (Promega, Madison, WI, USA) as an internal standard. At 48 h after transfection, cells were collected and luciferase activity was measured with Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s protocols. Luciferase activity was normalized against the activity of pRL-SV40 with the Renilla luciferase gene. Independent triplicate experiments were done for each plasmid construct.
EMSA assay
The DNA-protein interactions were detected using LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The biotin labeled and unlabeled double-stranded oligonucleotides corresponding to a 30-bp sequence 5′-BIO-AAGAC/TGGAATGGAATCCAGATGGGCGGGGG-3′ (SP1 binding site underlined) were synthesized by Viagene Biotech Inc (Viagene Biotech Inc, Tampa, Florida, USA). Nuclear extracts (10 μg) were isolated from PIG1 cells using Viagene Subcellular Protein Fraction Kit (Viagene Biotech Inc, Tampa, Florida, USA) and protein concentration was determined by BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA). For each gel shift reaction (15 μl), a total of 300 fmol labeled probes were combined with 10 μg of nuclear extract, 1.5 μg of poly (deoxyinosinic-deoxycytidylic acid) and 10 × binding buffer. For competition assays, a 100-fold molar excess of unlabeled either −129C or −129T probes was preincubated for 20 min at room temperature with nuclear extracts before addition of the labeled probes. For each supershift reaction (15 μl), 3.5 μl of anti-SP1 antibody (Abcam Inc., Cambridge, MA, USA) was incubated with nuclear extracts at 4 °C for 15 min, followed by an additional incubation for 20 min at room temperature with the labeled probes. After incubation, samples were separated on a native non-denaturing 6.5% polyacrylamide gel and then transferred to a nylon membrane. The positions of biotin-labeled probes in the membrane were detected by a chemiluminescent reaction with the stabilized streptavidin– horseradish peroxidase conjugate according to the manufacturer’s instructions and visualized by autoradiography.
ChIP assay
Before ChIP assay, PIG1 cells and LiBr cells were genotyped and sequenced to be CC and TT homozygous, respectively, which were performed similarly to our previous procedures (see Supplementary Fig. S1 online)17. ChIP assays were performed with ChIP-IT Express Kit (Active Motif, Carlsbad, USA) according to the manufacturer’s protocols. Briefly, 1 × 106 of PIG1 cells and LiBr cells were treated with 2.7% formaldehyde for 10 min, followed by 2.5 M glycine stop solution. Cells were then collected and centrifuged at 4 °C and resuspended in lysis buffer for 30 min. The extracts were sonicated, and the DNA was sheared using enzymatic shearing cocktail until the DNA fragments were 200 ∼ 500-bp in size. Sheared chromatin (10 μl) was incubated with protein G magnetic beads and 3 μg of rabbit polyclonal anti-SP1 antibody (Abcam Inc., Cambridge, MA, USA), rabbit control IgG (Santa Cruz Biotechnology) or rabbit polyclonal anti-RNA polymerase II antibody (Abcam Inc., Cambridge, MA, USA) overnight at 4 °C. After extensive washing of the beads, the samples were reverse cross-linked and treated with ribonuclease A and proteinase K. After DNA purification, the immunoprecipitated chromatin fraction was amplified by PCR with primers specific for the AHR promoter (see Supplementary Table S2 online) and control primers for the GAPDH promoter. The PCR products were analyzed on a 2.0% agarose gel.
Real-time quantitative RT-PCR
The total RNA was extracted from the PBMCs of vitiligo patients and controls with Trizol Reagent (Invitrogen, Inc.) and then converted to cDNA using PrimeScriptTM RT Master Mix (TaKaRa, Inc.). The mRNA levels of AHR, SP1 and ACTB were measured by real-time quantitative PCR using specific primers (see Supplementary Table S2 online). Fold changes were normalized by the level of ACTB expression, and each assay was done in triplicate.
Western blotting
SP1 and AHR protein expressions in PIG1 and LiBr cells were examined by Western blots analysis. Western blots (50 μg protein/lane) were performed as described previously30, and probed with monoclonal antibodies specific for AHR or SP1 proteins (Abcam Inc., Cambridge, MA, USA). Hybridization signals for AHR and SP1 were normalized to hybridization signal for β-actin (Abcam Inc., Cambridge, MA, USA).
Measurement of cytokine concentrations
The amounts of TNF-α, IFN-γ, IL-10, TGF-β1, IL-17A and IL-22 in the serum from each participant were determined using a corresponding ELISA kit (R&D systems, Bender MedSystems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Statistical analysis
Student’s t test was performed to evaluate the difference in luciferase activities between different constructs. Unpaired t test with Welch’s correction was used to assess the different DNA-protein binding affinities between −129T and −129C probes. Non-parametric Mann–Whitney U-test was applied to analyze the results of peripheral AHR and SP1 mRNA expressions and serum cytokine concentrations between vitiligo patients and controls with different AHR −129 genotypes. Correlation studies were performed using Pearson’s correlation test. All tests were two-sided by using SPSS software (Version 13.0, SPSS Inc., Chicago, USA). Statistical significance was defined as P < 0.05.
Additional Information
How to cite this article: Wang, X. et al. AHR promoter variant modulates its transcription and downstream effectors by allele-specific AHR-SP1 interaction functioning as a genetic marker for vitiligo. Sci. Rep. 5, 13542; doi: 10.1038/srep13542 (2015).
Supplementary Material
Acknowledgments
We thank Caroline Le Poole for providing the PIG1 immortalized human epidermal melanocyte cell line. This work was supported by the National Natural Science Foundation of China (No. 81130032 and No. 81172749).
Footnotes
Author Contributions All authors contributed significantly to this work. X.W., K.L. and L.L. conceived and designed the experiments. X.W., P.S. and S.G. performed the experiments. X.W., K.L. and L.L. analyzed the data. Q.S., Z.J. and G.W. contributed reagents/materials/analysis tools. X.W., C.L. and T.G. contributed to the writing of the manuscript. All authors reviewed the manuscript and approved the final draft.
References
- Ezzedine K., Eleftheriadou V., Whitton M. & van Geel N. Vitiligo. Lancet 10–1016 (2015). [DOI] [PubMed] [Google Scholar]
- Alikhan A., Felsten L. M., Daly M. & Petronic-Rosic V. Vitiligo: a comprehensive overview Part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J Am Acad Dermatol 65, 473–491 (2011). [DOI] [PubMed] [Google Scholar]
- Richmond J. M., Frisoli M. L. & Harris J. E. Innate immune mechanisms in vitiligo: danger from within. Curr Opin Immunol 25, 676–682 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura R. et al. Mechanisms underlying the dysfunction of melanocytes in vitiligo epidermis: role of SCF/KIT protein interactions and the downstream effector, MITF-M. J Pathol 202, 463–475 (2004). [DOI] [PubMed] [Google Scholar]
- Lee A. Y., Kim N. H., Choi W. I. & Youm Y. H. Less keratinocyte-derived factors related to more keratinocyte apoptosis in depigmented than normally pigmented suction-blistered epidermis may cause passive melanocyte death in vitiligo. J Invest Dermatol 124, 976–983 (2005). [DOI] [PubMed] [Google Scholar]
- Laddha N. C. et al. Vitiligo: interplay between oxidative stress and immune system. Exp Dermatol 22, 245–250 (2013). [DOI] [PubMed] [Google Scholar]
- Boissy R. E. & Spritz R. A. Frontiers and controversies in the pathobiology of vitiligo: separating the wheat from the chaff. Exp Dermatol 18, 583–585 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney J. A. & Rosen A. Apoptosis and autoimmunity. Curr Opin Immunol 17, 583–588 (2005). [DOI] [PubMed] [Google Scholar]
- Stromberg S. et al. Transcriptional profiling of melanocytes from patients with vitiligo vulgaris. Pigment Cell Melanoma Res 21, 162–171 (2008). [DOI] [PubMed] [Google Scholar]
- Schmidt J. V. & Bradfield C. A. Ah receptor signaling pathways. Annu Rev Cell Dev Biol 12, 55–89 (1996). [DOI] [PubMed] [Google Scholar]
- Burbach K. M., Poland A. & Bradfield C. A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci USA 89, 8185–8189 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigent L. et al. The aryl hydrocarbon receptor is functionally upregulated early in the course of human T-cell activation. Eur J Immunol 44, 1330–1340 (2014). [DOI] [PubMed] [Google Scholar]
- Quintana F. J. et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71 (2008). [DOI] [PubMed] [Google Scholar]
- Luecke S. et al. The aryl hydrocarbon receptor (AHR), a novel regulator of human melanogenesis. Pigment Cell Melanoma Res 23, 828–833 (2010). [DOI] [PubMed] [Google Scholar]
- Jux B. et al. The aryl hydrocarbon receptor mediates UVB radiation-induced skin tanning. J Invest Dermatol 131, 203–210 (2011). [DOI] [PubMed] [Google Scholar]
- Schallreuter K. U. et al. Blunted epidermal L-tryptophan metabolism in vitiligo affects immune response and ROS scavenging by Fenton chemistry, part 2: Epidermal H2O2/ONOO(-)-mediated stress in vitiligo hampers indoleamine 2,3-dioxygenase and aryl hydrocarbon receptor-mediated immune response signaling. Faseb J 26, 2471–2485 (2012). [DOI] [PubMed] [Google Scholar]
- Wang X. W. et al. The association of functional polymorphisms in the aryl hydrocarbon receptor (AHR) gene with the risk of vitiligo in Han Chinese populations. Br J Dermatol 166, 1081–1087 (2012). [DOI] [PubMed] [Google Scholar]
- Eguchi H., Hayashi S., Watanabe J., Gotoh O. & Kawajiri K. Molecular cloning of the human AH receptor gene promoter. Biochem Biophys Res Commun 203, 615–622 (1994). [DOI] [PubMed] [Google Scholar]
- Kadonaga J. T., Courey A. J., Ladika J. & Tjian R. Distinct regions of Sp1 modulate DNA binding and transcriptional activation. Science 242, 1566–1570 (1988). [DOI] [PubMed] [Google Scholar]
- Tan N. Y. & Khachigian L. M. Sp1 phosphorylation and its regulation of gene transcription. Mol Cell Biol 29, 2483–2488 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racky J., Schmitz H. J., Kauffmann H. M. & Schrenk D. Single nucleotide polymorphism analysis and functional characterization of the human Ah receptor (AhR) gene promoter. Arch Biochem Biophys 421, 91–98 (2004). [DOI] [PubMed] [Google Scholar]
- Englert N. A. et al. Genetic and epigenetic regulation of AHR gene expression in MCF-7 breast cancer cells: role of the proximal promoter GC-rich region. Biochem Pharmacol 84, 722–735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulero-Navarro S. et al. The dioxin receptor is silenced by promoter hypermethylation in human acute lymphoblastic leukemia through inhibition of Sp1 binding. Carcinogenesis 27, 1099–1104 (2006). [DOI] [PubMed] [Google Scholar]
- Rohs R. et al. Origins of specificity in protein-DNA recognition. Annu Rev Biochem 79, 233–269 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella S., Cascio D. & Johnson R. C. The shape of the DNA minor groove directs binding by the DNA-bending protein Fis. Genes Dev 24, 814–826 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum J. A., Pang B. & Tullius T. D. Construction of a genome-scale structural map at single-nucleotide resolution. Genome Res 17, 947–953 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop E. P. et al. A map of minor groove shape and electrostatic potential from hydroxyl radical cleavage patterns of DNA. ACS Chem Biol 6, 1314–1320 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi R. et al. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell 131, 530–543 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R. et al. The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T. et al. Overexpression of antioxidant enzymes upregulates aryl hydrocarbon receptor expression via increased Sp1 DNA-binding activity. Free Radic Biol Med 49, 487–492 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper P. A., Riddick D. S. & Okey A. B. Regulating the regulator: factors that control levels and activity of the aryl hydrocarbon receptor. Biochem Pharmacol 72, 267–279 (2006). [DOI] [PubMed] [Google Scholar]
- Takahashi Y., Itoh S., Shimojima T. & Kamataki T. Characterization of Ah receptor promoter in human liver cell line, HepG2. Pharmacogenetics 4, 219–222 (1994). [DOI] [PubMed] [Google Scholar]
- Lin X., Yang H., Zhou L. & Guo Z. Nrf2-dependent induction of NQO1 in mouse aortic endothelial cells overexpressing catalase. Free Radic Biol Med 51, 97–106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. et al. Inhibition of AHR transcription by NF1C is affected by a single-nucleotide polymorphism, and is involved in suppression of human uterine endometrial cancer. Oncogene 32, 4950–4959 (2013). [DOI] [PubMed] [Google Scholar]
- Saffer J. D., Jackson S. P. & Annarella M. B. Developmental expression of Sp1 in the mouse. Mol Cell Biol 11, 2189–2199 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., Maltepe E., Lu M. M., Simon C. & Bradfield C. A. Expression of ARNT, ARNT2, HIF1 alpha, HIF2 alpha and Ah receptor mRNAs in the developing mouse. Mech Dev 73, 117–123 (1998). [DOI] [PubMed] [Google Scholar]
- Do M. T. et al. Metformin suppresses CYP1A1 and CYP1B1 expression in breast cancer cells by down-regulating aryl hydrocarbon receptor expression. Toxicol Appl Pharmacol 280, 138–148 (2014). [DOI] [PubMed] [Google Scholar]
- Jin Y. et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N Engl J Med 362, 1686–1697 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimbow K., Chen H., Park J. S. & Thomas P. D. Increased sensitivity of melanocytes to oxidative stress and abnormal expression of tyrosinase-related protein in vitiligo. Br J Dermatol 144, 55–65 (2001). [DOI] [PubMed] [Google Scholar]
- Funasaka Y. et al. c-Kit-kinase induces a cascade of protein tyrosine phosphorylation in normal human melanocytes in response to mast cell growth factor and stimulates mitogen-activated protein kinase but is down-regulated in melanomas. Mol Biol Cell 3, 197–209 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S. et al. In utero manipulation of coat color formation by a monoclonal anti-c-kit antibody: two distinct waves of c-kit-dependency during melanocyte development. Embo J 10, 2111–2118 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischman R. A., Saltman D. L., Stastny V. & Zneimer S. Deletion of the c-kit protooncogene in the human developmental defect piebald trait. Proc Natl Acad Sci USA 88, 10885–10889 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel L. B. & Spritz R. A. Mutation of the KIT (mast/stem cell growth factor receptor) protooncogene in human piebaldism. Proc Natl Acad Sci USA 88, 8696–8699 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros L., Cassuto J. P. & Ortonne J. P. Imatinib mesilate (Glivec): a systemic depigmenting agent for extensive vitiligo? Br J Dermatol 153, 691–692 (2005). [DOI] [PubMed] [Google Scholar]
- Larribere L. et al. PI3K mediates protection against TRAIL-induced apoptosis in primary human melanocytes. Cell Death Differ 11, 1084–1091 (2004). [DOI] [PubMed] [Google Scholar]
- Eby J. M. et al. Immune responses in a mouse model of vitiligo with spontaneous epidermal de- and repigmentation. Pigment Cell Melanoma Res 27, 1075–1085 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak P. Y., Adiloglu A. K., Ceyhan A. M., Tas T. & Akkaya V. B. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J Am Acad Dermatol 60, 256–260 (2009). [DOI] [PubMed] [Google Scholar]
- Dwivedi M. et al. Regulatory T cells in vitiligo: Implications for pathogenesis and therapeutics. Autoimmun Rev 14, 49–56 (2015). [DOI] [PubMed] [Google Scholar]
- Wang C., Ye Z., Kijlstra A., Zhou Y. & Yang P. Activation of the aryl hydrocarbon receptor affects activation and function of human monocyte-derived dendritic cells. Clin Exp Immunol 177, 521–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi R. et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol 11, 846–853 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N. P. et al. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS One 6, e23522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana F. J. & Sherr D. H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev 65, 1148–1161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A., Naka T., Nohara K., Fujii-Kuriyama Y. & Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci USA 105, 9721–9726 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkvliet N. I. et al. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3 + T cells in pancreatic lymph nodes. Immunotherapy 1, 539–547 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funatake C. J., Marshall N. B., Steppan L. B., Mourich D. V. & Kerkvliet N. I. Cutting edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4 + CD25 + cells with characteristics of regulatory T cells. J Immunol 175, 4184–4188 (2005). [DOI] [PubMed] [Google Scholar]
- Elizondo G., Rodriguez-Sosa M., Estrada-Muniz E., Gonzalez F. J. & Vega L. Deletion of the aryl hydrocarbon receptor enhances the inflammatory response to Leishmania major infection. Int J Biol Sci 7, 1220–1229 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarquist J. et al. Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res 23, 276–286 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben, Ahmed M. et al. Functional defects of peripheral regulatory T lymphocytes in patients with progressive vitiligo. Pigment Cell Melanoma Res 25, 99–109 (2012). [DOI] [PubMed] [Google Scholar]
- Dwivedi M., Laddha N. C., Arora P., Marfatia Y. S. & Begum R. Decreased regulatory T-cells and CD4(+)/CD8(+) ratio correlate with disease onset and progression in patients with generalized vitiligo. Pigment Cell Melanoma Res 26, 586–591 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.